

Abstract
Gain-of-function mutations in the genes encoding Janus kinases have been discovered in various haematologic diseases. Jaks are composed of a FERM domain, an SH2 domain, a pseudokinase domain and a kinase domain, and a complex interplay of the Jak domains is involved in regulation of catalytic activity and association to cytokine receptors. Most activating mutations are found in the pseudokinase domain. Here we present recently discovered mutations in the context of our structural models of the respective domains. We describe two structural hotspots in the pseudokinase domain of Jak2 that seem to be associated either to myeloproliferation or to lymphoblastic leukaemia, pointing at the involvement of distinct signalling complexes in these disease settings. The different domains of Jaks are discussed as potential drug targets. We present currently available inhibitors targeting Jaks and indicate structural differences in the kinase domains of the different Jaks that may be exploited in the development of specific inhibitors. Moreover, we discuss recent chemical genetic approaches which can be applied to Jaks to better understand the role of these kinases in their biological settings and as drug targets.
Keywords: Janus kinases, Jak mutations, inhibitors, structural models, chemical genetics, analogue-sensitive kinases
Introduction
In its contribution to tumourigenesis the protein kinase family is overrepresented compared to other protein families [1, 2]. Mutations of kinase genes (point mutations, gene amplification, deletion, insertions and translocations) in cancer are common. In some disease settings, certain kinases are known to be mutated with high incidence. In melanoma BRAF is often mutated (up to 70%) [3], in non-small lung cancer EGFR is associated with the disease [4] and recently some myeloproliferative neoplasms were described to harbour Jak2 mutations with high incidence [5]. Based on the structural knowledge about Janus kinases, we discuss the recently discovered disease-driver mutations in Jaks and the currently available Jak inhibitors. We address chemical genetics approaches which may be applied to Jaks to further elucidate their role in biology as well as relevant drug targets.
Jaks in disease
The family of Janus kinases comprises four mammalian members: Jak1, Jak2, Jak3 and Tyk2. While Jak1, Jak2 and Tyk2 are ubiquitously expressed, Jak3 is confined mainly to cells of the haematopoietic system [6–8]. Janus kinases (Jaks) as mediators of cytokine signalling are involved in a variety of biological processes including haematopoiesis and the regulation of the immune system. The Jak binding preference of cytokine receptors (see Table S1; [7, 9–12]) and the specificity of the various signalling proteins for phosphotyrosine motifs within this receptor, along with other factors, determines the signalling characteristics of the different cytokines.
Evidently, Jaks are involved in inflammatory and immune disorders in which cytokines play crucial roles (e.g. rheumatoid arthritis and psoriasis) [13, 14] and in cytokine-dependent cancers such as multiple myeloma. Jak3 mutations and deletions are described to lead to severe combined immunodeficiency (SCID) [15]. Activating Jak2 fusion proteins (TEL-Jak2, PCM1-Jak2, ETV6-Jak2 and SSBP2-Jak2) have been described to evoke lymphoid and myeloid leukaemia and MPN-U [16–24]. In 2005, an acquired gain-of-function mutation affecting the kinase-like domain of Jak2, V617F, was found in patients with polycythemia vera, essential thrombocythemia and primary myelofibrosis [25–29] (incidence: PV (99%), ET (50%), PMF (50%); estimated 2–5 new cases per 100,000 per year; estimated cases for PV, ET and PMF is 80,000–100,000 in the United States) [30]. Gain of function mutations of Jak3 were found in ALL and AMKL patients while activating mutations in Jak1 have also been described in ALL (Table 1).
Table 1.
