

Abstract
The enhanced release of reactive oxygen species by excessively activated polymorphonuclear leucocytes (PMN) is a key step in the pathogenesis of sepsis. Potent action of adenosine in inhibiting cytotoxic PMN functions has been documented. Recent data, however provide evidence that in sepsis a diminished capability of adenosine to inhibit the generation of oxygen radicals by PMN occurs. Here, we investigated the underlying mechanisms in an in vitro sepsis model and in PMN of sepsis patients. We report that lipopolysaccharide (LPS)-incubation of human PMN elicited the same increase in the half-maximal inhibitory concentration (IC50) of adenosine as observed in patients with septic shock. Coupling to adenylyl cyclase was impaired as well, as indicated by a decreased potency of adenosine to stimulate cyclic adenosine monophosphate (cAMP) accumulation. Ligand-binding studies conducted with native, LPS-stimulated PMN, and with PMN of sepsis patients revealed that, despite an increased adenosine A2A receptor (A2AR) expression, the receptor function declines due to a diminished ligand-binding affinity most likely caused by allosteric modulators within the inflammatory environment. A2AR function obviously is highly dependent upon the cellular environment and thus, further functional characterization of A2AR responses in sepsis may be a promising approach to develop new adenosine or A2AR agonists based therapeutic strategies.
Keywords: sepsis, human PMN, A2A receptor, ligand affinity
Introduction
Sepsis describes a complex clinical syndrome resulting from excessive and uncontrolled host responses to infection. Many of the components of the innate immune response that are initially recruited to fight infection can, under some circumstances, cause life-threatening cell and tissue damage leading to multiple organ failure, the clinical hallmark of sepsis [1, 2].
Polymorphonuclear leucocytes (PMN) or neutrophils serve as the ‘first line’ of innate defence against infection by eliminating pathogens. They contribute to the pathophysiology of sepsis as they are potent in their antimicrobial defences but non-selective in their targets and fulfil the task of eradicating an infection or responding to local tissue injury at the cost of significant collateral damage [3]. In sepsis, circulating PMN develop a dysfunction syndrome characterized by the impairment of potentially microbicidal functions, whereas potentially tissue toxic functions get enhanced: Spontaneous respiratory burst activity is enhanced in resting PMN and even more after activation with soluble stimuli like fMLP (N-formyl-methionyl-leucyl-phenylalanine) and lipopolysaccharide (LPS), whereas distinct microbicidal functions of PMN like phagocytosis and the associated radical production elicited by unopsonized zymosan particles are suppressed [4, 5].
Adenosine is an endogenous purine nucleoside that is released from metabolically active cells and formed extracellularly by degradation of ATP [6]. Thus, extracellular adenosine accumulates in inflamed and hypoxic tissues [7], and elevated adenosine plasma levels have been detected in patients with sepsis and septic shock [8]. Adenosine is a potent mediator that modulates numerous cell functions and exerts its effects via binding to four subtypes of G-protein coupled adenosine receptors: A1, A2A, A2B and A3[9]. Activation of high-affinity A1 and low-affinity A3 receptor inhibits adenylyl cyclase activity, whereas activation of high-affinity A2A and low-affinity A2B adenosine receptors causes accumulation of intracellular cAMP, which has strong immuno-suppressive effects [10–12]. Tissue hypoxia-associated accumulation of extracellular adenosine and subsequent signalling through the A2A receptors (A2AR) are well known as important mechanisms inhibiting overactive immune cells, limiting inflammation and protecting normal tissues from excessive collateral damage. Consequently, up-regulation of A2A receptors, as shown for PMN of sepsis patients, for human granulocytes in vitro stimulated with LPS or tumour necrosis factor (TNF)-α[13, 14], and for human peripheral blood cells in patients with chronic heart failure [15] might be a physiological mechanism to increase the anti-inflammatory effects of adenosine and to control inflammation. Furthermore, recent findings suggest that in activated human PMN, adenosine, acting via A2A receptors, selectively inhibits the unwanted release of tissue-toxic oxygen radicals without compromising phagocytosis or the phagocytosis-associated production of oxygen radicals. Thus, the adenosine-A2AR-pathway is indispensable to hold the balance between the microbicidal response to infectious agents on the one hand, and the protection of own tissues from an overwhelming inflammatory reaction on the other hand. This has conclusively been shown in mouse models of inflammation: A2AR inactivation has been shown to be detrimental in states of acute hyperinflammation [16], whereas it revealed as beneficial in situations where the inflammatory process is appropriate in order to respond to microbial challenges [17].
In septic patients, the equilibrium between pro- and anti-inflammation obviously is disturbed: Despite an increased expression of A2A receptor mRNA, the favourable tissue protective effects of adenosine diminish, as the dose–response curves for the effect of adenosine shift towards increased IC50 values, which means higher plasma adenosine concentrations are necessary to achieve a comparable inhibition of the toxic H2O2 release. Furthermore, the maximum inhibition decreases [8]. These findings indicate a diminished anti-inflammatory potency of adenosine in sepsis that may contribute to the observed PMN dysfunction syndrome, thereby enhancing inflammatory reactions and facilitating further inflammatory tissue damage.
We hypothesized that an A2A receptor dysfunction might account for the decreased anti-inflammatory action of adenosine in septic patients. In this study we investigated these issues in an in vitro model comparing native to LPS-stimulated human granulocytes. Furthermore, we confirmed the clinical impact of our in vitro data by investigating the detected changes in A2AR para meters in PMN of septic patients compared to healthy volunteers. Our results indicate that in sepsis, the up-regulation of A2A receptors as a physiological mechanism to control inflammation is unaffected, whereas the A2A-receptor affinity is significantly decreased, most likely due to allosteric effects arising in the highly inflamed environment. This leads to the observed diminished anti-inflammatory potency of adenosine promoting the further exacerbation of septic processes.
