

Abstract
Reduced intrahepatic nitric oxide (NO) bioavailability and increased cyclooxygenase-1 (COX-1)-derived vasoconstrictor prostanoids modulate the hepatic vascular tone in cirrhosis. We aimed at investigating the reciprocal interactions between NO and COX in the hepatic endothelium of control and cirrhotic rats. NO bioavailability (DAF-FM-DA staining), superoxide (O2−) content (DHE staining), prostanoid production (PGI2 and TXA2 by enzyme immunoassays) as well as COX expression (Western Blot), were determined in hepatic endothelial cells (HEC) from control and cirrhotic rats submitted to different experimental conditions: COX activation, COX inhibition, NO activation and NO inhibition. In control and cirrhotic HEC, COX activation with arachidonic acid reduced NO bioavailability and increased O2− levels. These effects were abolished by pre-treating HEC with the COX inhibitor indomethacin. In control, but not in cirrhotic HEC, scavenging of O2− by superoxide dismutase (SOD) incubation partially restored the decrease in NO bioavailability promoted by COX activation. NO supplementation produced a significant and parallel reduction in PGI2 and TXA2 production in control HEC, whereas it only reduced TXA2 production in cirrhotic HEC. By contrast, in control and cirrhotic HEC, NO inhibition did not modify COX expression or activity. Our results demonstrate that NO and COX systems are closely interrelated in HEC. This is especially relevant in cirrhotic HEC where COX inhibition increases NO bioavailability and NO supplementation induces a reduction in TXA2. These strategies may have beneficial effects ameliorating the vasoconstrictor/vasodilator imbalance of the intrahepatic circulation of cirrhotic livers.
Keywords: cyclooxygenase, nitric oxide, superoxide, portal hypertension, endothelium
Introduction
In cirrhotic livers, increased resistance to portal blood flow is the primary factor in the pathophysiology of portal hypertension [1, 2]. This increase in intrahepatic resistance is determined by architectural alterations of the liver as well as by a dynamic component, which is due to an increased production of COX-1-derived vasoconstrictors, such as thromboxane (TXA2) [3, 4], in the setting of an insufficient availability of the vasodilator NO [5, 6].
Previous studies from our group have demonstrated that HEC, which overexpress COX-1 [7], are an important source of vasoconstrictor prostanoids, mainly TXA2 [8]. In addition, COX contributes to the increased oxidative stress found in cirrhotic rat livers [9]. On the other hand, reduced NO bioavailability in cirrhotic rat livers is not only attributable to a decreased nitric oxide synthase activity but also to an increased scavenging by superoxide anions (O2−) [10]. Particularly, NO availability is modulated by O2− in HEC [9]. As expected, the beneficial effects of COX inhibitors improving endothelial dysfunction in cirrhotic rat livers was attenuated when NO synthesis was previously inhibited [3, 11].
In endothelial cells from different vascular beds, there are evidence supporting direct and indirect interactions between NO and COX systems [12–14] as NO could positively or negatively modulate COX activity and modify eicosanoid production [15–18]. Conversely, COX-derived prostanoids, such as TXA2, are known to modulate NO-synthase activity, downregulating its phosphorylation and decreasing its activity [19]. Such interactions may also be present in the cirrhotic liver; specifically we hypothesize that NO and COX systems are interrelated in HEC. The current study was aimed at investigating possible reciprocal modulation between NO and COX pathways in the hepatic endothelium of control and cirrhotic rats.
Materials and methods
Induction of cirrhosis by carbon tetrachloride
Male Wistar rats weighing 50–75 g underwent inhalation exposure to Carbon Tetrachloride (CCl4). Phenobarbital (0.3 g/l) was added to the drinking water as previously described [3]. A high yield of micronodular cirrhosis was obtained after approximately 12–15 weeks of CCl4 inhalation. When the cirrhotic rats developed ascites, administration of phenobarbital was stopped, and the subsequent experiments were performed 1 week later. Control animals received only phenobarbital. The animals were kept in environmentally controlled animal facilities at the Institut d'Investigacions Biomèdiques August Pi i Sunyer. All experiments were approved by the Laboratory Animal Care and Use Committee of the University of Barcelona and were conducted in accordance with Guide for the Care and Use of Laboratory Animals (National Institutes of Health, NIH Publication 86-23, revised 1996).
Isolation and culture of hepatic endothelial cells
Hepatic endothelial cells were isolated from control and cirrhotic rats as previously described [9]. Briefly, after collagenase perfusion of the livers and isopycnic sedimentation of the resulting dispersed cells through a two-step density gradient of Percoll, pure monolayer cultures of HEC were established by selective attachment on a substrate of rat tail collagen type I. Afterwards, cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 and studies were performed on cells from the first passage, 12 hrs after their isolation, to preserve their typical phenotype [20].
Experimental protocols
Effects of COX activity on NO bioavailability and O2− production in control and cirrhotic HEC
Hepatic endothelial cells isolated from control and cirrhotic rats were pre-incubated for 15 min. with vehicle or with the non-selective COX inhibitor, indomethacin (10 μM). Then, arachidonic acid (AA, 40 μM) or its vehicle (ethanol 0.1%) was added. After 20 min., in situ NO bioavailability and O2− levels were assessed with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM-DA; Molecular Probes Inc., Eugene, OR, USA) and dihydroethidium (DHE; Molecular Probes Inc.), respectively, as described below.