Patient-derived mutations in Janus Kinases identified in haematological diseases*
| Jak | Domain | Mutation | Validated effecta | Diseaseb | Corresponding mutants in other Jaks | Ref. | |
|---|---|---|---|---|---|---|---|
| Prl | Sig | ||||||
| Jak2 | Pseudokinase | M535I (Exon12) | + | – | ch-AMKL | Jak3-M511 | [82, 202] |
| F537I (Exon12) | ni | ni | PV | / | [203] | ||
| K539L (Exon12) | + | + | PV, IE | / | [40, 204, 205] | ||
| F537-K539delinsL (Exon12) | + | + | PV, IE | / | [40, 204–207] | ||
| H538-K539delinsL (Exon12) | ni | ni | PV, IE | / | [207, 208] | ||
| H538Q + K539L (Exon12) | +c | +c | PV, IE | / | [40, 205] | ||
| H538D + K539L + I546S (Exon12) | ni | ni | PV | / | [205] | ||
| H538-K539del (Exon12) | ni | ni | PV | / | [205] | ||
| V536-F547dup (Exon12) | ni | ni | PV | / | [205] | ||
| V536-I546dup11 (Exon12) | ni | ni | PV/IE | / | [207] | ||
| F537-I546dup10 + F547L (Exon12) | ni | ni | PV/IE | / | [207] | ||
| I540-E543delinsMK (Exon12) | ni | ni | PV/IE | / | [207, 209] | ||
| R541-E543delinsK (Exon12) | ni | ni | PV, IE | / | [207–209] | ||
| N542-E543del (Exon12) | + | + | PV, IE | / | [40, 204–208] | ||
| E543-D544del (Exon12) | ni | ni | PV, IE | / | [204, 205, 207, 210] | ||
| D544-L545del (Exon12) | ni | ni | PV | / | [205] | ||
| K607N (Exon14) | ni | ni | AML | / | [211] | ||
| L611S (Exon14) | + | + | ch-B-ALL | / | [82, 212, 213] | ||
| C616Y +V617F (Exon14) | ni | ni | PV | / | [214] | ||
| V617F (Exon14) | + | + | PV, ET, PM, HES, CMML, SM, CNL, JMML, RARS, RA RCMD, RAEB, AML, IE, RARS-T, MDS/MPN-U, MPN-U | Jak1-V658 | [25–29, 82, 208, 211, 215–221] | ||
| C618R+V617F (Exon14) | ni | ni | PV | CR: Jak3-A593 | [222, 223] | ||
| D620E (Exon14) | ni | ni | PV, leukocytosis | / | [224, 225] | ||
| Δexon15+16 (ΔN622-D710) | ni | ni | MDS/MPN-U | / | [226] | ||
| I682F (Exon16) | + | + | ch-B-ALL | / | [97] | ||
| I682AQG (Exon16) | ni | ni | DS-B-ALL | / | [97] | ||
| R683G (Exon16) | + | + | DS-B-ALL, B-ALL | Jak1-R724 Jak3-R657 | [97, 188, 227, 228] | ||
| R683S (Exon16) | + | + | DS-B-ALL, B-ALL | Jak1-R724 Jak3-R657 | [82, 97, 188, 227, 228] | ||
| R683T (Exon16) | ni | ni | DS-B-ALL | Jak1-R724 Jak3-R657 | [227] | ||
| R683K (Exon16) | + | + | DS-B-ALL | Jak1-R724 Jak3-R657 | [188] | ||
| ΔIREED ( = I682-D686del) (Exon16) | + | + | DS-B-ALL | / | [229] | ||
| I682delinsMPAP (Exon16) | ni | ni | DS-B-ALL | / | [188] | ||
| L681+I682delinsTPYEGMPGH (Exon16) | ni | ni | DS-B-ALL | / | [188] | ||
| Kinase | R867Q | ni | ni | ch-B-ALL | / | [97] | |
| D873N | + | + | ch-B-ALL | / | [97] | ||
| T875N | + | + | AMKL-cell line | / | [39] | ||
| P933R | + | + | ch-B-ALL | / | [97] | ||
| Jak1 | FERM | I62V | ni | ni | B-ALL, T-ALL | / | [186] |
| K204M | ni | ni | B-ALL | / | [186] | ||
| R360W | ni | ni | T-ALL | / | [186] | ||
| SH2 | T478S | – | + | AML | / | [230, 231] | |
| S512L | ni | ni | T-ALL | / | [186] | ||
| Pseudo-kinase | V623A | – | + | AML | / | [230, 231] | |
| L624_R629>W | ni | ni | ch-B-ALL | / | [97] | ||