Materials and methods
Patients
The study was approved by the University of Munich Institutional Ethics Committee and informed consent for blood withdrawal was obtained from healthy volunteers, from patients or their relatives, respectively. Patients were considered eligible for this study if they met the criteria for severe sepsis or septic shock as defined by the members of the American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference Committee [18]. Healthy volunteers of comparable age and sex served as controls.
Materials
Fluorescence-activated cell sorting (FACS) Lysing Solution was obtained from Becton Dickinson (Heidelberg, Germany). Hanks buffered salt solution (HBSS) was manufactured from soluble ingredients by the hospital's own pharmacy. Dihydrorhodamine 123 (DHR 123) was obtained from MoBiTec GmbH (Göttingen, Germany). Hydrochloric acid (HCL) was bought from Merck GmbH (Darmstadt, Germany). The following agents were purchased from Sigma-Aldrich GmbH (Taufkirchen, Germany): Ficoll-Histopaque, fMLP, LPS (E. coli, EO55:B5), and the specific A2AR-agonist 2-[p-(2-carboxyethyl)-phenetyl-amino]-5′-N-ethylcarboxamidoadenosine (CGS 21680). The radiolabelled A2AR-antagonist 4-(2-[7-amino-2-(2-furyl)-[1 2 4]-traizolo-[2 3-a]-[1 3 5]-triazin-5-ylamino]ethyl)phenol) ([3H] ZM 241385, 27.4 Ci/mmol) was purchased from American Radiolabeled Chemicals, Inc. (Saint Louis, MO, USA). PCR primers were synthesized by MWG Biotech (Ebersberg, Germany).
Preparation of blood peripheral neutrophils
Granulocytes were separated from whole blood of sepsis patients and from healthy volunteers as previously described [19] using a continuous Percoll density gradient. PMN leucocytes were harvested and washed with Hanks’ buffered salt solution number of cells was counted by a Coulter Counter® T540 (Coulter Electronics, Hialeah, FL, USA).
Determination of adenosine concentrations
Concentrations of the purine nucleoside adenosine were analysed by dual-column switching high-affinity performance/reversed phase high-performance liquid chromatography technique using an internal surface nitrophenylboronic acid precolumn as previously described [8].
Cell stimulation
Cells were cultured in RPMI-1640 medium (Biofluids, Rockville, MD, USA) supplemented with 10% (v/v) foetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate and 1 mM Hepes in 12-well plates at a density of 5 × 106 cells/well. For stimulation, one of the following factors was added to each well: 10 μg/ml LPS, 100 pg/ml IL-1, 500 pg/ml TNF-α, 15 ng/mg IFN-γ and 5 ng/ml IL-8. After incubation for 6 hrs, cells were harvested by centrifugation and washed twice with HBSS.
Preparation of neutrophil membranes
Human neutrophils were suspended in 1.5-ml Tris buffer (50 mM Tris HCl pH 7.4 containing 10 mM MgCl2), sonicated on ice for 2 × 15 sec. at minimum power, and centrifuged at 800 ×g for 5 min. at 4°C. The cell pellet was resuspended in 1.5-ml Tris buffer, sonicated and centrifuged a second time. Supernatants were collected and centrifuged at 20,000 ×g for 30 min. at 4°C. The resulting pellet was suspended again at a concentration of 150–180 μg protein/100 μl and this homogenate was used for the binding assays. Protein concentrations were determined by Bradford protein assay (Bio-Rad Laboratories, München, Germany) with bovine albumin as reference standard.
RNA isolation and cDNA RT
Total cellular RNA was isolated from native and LPS-treated granulocytes using the RNaequous Kit (Ambion, Austin, TX, USA) with subsequent DNAse treatment (TURBO DNase, Ambion) following the manufacturer's protocol. RNA was quantified using a spectrophotometer, and 1000 ng from each samples were transcribed into cDNA. The RT reaction was carried out using mixed random and oligo-dT primers and Superscript III RT (Invitrogen, Carlsbad, CA, USA), as per manufacturer's instructions.
RT-PCR
RT-PCR was performed using the primer pairs listed in Table 1. The PCR was performed under the following conditions: 94°C for 5 min. denaturing, 35 cycles of 94°C for 30 sec., 58°C for 30 sec., 72°C for 45 sec. and a final extension at 72°C for 10 min. The PCR products were separated on 2% agarose gels and stained with ethidium bromide.
1.
Influence of inflammatory mediators on vitality and on spontaneous and fMLP-stimulated H2O2 production of human PMN: spontaneous and fMLP-stimulated oxidative activity, dead cells after stimulation and number of independent experiments
| Spontaneous activity [MFI] | fMLP [MFI] | PI-positive PMN [%] | n | |
|---|---|---|---|---|
| Control | 31 (± 0.85) | 59 (± 0.85) | 1.52 (± 0.14) | 31 |
| LPS | 33 (± 2.32) | 65 (± 4.88) | 3.91 (± 0.50) | 14 |
| TNF-α | 34 (± 1.51) | 59 (± 4.39) | 2.89 (± 0.39) | 9 |
| IFN-γ | 36 (± 2.37) | 65 (± 4.70) | 2.76 (± 0.37) | 7 |
| IL-1 | 33 (± 1.61) | 62 (± 3.37) | 1.77 (± 0.18) | 12 |
| IL-8 | 30 (± 1.76) | 55 (± 5.57) | 1.74 (± 0.36) | 8 |
Human PMN were incubated in culture medium with different cytokines or without stimulation (control) for 6 hrs. Spontaneous and fMLP (10−7 mol/l)-stimulated H2O2 production and proportion of propidium iodide (PI)-positive cells were assessed by flow cytometric analysis. Data represent the mean (± S.E.M.) of n independent experiments, MFI: mean fluorescence intensity
Determination of fMLP-induced production of hydrogen peroxide in human neutrophils
Isolated PMN (1 × 105) were incubated with DHR 123 (1 μM) in the presence or absence of different concentrations of adenosine (10−12 to 10−5 M) in 1 ml of HBSS at 37°C for 5 min. Then PMN were stimulated with fMLP (10−7 M). After 15 min. activation of cells was stopped by putting tubes on ice. Production of H2O2 was determined by flow cytometry using a Becton Dickinson FACScan (Becton Dickinson, San Jose, CA, USA) equipped with an argon laser emitting light at 488 nm, as previously described [5]. Dead cells were excluded from analyses after identification by propidium iodide staining (3 × 10−5 M).