To characterize whether O2− derived from COX modulates NO bioavailability, a different group of HEC from control or cirrhotic rats were treated with vehicle (phosphate-buffered saline; PBS), with AA alone or with AA plus the superoxide scavenger, superoxidedismutase (SOD: 300U). This dose of SOD has been shown to markedly attenuate the marked increase in O2− produced by the SOD inhibitor, diethyldithiocarbamate [9]. In parallel, control HEC were treated with the NO synthase (NOS) inhibitor Nω-Nitro-l-arginine methyl ester hydrochloride, L-NAME (1.5 mM) as a negative control. Vascular endothelial growth factor (VEGF, 40 ng/ml) was added to cirrhotic HEC in order to prime NO production. Fluorescence images were obtained every 2 min. for 40 min. with a laser scanning confocal microscope.
Effects of NO on COX activity in control and cirrhotic HEC
Effects of NO supplementation on COX activity in control and cirrhotic HEC: HEC isolated from control and cirrhotic rats were treated with the exogenous NO donor, sodium nitroprusside (SNP, 25 μM) or its vehicle (PBS) for 1 hr, and then incubated with AA for 20 min. Culture media samples were collected, stored at −80°C and assayed for prostanoid levels. Cells were lysed and processed for Western blot analysis as previously described [21].
Effects of NO inhibition on COX activity in control and cirrhotic HEC: HEC isolated from control and cirrhotic rats were pre-incubated with the NOS inhibitor L-NAME or its vehicle (PBS) for 1 hr. Then, the calcium ionophore A23187 (2 μM), which by increasing calcium levels activates both COX and NOS, or its vehicle (Dimethyl sulfoxide 0.01%) were added. After 1 hr of treatment, culture media samples were collected and stored at −80°C until assayed for prostanoid levels. Cells were lysed and processed for Western blot analysis.
Measurement of NO levels and O2− content in HEC
In situ NO levels or O2− levels in HEC were assessed with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM-DA; Molecular Probes Inc.) or with the oxidative fluorescent dye dihydroethidium (DHE; Molecular Probes Inc.) as described [9, 22].
Briefly, isolated HEC were washed in RPMI-1640 without phenol red and loaded with DAF-FM-DA (10 μM for 20 min. at 37°C) or DHE (10 μM for 20 min. at 37°C). Then, HEC were rinsed three times with PBS, kept in the dark, and maintained at 37°C with a warm stage on a laser scanning confocal microscope (model TCS-SL DMIRE2; Leica, Wetzlar, Germany). Fluorescence images were obtained with a 488-nm (excitation) and 505- to 530-nm (emission) filter set for DAF-FM-DA, and 610-nm (emission filter) set for DHE with a 40 × 1.3 oil objective. Quantitative analysis was obtained by averaging of the peak relative fluorescent intensity (optical density arbitrary units) of each confocal microscope image (Image J 1.43m software, National Institutes of Health) and normalization of the fluorescent result by the total number of cultured cells counted from each corresponding digitalized phase contrast microscope image.
Analysis of prostanoids
Prostacyclin (PGI2) and TXA2 were quantified in duplicate as their stable metabolites, 6-keto PGF1α and TXB2, respectively, as previously described [8] by using enzyme immunoassay kits. All assays contained media controls to exclude any effect of the reagents on the immunoassays.
Western blot analysis of COX-1, prostacyclin synthase and thromboxane synthase protein expression in HEC
Aliquots from each sample containing equal amounts of protein (10 μg) were run on a sodium dodecyl sulfate-polyacrylamide gel and transferred to a nitrocellulose membrane. After the transfer, the blots were blocked for 1 hr and were probed with a mouse anti-COX-1 antibody (5 μg/ml), rabbit anti-prostacyclin synthase (PGIS) (2 μg/ml) or rabbit anti-thromboxane synthase (TXAS) (1 μg/ml) overnight at 4°C followed by incubation with their associated horseradish peroxidase–conjugated secondary antibody (1:10,000; Stressgen Victoria, British Columbia, Canada) for 1 hr at room temperature. Blots were revealed by chemiluminiscence. Protein expression was determined by densitometric analysis with the Science Laboratory Image Gauge (Fuji Photo Film GmbH, Düsseldorf, Germany). After stripping, blots were assayed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, Santa Cruz, CA, USA) expression as a standardization of the sample loading. Quantitative densitometric values of all proteins were normalized to GAPDH.
Drugs and reagents
AA, enzyme immunoassays kits, COX-1, PGIS and TXAS antibodies were obtained from Cayman Chem Co (Tallin, Estonia). Collagen type I was from Invitrogen (El Prat de Llobregat, Barcelona, Spain). Collagenase was from Roche Diagnostics (Mannheim, Germany). Percoll was from Amersham Biosciences (Uppsala, Sweden). Reagents for cell culture were provided by Biological Industries Ltd (Kibbutz Beit Haemek, Israel). L-NAME, A23187, SNP, indomethacin and other chemical compounds were purchased from Sigma-Aldrich (Tres Cantos, Madrid, Spain).
Statistical analysis
Statistical analysis was performed using the SPSS 16.0 for Windows statistical package (SPSS Inc., Chicago, IL, USA). All results are expressed as mean ± S.E.M. Comparisons between groups were performed with the Student's t-test or Mann–Whitney t-test for unpaired data or anova for repeated measures when adequate. Differences were considered significant at a P-value <0.05.
Results
Effects of COX activity on NO bioavailability and O2− production in control and cirrhotic HEC
In the first set of experiments, we investigated whether activation or inhibition of COX pathway modulates NO bioavailability.