| A634D | + | + | B-ALL, T-ALL | / | [186] | ||
| S646F | + | + | ch-B-ALL | / | [97] | ||
| L653F | ni | ni | ch-T-ALL | / | [186] | ||
| V658F | + | + | T-ALL, AML, ch-B-ALL | Jak2-V617 | [96–98] | ||
| R724H | + | + | B-ALL, T-ALL | Jak2-R683 Jak3-R657 | [186] | ||
| L783F | ni | ni | T-ALL | / | [98] | ||
| Kinase | R879C | – | – | T-ALL | / | [186] | |
| R879H | ni | ni | T-ALL | / | [186] | ||
| R879S | ni | ni | T-ALL | / | [186] | ||
| Jak3 | FERM | I87T | + | + | TMD | / | [232, 233] |
| P132T | + | + | AMKL, AML | / | [191, 231] | ||
| P151R + 2851–3442del592 | nid | nid | DS-TMD | / | [234] | ||
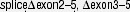 |
nie | nie | DS-TMD | / | [234] | ||
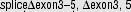 |
nie | nie | DS-AMKL | / | [234] | ||
| SH2 | P434R | ni | ni | DS-TMD | / | [234] | |
| SH2-Psk-linker | Q501H + R657Q | +f | +f | MGS cell line (DS-AMKL) | / | [232] | |
| Pseudo-kinase (Psk) | M511I + V722I | ni | ni | DS-ML | Jak2-M535 | [235] | |
| A572V | + | + | cell line CMK (AMKL) | / | [191, 202] | ||
| A573V | + | + | DS-AMKL, CMY cell line (AMKL) | / | [202, 234] | ||
| M576L | ni | ni | AMKL | / | [233] | ||
| A593T + A573V | ni | ni | DS-AMKL | Jak2-C618 | [233] | ||
| R657Q + Q501H | +f | +f | MGS cell line (DS-AMKL) | RQ: Jak1-R724, Jak2-R683 | [232] | ||
| V722I | + | + | DS-AMKL | / | [191, 202] | ||
| Psk-K-linker | S789P | ni | ni | ch-B-ALL | / | [97] | |
| Kinase (K) | 2851–3442del592 | nid | nid | DS-AMKL | / | [234] | |
| Tyk2 | FERM | G363S | ni | ni | AML | / | [231] |
SCID mutations and Jak fusion proteins were not included.
Effect demonstrated by introducing the mutant into a wild-type cellular system. Prl: monitoring of proliferation, Sig: monitoring of activated signalling components, ni: not investigated, (+): increased activity compared to wild-type kinase, (−): no effect compared to wild-type kinase.
Abbreviations used: ALL: acute lymphoblastic leukaemia, AMKL: acute megakaryoblastic leukaemia, AML: acute myeloid leukaemia, B-ALL: B cell precursor ALL, ch: childhood, CML: chronic myelogenous leukaemia, CMML: chronic myelomonocytic leukaemia, CNL: chronic neutrophilic leukaemia, DS: Down syndrome, ET: essential thrombocythemia, HES: hypereosinophilic syndrome, IE: idiopathic erythrocytosis, JMML: juvenile myelomonocytic leukaemia, MDS/MPN-U: unclassifiable myelodysplastic syndrome/myeloproliferative neoplasms, ML: myeloid leukaemia, MPN-U: unclassifiable or atypical myeloproliferative neoplasms, PM: primary myelofibrosis, PV: polycythemia vera, RA: refractory anaemia, RAEB: refractory anaemia with excess of blasts, RARS: refractory anaemia with ringed sideroblasts, RARS-T: RARS with thrombocytosis, RCMD: refractory cytopenia with multilineage dysplasia, SM: systemic mastocytosis, T-ALL: T cell ALL, TMD: transient myeloproliferative disorder.
The effect of the H538Q mutation alone was not investigated.
2851–3442del592 results in a deletion of 192 c-terminal aa of the kinase domain and will thus lead to an inactive kinase.