Measurement of cyclic AMP levels in human neutrophils
Human neutrophils (1 × 106 cells) were suspended in 0.9 ml HBSS, containing 0.5 mM Ro 20–1724 as phosphodiesterase inhibitor and, for the CGS 21680 experiments, 1.0 IU adenosine deaminase/ml and were pre-incubated for 5 min. in a shaking water bath at 37°C. Then eight different concentrations of adenosine or CGS 21680 were added to the mixtures and incubations continued for a further 5 min. Hereafter, fMLP in a concentration of 10−7 M was added and the incubations were continued for another 2 min. The reaction was stopped by the addition of 12.5 μl 30% HCl and transferred to −70°C. After thawing and centrifugation at 10,000 g to remove cellular debris, supernatants were assayed for cAMP using a cAMP EIA kit from Biomol (Hamburg, Germany).
[3H]-ZM241385 binding assays
Saturation-binding experiments were performed by incubating PMN membrane preparations with 12–14 different concentrations of [3H] ZM 241385 ranging from 0.125 to 12 nM. Non-specific binding was determined in the presence of 1 μM of ZM 241385. After 60 min. incubation at 4°C samples were filtered through Whatman GF/B glass fibre filters (Springfield Mill, UK) using a Brandel cell harvester (Gaithersburg, MD, USA). The filter bound radioactivity was counted on a scintillation counter 1219 LKB (efficiency 55%).
Statistical analysis
All data were analysed using SigmaPlot 9.0 and SigmaStat 3.5 software. If not stated otherwise, values given represent means ± S.E.M. Intergroup comparisons were performed by unpaired t-test or Mann-Whitney test, if data were normally or not normally distributed, respectively. Two-tailed levels of statistical significance are indicated by *:P< 0.05.
Results
Down-regulation of the adenosine-induced inhibition of H2O2 production in ex vivo stimulated neutrophils
A reduced anti-inflammatory potency of adenosine in PMN of sepsis patients has been reported previously [5]. Intending to establish an appropriate in vitro model to further investigate these findings, we examined whether the changes in adenosine-mediated effects also occur in neutrophils in vitro incubated with LPS, TNF-α IL-8, IFN-γ and IL-1. Stimulation times and mediator concentrations were tested out previously (for reproducible maximum effects and high cell viability, data not shown). Treatment with these mediators did neither affect the spontaneous nor the fMLP-stimulated H2O2 production of PMN (Table 1).
We determined the effects of increasing adenosine concentrations on fMLP-stimulated H2O2 production in native compared to LPS- or cytokine-treated PMN. As shown in Fig. 1, adenosine inhibited the generation of H2O2 in a dose-dependent manner, reaching maximum inhibitory effects at a concentration of 10−6M in untreated neutrophils, whereas maximum inhibition in LPS-incubated PMN was accomplished at 10−5M. IC50 values changed from 75 to 332 nM in LPS-treated PMN. These results completely agree with the data described for PMN from patients with septic shock. Incubation with TNF-α, IL-8, IFN-γ and IL-1 did not change the effects of adenosine on the H2O2 production significantly (Table 2). To exclude any artefacts provoked by unphysiological adenosine levels potentially arising during cell stimulation, we determined the adenosine concentrations being present in the culture medium alone and in supernatants after incubating PMN with or without LPS. As shown in Fig. 2, cell culture medium itself contained 40-nM adenosine. After incubation for 6 hrs with and without LPS, adenosine concentrations slightly increased to 49 and 70 nM, respectively. The comparison of these adenosine levels with plasma adenosine concentrations found in healthy volunteers (42 nM) and septic patients (120 nM) (we already described in one of our previously published studies [8]) clearly showed that our in vitro model lies within physiologically relevant adenosine ranges.
1.
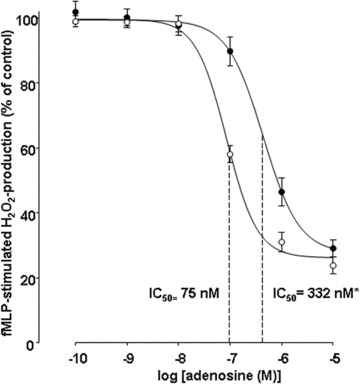
Effect of LPS incubation on the adenosine induced inhibition of the fMLP-stimulated H2O2 production in human PMN. Human PMN were incubated with either culture medium alone (○) or containing 10 μg/ml LPS (•) for 6 hrs. Hereafter, dose–response curves for the inhibition of the fMLP (10T−7 mol/l)-stimulated H2O2 production by increasing concentrations of adenosine were assessed by flow cytometric analysis. Data represent the mean (±S.E.M.) of n= 31 (Control) and n= 14 (LPS) independent experiments (P< 0.0001, one-way anova followed by Student's t-test). Curves were calculated using SigmaPlot 9.0 software. Dashed lines indicate IC50 values.
2.