COX activation reduces NO bioavailability and increases oxidative stress in control and cirrhotic HEC
To investigate the effects of COX activation on NO bioavailability, HEC from control and cirrhotic rats were treated with AA. AA administration promoted a significant and marked decrease in NO bioavailability both in HEC from control (Fig. 1A) and from cirrhotic rats (Fig. 1B).
Fig 1.
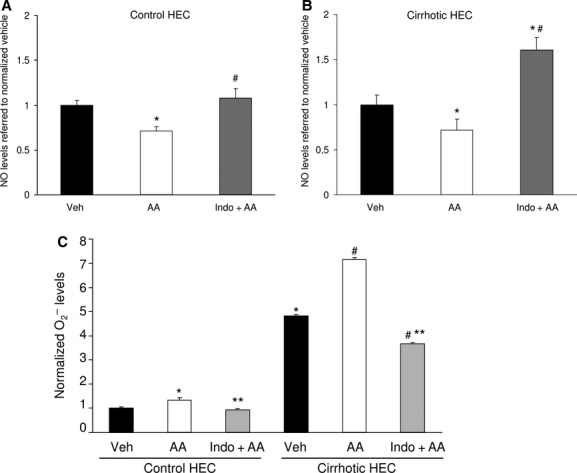
Effects of COX activity on nitric oxide (NO) bioavailability and superoxide (O2−) levels in hepatic endothelial cells (HEC) from control and cirrhotic rats. Fluorescent detection of intracellular NO bioavailability by 4-amino-5-methylamino-2′,7′difluorofluorescein diacetate (DAF-FM-DA) staining and O2− by dihydroethidium (DHE) staining in HEC. Arachidonic acid (AA) administration promoted a significant decrease in NO bioavailability in control (A) and cirrhotic (B) HEC which was prevented by coincubation with indomethacin. Besides, in cirrhotic HEC, indomethacin treatment increased NO bioavailability above baseline values. The fluorescence intensity of DAF-FM-DA in arbitrary units was normalized by the total number of cells. The data shown are from 5296 individual vehicle (Veh) control HEC, 5322 AA control HEC, 3124 indomethacin+AA (Indo+AA) control HEC, 1082 individual Veh cirrhotic HEC, 1280 AA cirrhotic HEC, and 1228 Indo+AA cirrhotic HEC obtained from three independent experiments. The mean NO levels of HEC treated with vehicle was considered one, either for control or cirrhotic rats (*P < 0.05 versus Veh, #P < 0.05 versus AA). (C) O2− was significantly higher in HEC from cirrhotic than in control rats. Arachidonic acid (AA) promoted a significant increase in O2− in control and cirrhotic HEC which was prevented by indomethacin. In cirrhotic HEC, indomethacin treatment decreased O2− below baseline values. The fluorescence intensity of DHE in arbitrary units was normalized by the total number of cells. The data shown are from 5642 individual Veh control HEC, 4713 AA control HEC, 4682 Indo+AA control HEC, 2616 individual Veh cirrhotic HEC, 1399 AA cirrhotic HEC, and 1101 Indo+AA cirrhotic HEC obtained from four independent experiments (*P < 0.05 versus control Veh, #P < 0.05 versus cirrhotic Veh, **P < 0.05 versus its own AA-condition).
COX activation also promoted a significant increase in O2− content in HEC from control or cirrhotic rats, being this increase much higher in the later (Fig. 1C).
COX inhibition increases NO bioavailability and reduces oxidative stress in control and cirrhotic HEC
To further explore the role of COX modulating the NO–O2− relationship, COX was inhibited before AA administration. Indomethacin prevented the decrease in NO bioavailability produced by AA in HEC from both control (Fig. 1A) and cirrhotic rats (Fig. 1B). Remarkably, in cirrhotic HEC, indomethacin treatment increased NO bioavailability above baseline values, supporting the role of COX activation reducing NO bioavailability.
Furthermore, COX inhibition prevented the increase in O2− content caused by AA in HEC from control and cirrhotic rats (Fig. 1C). In cirrhotic HEC, which displayed significantly higher levels of O2− in comparison with control HEC (Fig. 1C), indomethacin decreased O2− levels below baseline values, suggesting that COX is a source of O2− in cirrhotic HEC.
Effects of COX-derived O2− modulating NO bioavailability in HEC
To assess whether O2− is the main determinant of the reduction of NO bioavailability caused by COX activation, we assessed NO bioavailability in control and cirrhotic HEC incubated with AA or with AA plus the superoxide scavenger, SOD.
In control HEC, AA administration promoted a significant decrease in NO bioavailability. This effect was attenuated when O2− was scavenged by SOD coincubation, indicating that O2− was reducing NO bioavailability (Fig. 2A).
Fig 2.