Results in an alternatively spliced protein with deletions within the FERM domain that will probably lead to an inactive kinase.
Q501H and R657Q show constitutive activity alone and a stronger effect in combination.
Jak2-V617F signal transduction
In murine bone marrow transfer models, introduction of the Jak2-V617F mutation is sufficient to induce a PV phenotype [31–34]. Cytokine receptor binding is necessary for the transforming potential of the Jak2-V617F mutant [31, 35], in obvious contrast to the cytosolic TEL-Jak2 protein. The presence of the EpoR, TpoR or GCSFR is required for cytokine-independent growth in Ba/F3 cells. Interestingly, these cytokine receptors form homodimers upon ligand binding, thus bringing two constitutively activated Jak2-V617F into close proximity. Furthermore mutations in the TpoR were described in ET and PMF, and are known to activate signal transduction [36–38].
The constitutively active Jak2 mutants have been shown to lead to cytokine receptor-dependent constitutive activation of various signalling proteins, such as STATs, MAPKinases and PI3K/AKT [25, 35, 39, 40]. Jak2-V617F was also described to promote G1/S phase transition trough a redox-dependent regulation of cyclinD1 and p27 [41]. Signalling through Jak2-V617F has been described to lead to genetic instability [42]. SOCS1 and SOCS3 mRNA up-regulation has recently been reported in patients with Jak2-V617F-associated myeloproliferative disorders [43, 44]. These proteins are known to down-regulate Jak activity and mediate their degradation [45–48]. It was reported that Jak2-V617F may escape the direct inhibition by SOCS3 [49]. In contrast, SOCS3 was recently shown to negatively regulate Jak2-V617F activity via its kinase inhibitory region and SH2 domain and to mediate Jak2-V617F ubiquitination and degradation [50]. Despite this negative regulation, the constitutive signalling capacity of Jak2-V617F was not totally abrogated and higher levels of steady state Jak2-V617F were observed to have higher levels of constitutive signalling [50]. In patients with PV and PMF, the Jak2-V617F mutation frequently progresses to homozygosity through mitotic recombination, which is less frequently observed in patients with ET [26] and in these MPN the expression level was reported to reflect the allele load [51]. Animal studies also support the hypothesis that higher levels of Jak2-V617F lead from a thrombocytic to an erythrocytic and a fibrotic phenotype [32, 51, 52]. Thus, mechanisms interfering with the negative regulation and degradation of activated Jaks could considerably contribute to the development and progression of MPD and Jak2-V617F-positive leukaemia by increasing the levels of constitutively active Jak2 mutants. Epigenetic silencing of SOCS3 and SOCS1 was recently reported in about 40% of patients with Ph-negative chronic myeloid disorders [53, 54]. Negative regulation of Jak2-V617F by SOCS2 was also described and in the same study inactivation of the SOCS2 gene by hypermethylation was reported in Jak2-V617F positive leukaemic cell lines and in MPN patient cells [55]. The expression and potential mutation of SOCS proteins could be important clinical parameters in patients carrying constitutively active Jak2 proteins.
Structure/function: the potential interest of the Jak domains as drug targets
The domain structure of Jaks (MW: 120–140 kD) is shown in Figure 1. Due to the lack of crystallographic data, the structure–function relationship of the interaction between cytokine receptors and Janus kinases still remains largely elusive as does the exact sequence of events involved in Janus kinase activation. Sequence similarities between Jak family members have led to the description of seven Jak homology (JH) domains [56], which match the domain structure of Jaks only partially. Only the JH1 and JH2 domains correspond to the kinase and pseudokinase domain. The JH3 to JH7 regions are better described as a FERM and an SH2 domain [56, 57].
Fig 1.