Influence of inflammatory mediators on the adenosine induced inhibition of fMLP-stimulated H2O2 production in PMN: maximal Inhibitory effects, IC50, and number of independent experiments
| Max. inhibition (6 S.E.M.) [%] | IC50[nM] | n | |
|---|---|---|---|
| Control | 78.8 (± 1.5) | 80 (65–97) | 31 |
| LPS | 75.2 (± 2.5) | 332 (189–584)*** | 14 |
| TNF-α | 84.8 (± 3.6) | 106 (62–179) | 9 |
| IFN-γ | 86.8 (± 4.3) | 123 (72–213) | 7 |
| IL-1 | 78.6 (± 2.5) | 40 (25–64) | 12 |
| IL-8 | 76.3 (± 5.8) | 43 (20–96) | 8 |
Human PMN were incubated in culture medium with different cytokines or without stimulation (control) for 6 hrs. Hereafter, the inhibition of the fMLP (10−7 mol/l)-stimulated H2O2 production by increasing concentrations of adenosine was assessed by flow cytometric analysis. Data represent the mean (± S.E.M.) of n independent experiments (***P< 0.001, one-way anova followed by Student's t-test).
2.
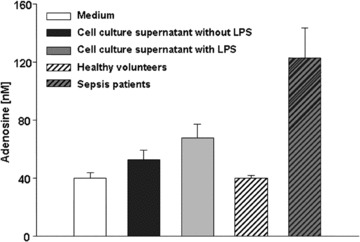
Adenosine concentrations of cell culture supernatants and adenosine plasma levels of healthy volunteers and of patients with septic shock. Human PMN were incubated with either culture medium alone or containing 10 μg/ml LPS for 6 hrs. Hereafter, cells were separated by centrifugation, and cell culture supernatants as well as cell culture medium alone as a control were subjected to high-performance liquid chromatography analysis for determination of adenosine levels. Patients’ plasma levels have been published previously [12]. Values are expressed as mean ± S.E.M.
Down-regulation of the adenosine-induced stimulation of the A2A receptor transduction system in ex vivo stimulated neutrophils
Based on our previously reported findings that A2A receptor mRNA expression increases upon LPS stimulation whereas the expression of the other adenosine receptors does not change significantly [13], we evaluated the stimulatory effect of adenosine and the selective A2AR agonist CGS 21680 on the adenylyl cyclase activity to test for changes in receptor function. In LPS-stimulated neutrophils, adenosine concentration-response curves were significantly shifted to the right with respect to native PMN (Fig. 3a). EC50 values changed from 165 nM in native to 801 nM in LPS-stimulated human granulocytes, indicating a decreased potency of adenosine to stimulate cAMP formation. Furthermore, in LPS-treated PMN the rise of cAMP levels only reached 90% of the maximum cAMP accumulation elicited in native PMN. The use of the selective A2AR agonist CGS 21680 exhibited identical results in stimulating adenylyl cyclase activity: dose–response curves shifted to the right and EC50 values increased from 64 nM to 893 mM after LPS treatment (Fig. 3b), thereby confirming that the differences in stimulatory effects were essentially A2A receptor mediated.
3.
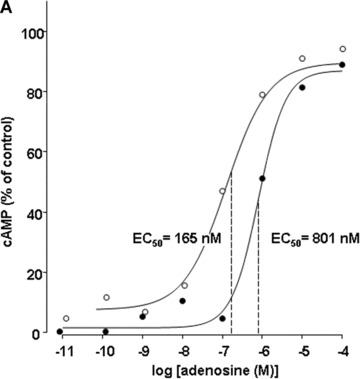
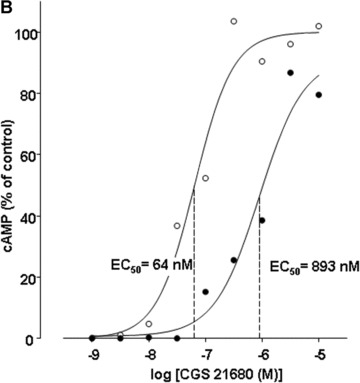
Effect of LPS incubation on stimulation of cyclic AMP levels by adenosine (A) and CGS 21680 (B) in human PMN. Human PMN were incubated with either culture medium alone (○) or with medium containing 10 μg/ml LPS (•) for 6 hrs. Hereafter, adenosine (A) and CGS 21680 (B) concentration-effect curves to stimulate accumulation of cAMP were obtained by ELISA. Basal cAMP values were 18 ± 4 and 21 ± 3 pmoles/106 in native and LPS-stimulated PMN, respectively. Corresponding maximal cAMP levels were determined in the presence of 10−4 M adenosine. Maximal cAMP levels were 85 ± 12 and 76 ± 10 pmoles/106 in native and LPS-stimulated PMN, respectively. Curves are representative of a single experiment measured in triplicates from a series of four independent experiments. Curves were calculated using SigmaPlot 9.0 software. Dashed lines indicate EC50 values.
In ex vivo stimulated neutrophils affinity of the A2A receptor is decreased
These results compelled us to focus on further studies of A2A adenosine receptors. In order to determine the effect of LPS stimulation on their binding parameters, we measured affinity and density of A2A adenosine receptors on human neutrophil membranes using the specific A2AR antagonist [3H] ZM 241385. In native PMN, the affinity (Kd) was 1.02 ± 0.22 nM and the receptor density (Bmax) was 64.2 ± 17.5 fmol/mg protein (Fig. 4a, Table 3). After LPS stimulation, neutrophil membranes exhibited increased Kd (2.48 ± 0.27) and Bmax (87.6 ± 26.9) values, indicating an increase in receptor density but a decrease in receptor affinity (Fig. 4b, Table 3). To exclude effects of the pro-inflammatory A1 receptors, binding parameters of A1 receptors using [3H]DPCPX were determined exhibiting no differences between native and LPS-stimulated PMN (data not shown).
4.