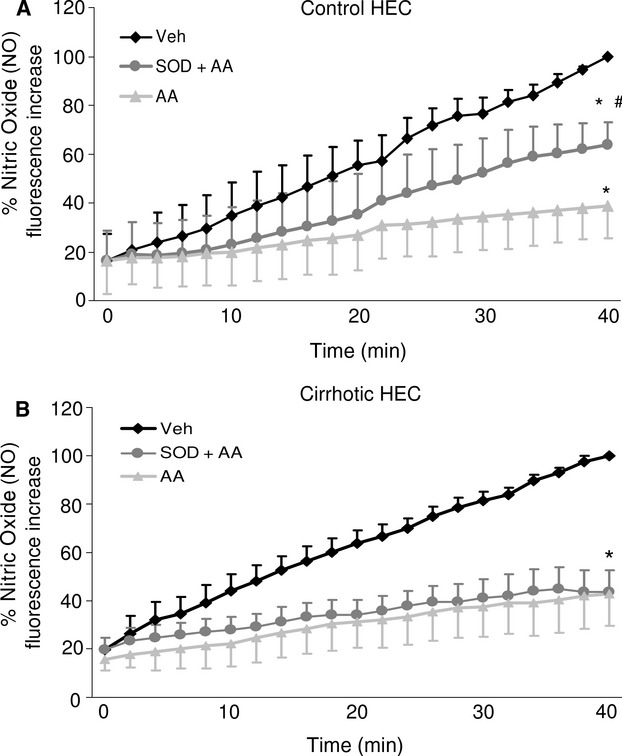
Effects of COX activation on NO production in control and cirrhotic hepatic endothelial cells (HEC). Time dependent increase in DAF-FM-DA fluorescence as a marker of intracellular NO bioavailability. (A) In control HEC, AA incubation induced a significant decrease in NO bioavailability which was partially restored by coincubation with SOD (*P < 0.05 versus vehicle, #P < 0.05 versus AA). (B) In cirrhotic HEC, AA incubation induced a significant decrease in NO bioavailability which was not restored by coincubation with SOD (*P < 0.05 versus vehicle). The fluorescence intensity of DAF-FM-DA was normalized by the total number of cells of each experimental condition. The data shown are from 409 individual vehicle control HEC, 317 AA control HEC and 256 SOD+AA control HEC, obtained from three independent experiments and 701 vehicle cirrhotic HEC, 767 AA cirrhotic HEC and 750 SOD+AA cirrhotic HEC obtained from five independent experiments.
In HEC from cirrhotic rats, AA administration promoted a marked decrease in NO production. However, contrary to control HEC, SOD coincubation did not attenuate the decrease in NO bioavailability suggesting that, in cirrhotic HEC, O2− was not a major determinant of decreased NO bioavailability after COX activation (Fig. 2B).
Effects of NO on COX pathway in control and cirrhotic HEC
In a second series of experiments, HEC from control and cirrhotic rats were incubated with a NO synthase inhibitor or with a NO donor to elucidate whether variations in NO levels modulate COX pathway.
NO supplementation induces a marked reduction of COX activity in control and cirrhotic HEC
In control HEC, NO supplementation did not modify COX-1 protein expression (Fig. 3A) or the basal level of PGI2 and TXA2 (data not shown). As expected, AA administration induced a significant increase in PGI2 and TXA2 [8] that was blunted when HEC were pre-incubated with the NO donor SNP (Fig. 3B).
Fig 3.
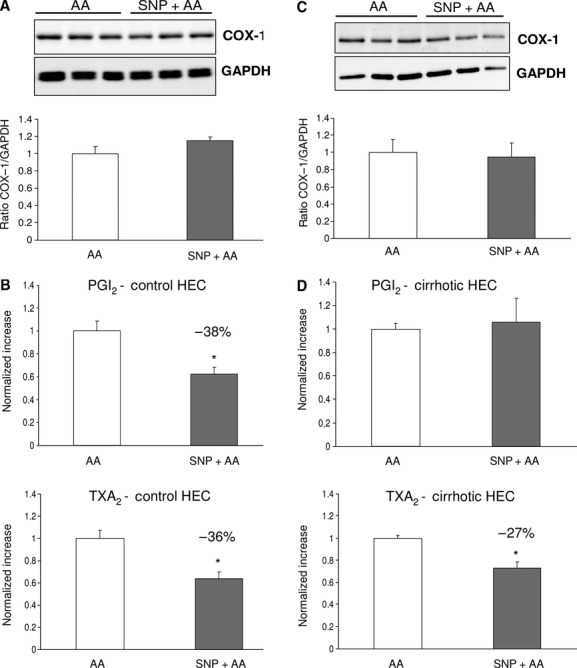
Effects of NO supplementation on COX pathway in control and cirrhotic hepatic endothelial cells (HEC). (A) Representative Western blot and analysis of COX-1 from control HEC treated with sodium nitroprusside (SNP) or its vehicle in the presence of AA. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed no differences between both groups. (n = 5 per group). (B) Prostacylin (PGI2) and Thromboxane (TXA2) production by HEC from control rats stimulated with AA in the presence or absence of the exogenous NO donor SNP (n = 6 per group). In control HEC, NO supplementation produced a significant and similar decrease in PGI2 (−38%) and TXA2 (−36%). (*P < 0.05 versus AA). (C) Representative Western blot and analysis of COX-1 from cirrhotic HEC treated with sodium nitroprusside (SNP) or its vehicle in the presence of AA. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed no differences between both groups. (n = 7 per group). (D) Prostacylin (PGI2) and Thromboxane (TXA2) production by HEC from cirrhotic rats stimulated with AA in the presence or absence of the exogenous NO donor SNP (n = 6 per group). In cirrhotic HEC, NO supplementation resulted in a significant reduction in TXA2 production (−27%) without significant changes in PGI2 (*P < 0.05 versus AA).
In cirrhotic HEC, NO supplementation did not modify COX-1 protein expression (Fig. 3C). However, it significantly reduced TXA2 levels without significant changes in PGI2 production (Fig. 3D), suggesting that NO supplementation could partially restore the pathological disequilibrium in the PGI2/TXA2 ratio described in cirrhotic HEC.