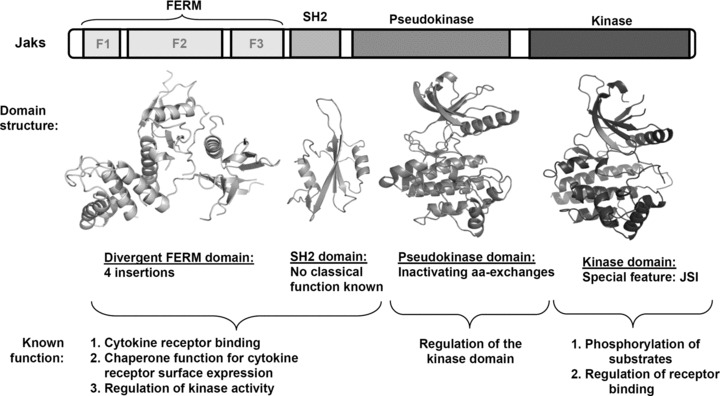
Domain structure of Janus kinases and general functions of the different domains. Model structures of the Jak1-FERM, -SH2 and pseudokinase domain as well as the solved crystal structure of the Jak2 kinase domain (PDB entry code: 2B7A) are represented.
The FERM domain
The N-terminal FERM domain promotes binding to the membrane-proximal box1/2 region of cytokine receptors [58–63]. The FERM domains are clover-shaped domains comprising three subdomains: subdomain F1 with a ubiquitin-like β-grasp fold, F2 with an acyl-CoA-binding-protein-like fold, and F3, which has a PH-domain (pleckstrin homology) fold [64]. Structural data of a growing number of solved FERM domains [64–68] have been the basis for structural modelling and for exploring the function of the postulated Jak FERM domain [57, 69–71], reviewed in Ref. [63]. Taken together, the involvement of rather long sequence stretches within the receptor and Jaks suggests that the interaction is mediated by multiple contact sites, which dictate the Jak position in a defined orientation, and which ultimately become critical for activation. The receptor-Jak interaction probably induces a restructuring of certain receptor residues into defined interaction interfaces. This ‘induced fit-like’ scenario seems probable since the length of the non-structured 65 amino acids of the receptor (23 nanometres) is about three to four times the dimension of the FERM domain (6 to 7 nanometres across). Alternatively, a non-structured cytoplasmic tail of a cytokine receptor would have to adopt a loop structure, which would require to wind repeatedly through the clefts or along the surface of the FERM domain [7, 63]. Whatever the binding mode really is, the involvement of several subdomains (FERM subdomains and SH2) of the Jak and long stretches of protein within the receptor harbours the potential for a very tight and long-lasting interaction. It seems to be a general phenomenon that the mere proximity of Jaks in receptor complexes is not sufficient for their activation, but needs further conformational changes induced by cytokine binding to its cognate receptors [72–75] (reviewed in [63]).
FERM domains of the ERM proteins (ezrin, radixin, moesin) are endowed with the ability to associate to membranes by binding phospholipids. Interestingly, the residues which mediate phospholipid binding in the FERM domain of radixin [65] are not conserved in Jaks, thus indicating that the Jaks are recruited to membranes solely by interaction with cytokine receptors.
Data obtained from Jak3 and Jak1 suggest that even the kinase domain may affect receptor binding [71, 76]. Staurosporine, a broad activity kinase inhibitor, was described to decrease Jak3 binding to the γc receptor chain [76].
The FERM domain as drug target
The FERM domain harbours great potential for the development of specific inhibitors targeting the Jak-receptor interaction, because activating mutations in Jaks are not able to promote signal transduction if the protein is dissociated from cytokine receptors [77, 78]. Interestingly, the intracellular regions of different cytokine receptors show limited similarity concerning the nature and the position of the residues involved in Jak binding, although they bind the same Janus kinase (for review see [63]). Hence, interfering with the receptor/Jak interaction by just disturbing the interaction site of a defined cytokine receptor might be a very specific way to inhibit cytokine signal transduction. What might be limiting the use of interaction inhibitors is the fact that one Jak can promote pathogenic signal through different receptors, i.e. for factor-independent growth of Ba/F3–Jak2-V617F cells the presence of the EpoR, TpoR or GCSFR is sufficient. If several cytokine receptor-Jak interactions have to be targeted, finding one compound inhibiting all will complicate the task. The lack of structural information on the FERM-cytokine receptor interface is a major hindrance to the rational development of such interaction-inhibitors. Nanomolar allosteric inhibitors for Jaks have not been described yet. Nevertheless, this approach might be superior concerning specificity compared to kinase inhibitors and could be a less costly alternative to protein drugs such as cytokine antagonists.