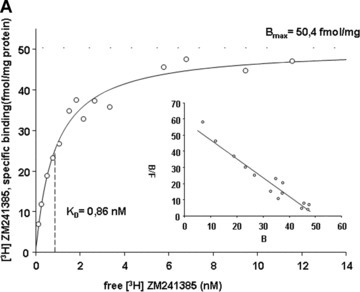
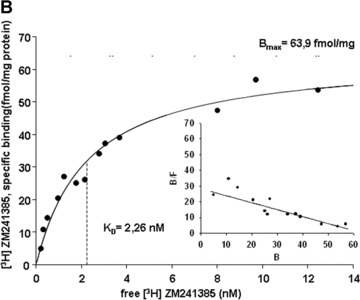
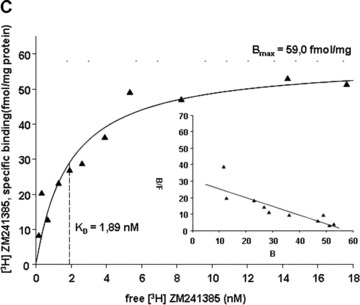
Saturation curves of [3H]-ZM 241385 binding to neutrophil membranes obtained from native (A, ○) and LPS-stimulated PMN (B, •), and for PMN of sepsis patients (C, ▴). Human PMN were incubated with either culture medium alone (A, ○) or with medium containing 10 μg/ml LPS (B, •) for 6 hrs; Membrane fractions from the pre-incubated PMN and from PMN of sepsis patients were prepared and incubated with increasing concentrations of the specific A2A receptor antagonist [3H]-ZM 241385. Non-specific binding was determined in the presence of 1 μM of ZM 241385. In the inset the Scatchard plot of the same data is shown. (B) and (F) denote bound and free ligand, respectively. See Table 3 for a summary of mean Kd and mean Bmax values for native and LPS-stimulated PMN, and for PMN of sepsis patients. Curves are representative of a single experiment measured in triplicates from a series of six independent experiments. Curves were calculated using SigmaPlot 9.0 software. Dashed lines indicate Kd values.
3.
[3H] ZM 241385 binding to neutrophil membranes of native human PMN, LPS-stimulated human PMN and PMN of sepsis patients: mean (±S.E.M.) KD-values, mean (±S.E.M.) Bmax-values and number of independent experiments
| KD [nM] | Bmax[fmol/mg protein] | n | |
|---|---|---|---|
| Control | 1.02 (± 0.22) | 64.2 (± 17.5) | 6 |
| LPS | 2.48 (± 0.27) | 87.6 (± 26.9) | 6 |
| Sepsis | 1.86 (± 0.32) | 70.2 (±15.5) | 6 |
Membrane fractions from the pre-incubated PMN and from PMN of sepsis patients were prepared and incubated with increasing concentrations of the specific A2A receptor antagonist [3H]-ZM 241385. Non-specific binding was determined in the presence of 1 μM of ZM 241385.
In neutrophils of sepsis patients affinity of the A2A receptor is decreased
In concordance with the in vitro data, sepsis patients exhibited an altered binding of [3H] ZM 241385 to their PMN membranes compared to control patients: Kd values were elevated from 1.02 ± 0.22 nM to 1.86 ± 0.31 nM and Bmax values from 64.2 ± 17.5 fmol/mg protein to 70.2 ± 17.3 fmol/mg protein (Fig. 4c, Table 3), indicating that sepsis patients exhibited an increased receptor density and a decreased receptor affinity compared to healthy volunteers.
Changes in the affinity of the A2A receptor are not due to differential expression of splice variants
The human A2AR gene consists of a complex organized 5′-UTR with five non-coding exons followed by two coding exons which are separated by a single intron leading to the translation of a 43-kD protein. We analysed whether so far undetected splice variants may lead to changes in the A2A receptor protein sequence which may account for the observed changes in receptor affinity. Therefore, we performed RT-PCR experiments with primer sets spanning the exon–intron boundaries to check for splice variants (primer pairs are listed in Table 4). All RT-PCR experiments did not result in differences between native and LPS-stimulated cells, and PMN of sepsis patients (Fig. 5).
4.
RT-PCR Primer Sequences to check for A2AR splice variants: sequences, annealing temperatures, expected amplification product lengths and accession number
| Primer name | Sequence | Tm[°C] | Product size [bp] |
|---|---|---|---|
| A2A-splice 3′-For | 5′-GACCGCTACATTGCCATCC-3′ | 58.8 | 298 |
| A2A-splice-3′-Rev | 5′-GCAAATAGACACCCAGCATGA-3′ | 57.9 | |
| A2A-splice 5′-For | 5′-GTGTGGCTCAACAGCAACCT-3′ | 59.4 | 301 |
| A2A-splice 5′-Rev | 5′-ACCCAGCAGATGGCAATGAT-3 | 57.3 |
Accession no. A2AR: NM_000675.
5.
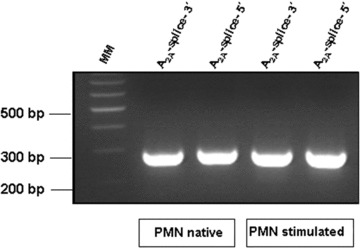
In human PMN the A2aR mRNA expression patterns of the coding region do not change after stimulation with LPS. A2aR transcripts of human PMN were analysed by conventional RT-PCR using transcript-specific primer pairs spanning the exonintron boundaries to check for splice variants. RNA was prepared from both native and LPS-stimulated (10 μg/ml LPS for 6 hrs) human PMN of the same donor, 6 independent experiments with six different donors were performed. The PCR products were resolved on 2% agarose gel and stained with ethidium bromide. A representative agarose gel is shown. MM: Molecular marker, for primer pairs see Table 4.