NO inhibition does not modify COX pathway in control and cirrhotic HEC
As expected, HEC from control rats incubated with the nitric oxide synthase inhibitor L-NAME, exhibited a significant reduction in NO bioavailability (data not shown). However, L-NAME pre-incubation did not modify COX-1 expression (Fig. 4A) or activity, as evaluated by prostanoid production (Fig. 4B). In HEC from control rats, A23187 treatment, which simultaneously activate COX and NOS pathways, produced a marked release of PGI2 and TXA2 that was not modified by NO inhibition.
Fig 4.
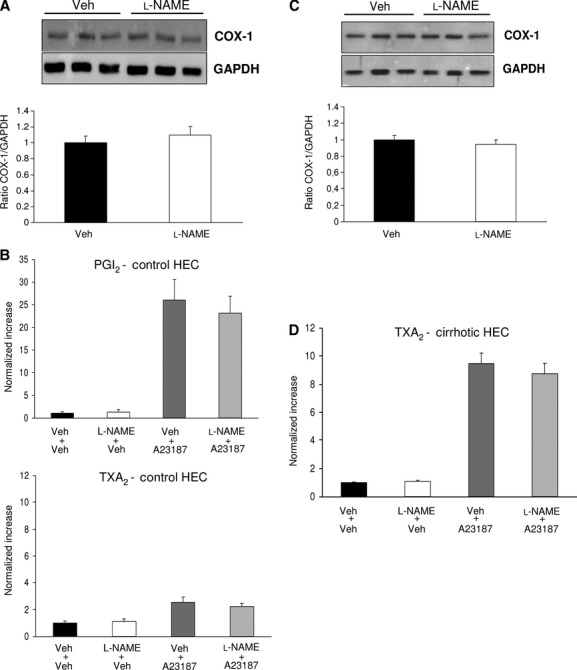
Effects of NO inhibition on COX pathway in control and cirrhotic hepatic endothelial cells (HEC). (A) Representative Western blot and analysis of COX-1 from control HEC treated with vehicle (Veh) or with L-NAME. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed no differences between both groups (n = 7 per group). (B) Prostacyclin (PGI2) and Thromboxane (TXA2) production in control HEC treated with Veh or L-NAME in the presence of the calcium ionophore A23187 or its Veh (n = 6 per group). The mean prostanoid levels of HEC treated with Veh+Veh was considered one. NO inhibition did not modify either basal or A23187-induced prostanoid (PGI2 and TXA2) production. (C) Representative Western blot and analysis of COX-1 from cirrhotic HEC treated with vehicle (Veh) or with L-NAME. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed no differences between both groups (n = 9 per group). (D) Thromboxane (TXA2) production by cirrhotic HEC treated with Veh or L-NAME in the presence of A23187 or its Veh (n = 9 per group). The mean prostanoid levels of HEC treated with Veh+Veh was considered one. NO inhibition did not modify either basal or A23187 induced TXA2 production.
Similarly, in HEC isolated from cirrhotic rats, NO inhibition did not modify COX-1 protein expression (Fig. 4C) or basal TXA2 production, the main prostanoid produced by cirrhotic HEC. HEC from cirrhotic rats incubated with A23187 exhibited a significant increase in TXA2 which was significantly higher than that observed in control HEC and that was not modified by NO inhibition (Fig. 4D).
PGIS and TXAS expression in control and cirrhotic HEC
A third set of experiments were aimed at elucidating the main prostanoid synthase involved in the observed NO-COX interactions. For that purpose, PGIS and TXAS protein expression were determined in HEC from control and cirrhotic rats. Cirrhotic HEC exhibited a significantly higher TXAS expression compared with control HEC (Fig. 5A) whereas PGIS expression remained unchanged (Fig. 5B).
Fig 5.
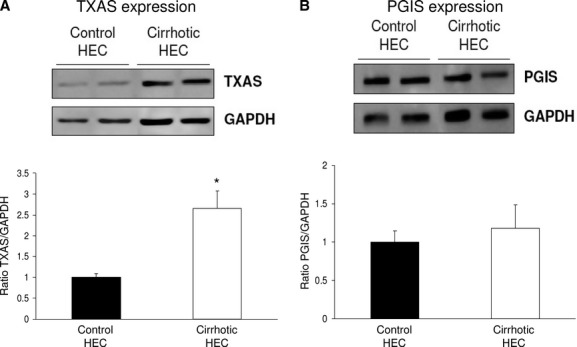
Prostacyclin and Thromboxane synthase protein expression in control and cirrhotic hepatic endothelial cells (HEC). (A) Representative Western blot and analysis of prostacyclin synthase (PGIS) from control and cirrhotic HEC. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed no differences between both groups (n = 4 per group). (B) Representative Western blot and analysis of thromboxane synthase (TXAS) from control and cirrhotic HEC. Densitometry quantification in arbitrary units, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), showed a significant increase in TXAS in cirrhotic HEC (n = 4 per group) (*P < 0.05 versus control HEC).
Discussion
Hepatic endothelial cells play a major role regulating intrahepatic vascular tone by the release of vasoactive substances that diffuse to hepatic stellate cells and possibly to other contractile structures causing their relaxation or constriction [23]. Among these vasoactive mediators, we and others have demonstrated a major role for NO and COX-derived prostanoids. In normal livers, HEC have a vasodilator phenotype with predominant production of NO and PGI2 and reduced amounts of TXA2. By contrast, cirrhotic livers exhibit endothelial dysfunction characterized by a change to a vasoconstrictor phenotype. In that regard, HEC from cirrhotic rats have been shown to produce large amounts of the COX-derived vasoconstrictor prostanoid TXA2 [8], while NO is markedly reduced [9].