The SH2 domain
The FERM domain is followed by a predicted SH2 domain which does not seem to fulfil a classical SH2 domain function, and which shows some unconventional features [63, 79]. There is a striking discrepancy in conservation between structural (conserved in all) and functional residues (conserved in only some Jaks) within the Jak SH2 domains. The essential functional arginine residue at position βB5, conserved to 99.8% in SH2 sequences, is only conserved to 80% in all Jak SH2 sequences. The divergent Jak SH2 domain is structurally important for binding to cytokine receptors [79] and for the up-regulation of cytokine receptor surface expression [79–82] but could not be shown to have a phosphotyrosine binding function [79].
The SH2 domain as drug target
SH2 domain inhibitors, interfering with the phosphotyrosine binding ability of SH2 domains, have been described [83–85] and nanomolar activities have been achieved in the case of the Grb3 SH2 domain. Because Jaks have a divergent SH2 domain for which no phosphotyrosine binding has been shown to date, this approach might have lower priority until new data on the SH2 domain are uncovered.
The pseudokinase domain
Of the 518 protein kinases, 48 family members contain pseudokinase domains [86]. Only five of these contain an additional functional kinase domain. These are the four Janus kinases and the serine/threonine kinase GCN2. Even pseudokinases with important sequence degeneration have a classical kinase fold quite close to the fold of their nearest functional relative [87].
Pseudokinases are kinase domains that lack conserved residues critical for kinase activity. The loss of functionality is thought to be mainly due to sequence degeneration in the G-loop (ATP binding), the VAIK motif (ATP orientation), the HRD motif in the catalytic loop and the DFG motif (Mg2+ binding and ATP orientation). The major defect in Jak pseudokinase domains is the loss of the catalytic aspartate residue in the catalytic HRD loop. In addition to this, the Jak pseudokinase domains have a non-optimal G-loop, although ATP binding is not necessarily precluded. In the VAIK motif, Jaks have a large residue instead of the conserved alanine, which could represent a hindrance to ATP binding. Furthermore, the pseudokinase domains in Jaks lack tyrosine residues in the activation loop whose phosphorylation would stabilize the activation loop in the active conformation of a functional kinase.
Pseudokinases have been described to have diverse functions, e.g. to be scaffolding domains or to interact with kinase domains to regulate kinase activity [88]. Pseudokinases and kinases from two different protein chains have been described to interact and the pseudokinase domain could thereby activate the kinase [89, 90]. In the four Jaks and in GCN2 the pseudokinase domains suppress the activity of the adjacent kinase domain [91–95]. Many mutations in the pseudokinase domain that lead to hyperactive Janus kinases have recently been discovered in patients (see Table 1). However, because no structural data are yet available, the mechanism by which the pseudokinase domain inhibits the kinase can only be speculated about.
Interestingly, the Jak2-V617F mutation was described to be transferable to Jak1 and Tyk2 but not to Jak3 [96]. Later, the corresponding Jak1-V658F mutant was identified in ALL and AML patients [97, 98]. Similarly, transfer of the Jak3-A572V mutation to Jak2 does not lead to activation of Jak2 (C. Haan, unpublished data). Although it is too premature to be sure, there might be a difference in how Jak3 activity is inhibited by the pseudokinase domain compared to the three others.
The pseudokinase domain as drug target
The lack of structural information on the pseudokinase–kinase domain interface and the lack of knowledge about the activation mechanism of Jaks preclude any attempts of rationally designing possible inhibitors which target this interaction. Different molecular models exist and are discussed later.
The pseudokinase domain might have the ability to bind ATP or inhibitors and, as in the case of kinase domains, this might impact on its conformation and on the activation state of the protein. In RNAseL the binding of nucleotides to the pseudokinase domain regulates the RNAse domain [99]. Potentially, a protein kinase inhibitor could preferentially bind the pseudokinase domain and have an impact on kinase activation. Since binding assays of inhibitors into kinase domains exist [100], it would be interesting to investigate binding of inhibitors to pseudokinase domains too.