Discussion
In view of the multitude of immunomodulatory effects of adenosine via occupancy of adenosine receptors [20, 21], expression and regulation of adenosine receptors in PMN of sepsis patients is an important topic with high clinical relevance which has not been analysed conclusively so far. In our previous studies, we have reported observations concerning the adenosine receptors in sepsis patients that, at the first sight, seem to be contradictory: We found a PMN dysfunction syndrome with enhanced tissue toxic functions going along with diminished anti-inflammatory potency of adenosine [8]. Although one would expect a down-regulation of anti-inflammatory A2A receptor expression or an up-regulation of pro-inflammatory A1-receptor expression to account for these phenomena, we found, in fact, an A2AR up-regulation and no changes in A1R expression [13]. The present study was accomplished in order to dissolve these apparently contradictory findings and to characterize the underlying mechanisms. First, by stimulating PMN with LPS, we established an appropriate in vitro model that mimics the diminished anti-inflammatory effects of adenosine found in PMN of sepsis patients. LPS stimulation is a widely used and accepted model to investigate some important aspects out of the multitude of mediators being engaged in the pathomechanisms of sepsis as activation of TLR4 (a member of IL-1/Toll receptor family) by its ligand LPS has been shown to be critical for the regulation of innate immune response [22, 23]. Challenge of PMN with LPS activates several mitogen-activated protein kinase signalling pathways, the extracellular signal- regulated kinase, c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase, which in turn induce gene transcription [24]. LPS also induces the activation of nuclear transcription factors, nuclear factor κB (NF- κB), activator protein-1 (AP-1), PU.1 and interferon regulatory factor-1 in PMN [25, 26].
In the current study, all in vitro stimulation experiments have been performed with LPS concentrations of 10 μg/ml. A saturable dose–response relationship of LPS on the PMN answer has already been pointed out in one of our previously published studies (stable maximum effects from 10 ng/ml up to 10 μg/ml with no adverse effects of high LPS concentrations) [27]. Because LPS physically adheres to the polystyrene material of reaction tubes [28], the μg/ml concentration range was used here in order to guarantee maximum stimulation conditions within the saturation range for all reactions. Moreover, even in vivo a wide range of endotoxin concentrations occur: While average plasma concentrations of endotoxin reported for sepsis patients are within the pg-ng/ml range [29], several clinical reports have documented endotoxemia within the range of μg/ml for selected patients with systemic meningococcal disease [29, 30].
We show that LPS incubation of human PMN for 6 hrs leads to a 4.4-fold increase of the IC50 of adenosine on the H2O2 production; In other words: 4.4-fold higher concentrations of adenosine are needed to achieve a comparable inhibition of H2O2 production in LPS-treated versus native human PMN. These results, together with the determined adenosine levels being present in the cell culture supernatants after stimulation, were completely in line with our previously published data for PMN of patients with septic shock [8] which – regarding our question – proved the LPS-in vitro model as suitable.
Recent studies have shown that treatment with TNF-α and other inflammatory mediators enhances the function and expression of adenosine receptors, and specially A2ARs, by increasing the receptor number and by modulating mechanisms of desensitization [31–34]. The changes in A2AR signalling we investigated in this study could not be provoked by those pro-inflammatory cytokines: Incubation with TNF-α, IL-8, IFN-γ and IL-1 did not significantly influence the anti-inflammatory effects of adenosine which indicates that the observed effects of LPS incubation are not due to the secondary induction of cytokines but may be related to one of the signalling pathways described above.
Because A2A receptors are well known to ‘put the brake on inflammation’[35], which means they inhibit overactive immune cells during acute inflammation thereby limiting inflammatory collateral tissue damage, it is reasonable to assume that the here reported adenosine effects were mainly A2AR mediated. This is further supported by our recently reported findings providing evidence that A2A receptor mRNA expression in human PMN increases upon LPS stimulation whereas the expression of the other adenosine receptors does not change significantly [13]. In the current study, we performed cAMP-assays with both adenosine and the specific A2A receptor agonist CGS 21680 which exhibited a good concordance thereby giving an experimental prove for A2A receptors to be the key players in the here examined scenario. Furthermore, these experiments showed that the attenuating effects of adenosine on H2O2 production were mediated by the signal transduction system typically associated to the A2AR as a prototypical Gs-coupled receptor: Treating PMN with LPS was followed by an impaired potency to stimulate cAMP accumulation, as indicated by a significant increase in the EC50 value.
In the presence of continued agonist activation of the A2AR, signalling is attenuated by a coordinated process of desensitization [33, 36, 37]. In sepsis, mechanisms of desensitization may also play a role in the network of A2AR regulatory processes. However, it seems unlikely that the here reported observations of a reduced anti-inflammatory potency of adenosine and A2A receptors are due to desensitization processes because (i) treatment times with LPS and/or with agonist were identical in all reactions tested and (ii) adenosine concentrations in control and LPS-treated PMN were comparable after a culturing time of 6 hrs. Therefore, the applied experimental setting excluded most influences of potentially occurring desensitization processes. However, desensitization from endogenous adenosine exposure occurring in the first minutes of exposure to LPS that may also influence the characteristics of A2A receptors even after 6 hrs of incubation cannot completely be ruled out.
To determine whether the changes in functional responses of A2A receptors observed in PMN of sepsis patients correlated with changes in binding parameters, we performed ligand-binding studies with membranes from native and LPS-incubated PMN, and from PMN of sepsis patients. A2A receptor density (Bmax) and dissociation constant (Kd) determined with [3H] ZM 241385 for native PMN were in good concordance with those previously described by Varani et al.[15, 38, 39]. After LPS incubation, we detected significant changes in ligand-binding parameters: Receptor number increased 1.4-fold which definitely excluded a reduced A2AR number being responsible for the observed diminished anti-inflammatory potency of adenosine. Surprisingly, although an increase in Bmax is expected to go along with a decrease in Kd[40], Kd actually increased (2.5-fold), thereby proving a significantly impaired ligand binding. In addition, saturation-binding experiments with the specific A1 receptor antagonist [3H]DPCPX showed that LPS did neither alter the affinity nor the expression of A1 receptors in PMN. In PMN of sepsis patients, ligand binding of the A2AR exhibited identical alterations: Receptor number increased 1.1-fold (the sample size did not allow reliable statistical analysis in the current study. Notwithstanding that, results exhibited a clear tendency towards an increase in Bmax in all of the 6 conducted experiments), whereas receptor affinity decreased by almost 50% (Kd increased 1.9-fold) as compared to PMN of healthy volunteers. Consequently, although more receptor molecules are available, higher concentrations of adenosine are needed to inhibit tissue-toxic H2O2 production. Up-regulation of A2A receptors during inflammatory processes is a useful physiological mechanism to protect own tissues from an overwhelming immune response which obviously in sepsis is overcompensated by a reduced receptor affinity thereby promoting further development of the septic disease.