A reciprocal regulation between NO and COX-derived products, such as TXA2, has been demonstrated in endothelial cells from different vascular beds [12, 24]. However, the interactions between these molecular pathways in the hepatic endothelium have not been investigated.
The present study shows, for the first time in HEC, that both systems are reciprocally related. Indeed, COX products are able to modify NO bioavailability, whereas NO content modulates COX-derived prostanoid production.
Our study clearly demonstrates that COX activation induces a reduction in intracellular NO bioavailability in both control and cirrhotic HEC. Remarkably, in cirrhotic HEC, COX activation further reduces the already low NO bioavailability. COX activation markedly increases the production of TXA2 and PGI2 but also leads to an increase in O2−. TXA2 has been shown to reduce NO synthase activity in endothelial cells through a receptor-mediated mechanism [19, 25]. In addition, we have recently demonstrated in HEC that O2− is able to interact with NO reducing its bioavailability [9]. Thus, COX activation could modulate NO bioavailability through the production of prostanoids or O2−. Our results suggest that the COX-derived product responsible for this decrease in NO availability is different in control and cirrhotic HEC. In control HEC, abolition of O2− content with SOD partially restored NO availability, indicating that COX-derived O2− was modulating, at least in part, NO availability. By contrast, in cirrhotic HEC, SOD administration was not able to restore at all the reduction in NO caused by COX activation. It may be argued that the used dose of SOD may have not sufficiently scavenged the higher increase in O2− observed in cirrhotic livers in comparison with controls. However, this is unlikely because the same dose of SOD has been previously shown to markedly attenuate the huge increase in O2− promoted in an experimental model of induced oxidative stress caused by direct SOD inhibition [9]. Altogether this suggests that, in cirrhotic HEC, COX-derived O2− is not the main determinant in the reduction of NO bioavailability. Although the exact mechanism remains conjectural, we propose that COX-derived TXA2, and not O2−, contributes to the reduced NO bioavailability found in cirrhotic HEC. This is supported by the fact that the production of TXA2, as well as TXAS expression, is markedly increased in HEC isolated from cirrhotic but not from control livers, where PGI2 production predominates [8].
The negative effect of COX activation on NO availability is further supported by our findings that COX inhibition restored NO bioavailability in control HEC, and that it even increased NO levels above baseline values in cirrhotic HEC. Therefore, our data strongly suggest that in cirrhotic HEC COX inhibition would contribute to improve hepatic vascular tone, not only by reducing TXA2 production but also by increasing NO production (Fig. 6A).
Fig 6.
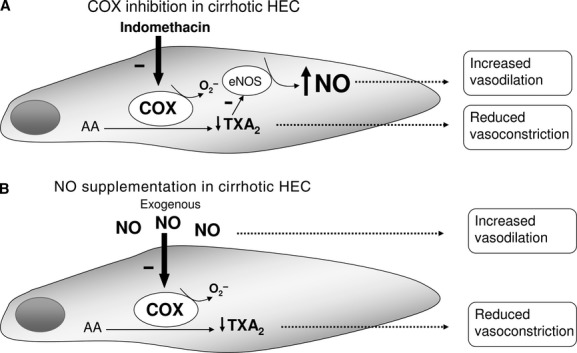
Suggested molecular mechanisms between NO and COX pathways in cirrhotic hepatic endothelial cells (HEC). (A) NO supplementation to cirrhotic HEC inhibits COX activity by reducing the vasoconstrictor prostanoid TXA2. This effect leads the cell to shift from a vasoconstrictor to a more vasodilatory phenotype suggesting that NO could promote hepatic vasodilation and reduce the exacerbated vasoconstriction in cirrhosis. (B) COX inhibition to cirrhotic HEC increases NO bioavailability. Therefore, COX inhibition would contribute to improve hepatic vascular tone, not only by reducing TXA2 production but also by increasing NO production.
Another important aspect of our study was to assess whether NO availability could influence prostanoid production in control and cirrhotic HEC. Interestingly, our data demonstrate that NO supplementation is able to inhibit COX activity, as shown by the observed reduction in prostanoid production, without modifying COX expression. The effect of NO in COX activity was different in HEC from control or from cirrhotic rats. In control HEC, NO supplementation reduced the vasodilator PGI2 and the vasoconstrictor TXA2, thus maintaining the predominant vasodilator phenotype. By contrast, in cirrhotic HEC, NO supplementation significantly reduced TXA2 levels, the main prostanoid produced by these cells, whereas it did not modify PGI2, thus restoring in part, the disequilibrium in the PGI2/TXA2 ratio observed in cirrhotic HEC. It is plausible to think that NO supplementation only reduced TXA2 levels, and not PGI2, due to the increased TXAS expression found in cirrhotic HEC. This effect leads the cell to shift from a vasoconstrictor to a more vasodilatory phenotype. Therefore, NO supplementation in cirrhosis would have a dual beneficial effect, restoring the decreased vasodilator content and attenuating the exaggerated vasoconstrictor production (Fig. 6B). Supporting this concept, it has been demonstrated that simvastatin, a treatment that increases NO availability [26], reduces TXA2 release in aged rats [27].