The kinase domain
The kinase activity is mediated by the C-terminal kinase domain. All protein kinases possess a catalytic domain that comprises approximately 300 amino acids. They share the bilobal kinase fold: The N-terminal lobe is composed of five β-strands and a single α-helix. The C-terminal lobe is predominantly α-helical and contains the regulatory activation loop (A-loop). The active site is located in the cleft between the two lobes. The inactive conformation of kinases seems to present more differences between kinases than the active conformation. For more and more kinases structures of the active and inactive conformations exist and have helped explain the conformational rearrangements that are observed during the transition from the inactive to the active state of the kinase [101]. The major events of kinase activation are largely conserved although there are subtle but important differences. For example, Jak3 is the only Jak family member in which an alanine residue directly precedes the DFG-motif (in contrast to a glycine residue in the other Jaks). This subtle difference could directly affect the conformation of the A-loop in the way that was already discussed for the inactive insulin receptor (GDFG-motif) and fibroblast growth factor receptor (ADFG-motif) kinase domains [102]. Applied to the Jaks this could mean that the A-loop in inactive Jak1, Jak2 and Tyk2 would adopt an ATP-competitive conformation and allow the accommodation of an A-loop tyrosine into the substrate binding site whereas the A-loop of inactive Jak3 would allow ATP binding and potentially preclude binding of an A-loop tyrosine to the substrate binding site.
Alignments of all published Janus kinase sequences show that all family members, with the exception of hopscotch, the Drosophila homologue, contain a 14–15 amino acid Jak-specific insertion (JSI) which was predicted to contain an α-helix and has been shown to be crucial for kinase activity [103, 104]. Such an insertion is absent in the pseudokinase domains of the Jaks. The published crystal structures of the Jak1, Jak2 and Jak3 kinase domains have proven the existence of this additional helix within the C-lobe of the Jak kinase domain which was termed αH-helix for Jak2 and FG-helix in the case of Jak3 [105–107]. The region encompassing the Jak2 αH-helix and the preceding 3/10 helix was also named Lip-region [105]. This special feature in Jaks is lining the substrate binding site of the kinase and lies in close proximity to the catalytic cleft of the enzyme. Mutations of structurally important residues within the JSI impair kinase activity while the mutation of an exposed residue, M1062, conferred constitutive activity [104]. Interestingly, mutations within the JSI differentially affect IFNγ and Epo signalling.
Another interesting feature of the Jak kinase domains is that they harbour a large methionine gatekeeper residue, reducing the size of the so-called ‘hydrophobic pocket II’[108], which often confers specificity of the binding to kinase inhibitors.
The sequential similarity of the Jak kinase domains is quite high and the solved structures of three of the Jaks also show little difference in and around the ATP binding pocket. Figure S1 highlights some of the non-conserved residues in and around the catalytic site and the predicted substrate-binding site of the Jaks which might be exploited for the design of more specific inhibitors. Interestingly, Jak3 is the only Janus kinase having a cysteine residue at position C909 in close proximity to the ATP binding pocket. In the case of the EGFR, which also carries a cysteine at this position, irreversible inhibitors were developed by attaching an electrophilic group to ATP-competitive inhibitors. The electrophilic group targets the cysteine residue and covalently attaches the inhibitor to the kinase domain [109, 110]. Irreversible inhibitors for EGFR are tested in clinical trials [111–114] and these trials will show how these compounds perform with regard to selectivity and toxicity. The toxic potential is hard to evaluate but the amount of possible off-kinase-targets potentially reacting with the electrophile is a risk [115]. Nevertheless, it could be worth a try to modify an existing Jak-specific inhibitor with an according electrophilic group to exploit the uniqueness of the cysteine of Jak3 within the Jak family.