Changes in the protein primary structure as a result of alternative splicing or conformational changes in the receptor protein caused by allosteric modulation may account for the reduced affinity of the A2AR in sepsis. The human A2AR gene consists of two coding exons separated by a single intron leading to the translation of a 45-kD protein [41]. In view of this genetic organization, the existence of splice variants leading to the differential expression of receptor protein with varying functions appears to be unlikely. Nevertheless, we performed RT-PCR analyses to definitely exclude the existence of such receptor isoforms that are typically generated by alternative splicing at the exon-intron boundaries or by usage of alternative startcodons. The latter is not likely because an in-frame stop codon 21 bp upstream of the start ATG avoids 5′ extension of the open reading frame. RT-PCR fragments amplified across the exonintron boundaries did not exhibit any length differences in native versus LPS incubated PMN which excluded splice variants being responsible for the altered receptor affinity.
GPCRs are per se allosteric, in that they possess binding sites for the endogenous ligand and the G protein. Additionally, binding sites for allosteric ligands exist, that act by inducing conformational changes that are transmitted from the allosteric site to the orthosteric site and/or to effector coupling sites [42]. It therefore is widely accepted that also the A2A receptor has more than one binding site [43–45]. Ligands binding to the allosteric site can alter the binding of ligands to the orthosteric site. Additionally, dimerization with other receptors (i.e. D1 receptor) [46] is discussed. The effects on the receptor affinity we found in sepsis can be explained by allosteric effects provoked by so far unknown mediators arising in the inflammatory environment: A negative allosteric regulator decreases the binding of the orthosteric ligand such that either the orthosteric agonist concentration–response curve will be shifted to higher agonist concentrations (i.e. rightwards) and/or the maximum response will be decreased [47].
In view of the multiplicity of mediators being involved in the activation of human PMN, the identification of the allosteric modulators giving rise to the phenomena reported here appears to be rather complex and may be subject of further studies.
In summary, the present study provides evidence that the significant decrease in the potency of adenosine to inhibit the generation of reactive oxygen species by PMN of sepsis patients is provoked by an impairment of A2A receptor signalling: Despite an increased receptor expression, the receptor function declines, due to a diminished ligand-binding affinity most likely caused by allosteric modulators within the inflammatory environment. This may contribute to the further exacerbation of inflammation and ensuing tissue damage in the course of sepsis.
It now appears that A2AR function is highly dependent upon the cellular environment in which it is expressed and thus further functional characterization of A2AR responses in sepsis may be a promising approach to develop new adenosine or A2A receptor agonists based therapeutic strategies.
Acknowledgments
This study was partially supported by a grant from the German National Research Foundation to MT, TH733/4-1.
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60:49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 3.Hoesel LM, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, Pianko MJ, Bernacki KD, Sarma JV, Ward PA. Harmful and protective roles of neutrophils in sepsis. Shock. 2005;24:40–7. doi: 10.1097/01.shk.0000170353.80318.d5. [DOI] [PubMed] [Google Scholar]
- 4.Martins PS, Kallas EG, Neto MC, Dalboni MA, Blecher S, Salomao R. Upregulation of reactive oxygen species generation and phagocytosis, and increased apoptosis in human neutrophils during severe sepsis and septic shock. Shock. 2003;20:208–12. doi: 10.1097/01.shk.0000079425.52617.db. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann I, Hoelzl A, Schliephake F, Hummel T, Chouker A, Peter K, Thiel M. Polymorphonuclear leukocyte dysfunction syndrome in patients with increasing sepsis severity. Shock. 2006;26:254–61. doi: 10.1097/01.shk.0000223131.64512.7a. [DOI] [PubMed] [Google Scholar]
- 6.Cronstein BN, Levin RI, Belanoff J, Weissman G, Hirschhorn R. Adenosine: an endogenous inhibitor of neutrophil-mediated injury to endothelial cells. J Clin Invest. 1986;78:760–70. doi: 10.1172/JCI112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Latini S, Bordoni F, Pedata F, Corradetti R. Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. Br J Pharmacol. 1999;127:729–39. doi: 10.1038/sj.bjp.0702591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann I, Hoelzl A, Schliephake F, Hummel T, Chouker A, Lysenko L, Thiel M. Effects of adenosine on functions of polymorphonuclear leukocytes from patients with septic shock. Shock. 2007;27:25–31. doi: 10.1097/01.shk.0000238066.00074.90. [DOI] [PubMed] [Google Scholar]
- 9.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–52. [PMC free article] [PubMed] [Google Scholar]
- 10.Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. doi: 10.1016/j.it.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Linden J. New insights into the regulation of inflammation by adenosine. J Clin Invest. 2006;116:1835–7. doi: 10.1172/JCI29125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–23. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreth S, Ledderose C, Kaufmann I, Groeger G, Thiel M. Differential expression of 5′-UTR splice variants of the adenosine A2A receptor gene in human granulocytes: identification, characterization, and functional impact on activation. FASEB J. 2008;22:3276–86. doi: 10.1096/fj.07-101097. [DOI] [PubMed] [Google Scholar]