Consistent with our data, it has been observed that other NO-releasing compounds inhibit COX activity in endothelial and smooth muscle cells from other vascular beds [28–31], an effect that has been explained because NO promoted the nitration of tyrosine residues of the COX molecule leading to a loss of enzyme function [13, 32, 33]. By contrast, in different experimental settings a stimulatory effect of NO on prostanoid production through a direct modification of cysteine residues located in the catalytic domain of the COX molecule has been shown [17]. These reported differences have been explained by different cell-specific responses [34] or by variations in the amount of NO [14, 18].
By contrast, NO reduction did not modify COX activity in control or in cirrhotic HEC. This has been previously shown in other endothelial cells, where NOS inhibition did not produce changes in prostanoid production [35] or COX expression [15].
In conclusion, our study demonstrates that NO and COX systems are closely interrelated in HEC. This is particularly relevant in cirrhotic HEC where the release of vasoactive substances is shifted towards vasoconstrictor prostanoids [8]. Our results indicate that in cirrhotic HEC, COX inhibition increases NO bioavailability, and that NO supplementation not only ameliorates the decreased NO availability but also reduces the exaggerated production of TXA2, without affecting the release of PGI2. This reinforces the rationale to use combined therapies directed to both, increase intrahepatic NO availability and block increased vasoconstrictor prostanoid pathway.
Acknowledgments
The work was carried out at the Centre Esther Koplowitz, Barcelona. We also thank Elisabeth Sánchez, for her valuable help. This study was supported by grants from the Ministerio de Educación y Ciencia (SAF 2010/17043) and from Instituto de Salud Carlos III (FIS PS09/01261) and FIS PI11/00235. Ciberehd is funded by Instituto de Salud Carlos III.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Authors contributions
Eugenio Rosado: study concept and design, study supervision, acquisition of data, analysis and interpretation of data, drafting of the manuscript.
Aina Rodríguez-Vilarrupla: study concept and design, study supervision, acquisition of data, analysis and interpretation of data, drafting of the manuscript.
Jorge Gracia-Sancho: study concept and design, study supervision, acquisition of data, analysis and interpretation of data, drafting of the manuscript, obtained funding.
Montserrat Monclús: technical support, acquisition of data.
Jaume Bosch: critical revision of the manuscript for important intellectual content, obtained funding.
Joan-Carles García-Pagán: study supervision, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, obtained funding.
References
- 1.Bosch J, Garcia-Pagan JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32:141–56. doi: 10.1016/s0168-8278(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 2.Groszmann RJ, Abraldes JG. Portal hypertension: from bedside to bench. J Clin Gastroenterol. 2005;39:S125–30. doi: 10.1097/01.mcg.0000155552.14396.3d. [DOI] [PubMed] [Google Scholar]
- 3.Graupera M, Garcia-Pagan JC, Abraldes JG, et al. Cyclooxygenase-derived products modulate the increased intrahepatic resistance of cirrhotic rat livers. Hepatology. 2003;37:172–81. doi: 10.1053/jhep.2003.50004. [DOI] [PubMed] [Google Scholar]
- 4.Graupera M, Garcia-Pagan JC, Pares M, et al. Cyclooxygenase-1 inhibition corrects endothelial dysfunction in cirrhotic rat livers. J Hepatol. 2003;39:515–21. doi: 10.1016/s0168-8278(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 5.Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35:478–91. doi: 10.1053/jhep.2002.31432. [DOI] [PubMed] [Google Scholar]
- 6.Loureiro-Silva MR, Cadelina GW, Groszmann RJ. Deficit in nitric oxide production in cirrhotic rat livers is located in the sinusoidal and postsinusoidal areas. Am J Physiol Gastrointest Liver Physiol. 2003;284:G567–74. doi: 10.1152/ajpgi.00452.2002. [DOI] [PubMed] [Google Scholar]
- 7.Graupera M, March S, Engel P, et al. Sinusoidal endothelial COX-1-derived prostanoids modulate the hepatic vascular tone of cirrhotic rat livers. Am J Physiol Gastrointest Liver Physiol. 2005;288:G763–70. doi: 10.1152/ajpgi.00300.2004. [DOI] [PubMed] [Google Scholar]
- 8.Gracia-Sancho J, Lavina B, Rodriguez-Vilarrupla A, et al. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J Hepatol. 2007;47:220–7. doi: 10.1016/j.jhep.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Gracia-Sancho J, Lavina B, Rodriguez-Vilarrupla A, et al. Increased oxidative stress in cirrhotic rat livers: a potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology. 2008;47:1248–56. doi: 10.1002/hep.22166. [DOI] [PubMed] [Google Scholar]
- 10.Van de Casteele M, Van Pelt JF, Nevens F, et al. Low NO bioavailability in CCl4 cirrhotic rat livers might result from low NO synthesis combined with decreased superoxide dismutase activity allowing superoxide-mediated NO breakdown: a comparison of two portal hypertensive rat models with healthy controls. Comp Hepatol. 2003;2:2. doi: 10.1186/1476-5926-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laleman W, Van Landeghem L, Van der Elst I, et al. Nitroflurbiprofen, a nitric oxide-releasing cyclooxygenase inhibitor, improves cirrhotic portal hypertension in rats. Gastroenterology. 2007;132:709–19. doi: 10.1053/j.gastro.2006.12.041. [DOI] [PubMed] [Google Scholar]