- 14.Fortin A, Harbour D, Fernandes M, Borgeat P, Bourgoin S. Differential expression of adenosine receptors in human neu-trophils: up-regulation by specific Th1 cytokines and lipopolysaccharide. J Leukoc Biol. 2006;79:574–85. doi: 10.1189/jlb.0505249. [DOI] [PubMed] [Google Scholar]
- 15.Varani K, Laghi-Pasini F, Camurri A, Capecchi PL, Maccherini M, Diciolla F, Ceccatelli L, Lazzerini PE, Ulouglu C, Cattabeni F, Borea PA, Abbracchio MP. Changes of peripheral A2A adenosine receptors in chronic heart failure and cardiac transplantation. FASEB J. 2003;17:280–2. doi: 10.1096/fj.02-0543fje. [DOI] [PubMed] [Google Scholar]
- 16.Németh ZH, Csdka B, Wilmanski J, Xu D, Lu Q, Ledent C, Deitch EA, Pacher P, Spolarics Z, Haskd G. Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J Immunol. 2006;176:5616–26. doi: 10.4049/jimmunol.176.9.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–20. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 18.Bone RC, Balk A, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 19.Thiel M, Imendorffer S, Chouker A, Groh J, Briegel J, Anthuber M, Kramling HJ, Arfors KE, Peter K, Messmer K. Expression of adhesion molecules on circulating polymorphonuclear leukocytes during orthotopic liver transplantation. Hepatology. 1998;28:1538–50. doi: 10.1002/hep.510280614. [DOI] [PubMed] [Google Scholar]
- 20.Haskó G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–9. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A(2A) receptors. Annu Rev Immun. 2004;22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 23.Janssens S, Beyaert R. Role of toll-like receptors in pathogen recognition. Clin Microbiol Rev. 2003;16:637–46. doi: 10.1128/CMR.16.4.637-646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 25.Liu SF, Ye X, Malik AB. In vivo inhibition of nuclear factor-kappa B activation prevents inducible nitric oxide synthase expression and systemic hypotension in a rat model of septic shock. J Immunol. 1997;159:3976–83. [PubMed] [Google Scholar]
- 26.Liu SF, Ye X, Malik AB. Inhibition of NF-kappaB activation by pyrrolidine dithiocarbamate in vivo prevents expression of proinflammatory genes. Circulation. 1999;100:1330–7. doi: 10.1161/01.cir.100.12.1330. [DOI] [PubMed] [Google Scholar]
- 27.Thiel M, Chouker A. Acting via A2 receptors, adenosine inhibits the production of tumor necrosis factor-alpha of endotoxin-stimulated human polymorphonuclear leukocytes. J Lab Clin Med. 1995;126:275–82. [PubMed] [Google Scholar]
- 28.Peula-GarcÌa JM, Molina-Bolivar JA, Velasco J, Rojas A, Galisteo-González F. Interaction of bacterial endotoxine (lipopolysaccharide) with latex particles: application to latex agglutination immunoassays. J Colloid Interface Sci. 2002;245:230–6. doi: 10.1006/jcis.2001.7958. [DOI] [PubMed] [Google Scholar]
- 29.Hurley JC. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev. 1995;8:268–92. doi: 10.1128/cmr.8.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650–2. doi: 10.1093/infdis/166.3.650. [DOI] [PubMed] [Google Scholar]
- 31.Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J Immunol. 2001;167:4026–32. doi: 10.4049/jimmunol.167.7.4026. [DOI] [PubMed] [Google Scholar]
- 32.Khoa ND, Montesinos MC, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol. 2003;171:3991–8. doi: 10.4049/jimmunol.171.8.3991. [DOI] [PubMed] [Google Scholar]
- 33.Khoa ND, Postow M, Danielsson J, Cronstein BN. Tumor necrosis factor-alpha prevents desensitization of Galphas-coupled receptors by regulating GRK2 association with the plasma membrane. Mol Pharmacol. 2006;69:1311–9. doi: 10.1124/mol.105.016857. [DOI] [PubMed] [Google Scholar]
- 34.McColl SR, St Onge M, Dussault AA, Laflamme C, Bouchard L, Boulanger J, Pouliot M. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J. 2006;20:187–9. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirkpatrick P. G-protein coupled receptors: putting the brake on inflammation. Nat Rev Drug Discov. 2002;1:99. [Google Scholar]
- 36.Klaasse EC, IJzerman AP, De Grip WJ, Beukers MW. Internalization and desensitization of adenosine receptors. Purinergic Signalling. 2008;4:21–37. doi: 10.1007/s11302-007-9086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153:379–88. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varani K, Gessi S, Merighi S, Iannotta V, Cattabriga E, Spisani S, Cadossi R, Borea PA. Effect of low frequency electromagnetic fields on A2A adenosine receptors in human neutrophils. Br J Pharmacol. 2002;136:57–66. doi: 10.1038/sj.bjp.0704695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varani K, Portaluppi F, Gessi S, Merighi S, Vincenzi F, Cattabriga E, Dalpiaz A, Bortolotti F, Belardinelli L, Borea PA. Caffeine intake induces an alteration in human neutrophil A2A adenosine receptors. Cell Mol Life Sci. 2005;62:2350–8. doi: 10.1007/s00018-005-5312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rovati GE. Ligand-binding studies: old beliefs and new strategies. Trends Pharmacol Sci. 1998;19:365–9. doi: 10.1016/s0165-6147(98)01242-5. [DOI] [PubMed] [Google Scholar]
- 41.Pierce KD, Furlong TJ, Selbie LA, Shine J. Molecular cloning and expression of an adenosine A2b receptor from human brain. Biochem Biophys Res Commun. 1992;187:86–93. doi: 10.1016/s0006-291x(05)81462-7. [DOI] [PubMed] [Google Scholar]
- 42.Gilchrist A. Modulating G-protein-coupled receptors: from traditional pharmacology to allosterics. Trends Pharmacol Sci. 2007;28:431–7. doi: 10.1016/j.tips.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 43.Zezula J, Freissmuth M. The A2A-adenosine receptor: a GPCR with unique features? Br J Pharmacol. 2008;153:184–90. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 45.Gao ZG, Ijzerman AP. Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol. 2000;60:669–76. doi: 10.1016/s0006-2952(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 46.Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferre S, Fuxe K. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277:18091–7. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- 47.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–74. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]