- 12.Mollace V, Muscoli C, Masini E, et al. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–52. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 13.Upmacis RK, Deeb RS, Hajjar DP. Oxidative alterations of cyclooxygenase during atherogenesis. Prostaglandins Other Lipid Mediat. 2006;80:1–14. doi: 10.1016/j.prostaglandins.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Goodwin DC, Landino LM, Marnett LJ. Effects of nitric oxide and nitric oxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J. 1999;13:1121–36. doi: 10.1096/fasebj.13.10.1121. [DOI] [PubMed] [Google Scholar]
- 15.Davidge ST, Baker PN, Laughlin MK, Roberts JM. Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circ Res. 1995;77:274–83. doi: 10.1161/01.res.77.2.274. [DOI] [PubMed] [Google Scholar]
- 16.Miyamoto A, Hashiguchi Y, Obi T, et al. Ibuprofen or ozagrel increases NO release and l-nitro arginine induces TXA(2) release from cultured porcine basilar arterial endothelial cells. Vascul Pharmacol. 2007;46:85–90. doi: 10.1016/j.vph.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 17.Salvemini D, Misko TP, Masferrer JL, et al. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA. 1993;90:7240–4. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidge ST, Pitt BR, McLaughlin MK, et al. Biphasic stimulation of prostacyclin by endogenous nitric oxide (NO) in endothelial cells transfected with inducible NO synthase. Gen Pharmacol. 1999;33:383–7. doi: 10.1016/s0306-3623(99)00033-6. [DOI] [PubMed] [Google Scholar]
- 19.Ashton AW, Ware JA. Thromboxane A2 receptor signaling inhibits vascular endothelial growth factor-induced endothelial cell differentiation and migration. Circ Res. 2004;95:372–9. doi: 10.1161/01.RES.0000138300.41642.15. [DOI] [PubMed] [Google Scholar]
- 20.DeLeve LD, Wang X, Hu L, et al. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G757–63. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- 21.Gracia-Sancho J, Laviña B, Rodriguez-Vilarrupla A, et al. Evidence against NADPH oxidase modulating hepatic vascular tone in cirrhosis. Gastroenterology. 2007;133:959–66. doi: 10.1053/j.gastro.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Caldero H, Rodriguez-Vilarrupla A, Gracia-Sancho J, et al. Tempol administration, a superoxide dismutase mimetic, reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol. 2010;54:660–5. doi: 10.1016/j.jhep.2010.07.034. [DOI] [PubMed] [Google Scholar]
- 23.Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. J Hepatol. 2007;46:927–34. doi: 10.1016/j.jhep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Cuzzocrea S, Salvemini D. Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney Int. 2007;71:290–7. doi: 10.1038/sj.ki.5002058. [DOI] [PubMed] [Google Scholar]
- 25.Liu CQ, Leung FP, Wong SL, et al. Thromboxane prostanoid receptor activation impairs endothelial nitric oxide-dependent vasorelaxations: the role of Rho kinase. Biochem Pharmacol. 2009;78:374–81. doi: 10.1016/j.bcp.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Abraldes JG, Graupera M, Zafra C, et al. Simvastatin improves sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol. 2005;42:62–3. doi: 10.1016/j.jhep.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 27.de Sotomayor MA, Perez-Guerrero C, Herrrera MD, et al. Improvement of age-related endothelial dysfunction by simvastatin: effect on NO and COX pathways. Br J Pharmacol. 2005;146:1130–8. doi: 10.1038/sj.bjp.0706420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onodera M, Morita I, Mano Y, Murota S. Differential effects of nitric oxide on the activity of prostaglandin endoperoxide H synthase-1 and -2 in vascular endothelial cells. Prostaglandins Leukot Essent Fatty Acids. 2000;62:161–7. doi: 10.1054/plef.2000.0136. [DOI] [PubMed] [Google Scholar]
- 29.Clancy R, Varenika B, Huang W, et al. Nitric oxide synthase/COX cross-talk: nitric oxide activates COX-1 but inhibits COX-2-derived prostaglandin production. J Immunol. 2000;165:1582–7. doi: 10.4049/jimmunol.165.3.1582. [DOI] [PubMed] [Google Scholar]
- 30.Upmacis RK, Deeb RS, Hajjar DP. Regulation of prostaglandin H2 synthase activity by nitrogen oxides. Biochemistry. 1999;38:12505–13. doi: 10.1021/bi983049e. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi K, Watanabe H, Tran QK, et al. Nitric oxide: inhibitory effects on endothelial cell calcium signaling, prostaglandin I2 production and nitric oxide synthase expression. Cardiovasc Res. 2004;62:194–201. doi: 10.1016/j.cardiores.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 32.Goodwin DC, Gunther MR, Hsi LC, et al. Nitric oxide trapping of tyrosyl radicals generated during prostaglandin endoperoxide synthase turnover. Detection of the radical derivative of tyrosine 385. J Biol Chem. 1998;273:8903–9. doi: 10.1074/jbc.273.15.8903. [DOI] [PubMed] [Google Scholar]
- 33.Shimokawa T, Kulmacz RJ, DeWitt DL, Smith WL. Tyrosine 385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J Biol Chem. 1990;265:20073–6. [PubMed] [Google Scholar]
- 34.Salvemini D. Regulation of cyclooxygenase enzymes by nitric oxide. Cell Mol Life Sci. 1997;53:576–82. doi: 10.1007/s000180050074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vassalle C, Domenici C, Lubrano V, L'Abbate A. Interaction between nitric oxide and cyclooxygenase pathways in endothelial cells. J Vasc Res. 2003;40:491–9. doi: 10.1159/000074550. [DOI] [PubMed] [Google Scholar]