The kinase domain as drug target
The potential of the kinase domain as a target for ATP competitive inhibitors is obvious and a variety of those inhibitors are described below. There is, however, one special feature in the Jak kinase domains which might be used to develop specific allosteric inhibitors. As mentioned above, the JSI region contains an extra α-helix which is lining the substrate binding site of Jak kinase domains and which was shown to be crucial for kinase activity [104]. It is an almost unique feature in kinases as such a long insertion at this position can only be found in the mitogen-activated protein kinase kinase 1 (MEK1). MEK1 has a longer insert of about 40 aa and the crystal structure of MEK1 shows that the insert also contains an additional α-helix which is located further away from the substrate binding site at the base of the C-lobe of the kinase domain [116]. As the JSI region shows some differences between the Jaks and is quite close to the substrate binding site, the JSI region and its surrounding may represent a suitable target site for allosteric inhibition (Fig. S1).
Summary
Although the exact sequence of events in the activation mechanism of the Jaks remains elusive, it is evident that the different domains influence their neighbouring domains structurally and functionally. The FERM and SH2 domains regulate binding to cytokine receptors and are involved in the regulation of surface expression of at least some cytokine receptors. The FERM domain has also been described to influence kinase activity. Even the structural integrity of the pseudokinase domain of Tyk2 is vital for high affinity binding of cytokines to the IFNAR [94, 117], suggesting a role of Tyk2 in ‘organizing’ the receptor complex. Data obtained from Jak3 and Jak1 show that also the kinase domain may affect receptor binding [71, 76]. All this is indicative of a complex interplay of the different Jak subdomains with each other and with the cytokine receptor, which very likely reflects different activation states, all of which might be susceptible to allosteric inhibitor treatment.
The growing family of ATP-competitive nanomolar Jak inhibitors
Only a small fraction of the kinome is targeted by selective inhibitors, although the situation is improving at least as far as the Janus kinase family is concerned. The specificity of inhibitors is thought to be of critical importance for its clinical utility and for its utility in research. The experience with kinase inhibitors in clinical trials has obviously changed this view since multikinase inhibitors have been approved for the use in patients with cancer and are now tested in many disease settings. Some were well tolerated with minor side effect even though they target many signalling pathways (e.g. sunitinib) [100]. Nevertheless, clinical trials will have to show if higher selectivity, which could translate into better tolerability, is indeed dispensable, especially in diseases where prolonged treatment is necessary. But specific inhibition of one Jak certainly does not guarantee that one biological response is targeted specifically. The whole variety of cytokine receptors use only four Jaks which means that even a specific inhibitor targets multiple cytokine pathways. Especially Jak1 is involved in signalling through many classes of cytokines. Table S1 summarizes the different cytokine families and the Jaks which are activated upon stimulation [7, 9–11].
The recent discovery of more and more Jak mutations in disease has sparked the development of a host of low nanomolar inhibitors with different selectivity within the Jak family, and some compounds are tested in clinical trials [5, 13]. Taken together, Jak inhibitors are tested for use in transplant rejection, inflammatory diseases (rheumatoid arthritis, psoriasis, and chronic obstructive pulmonary disease), haematologic disorders (MPN, leukaemia, multiple myeloma) and prostate cancer (for an overview, see e.g. http://clinicaltrials.gov). While Jak3, Jak2, and Jak1 are in the main focus in Jak inhibitor development, Tyk2 might also be an interesting target. A naturally occurring point mutation in the pseudokinase domain of Tyk2 impairs IL-12 and IFN-mediated signalling and was associated with resistance to collagen-induced arthritis in a murine model [118]. Moreover, it has recently been shown that polymorphisms at the Tyk2 locus are associated with systemic lupus erythematosus [119]. In this regard, an inhibitor of Tyk2 may become a potential future drug for autoimmune diseases.
CP690,550, initially thought to be Jak3-specific, is now recognized to have significant activity also against other Jaks. The compound is tested for use in transplant rejection, psoriasis and rheumatoid arthritis, all disorders in which a suppression of immune responses is desired, and has proved active in human disease [13]. An increasing number of Jak inhibitors enter clinical trials (R348, INCB18424, TG101348, XL019, CEP701, SB1518, AT9283, AZD-1480, VX680,…). Some trials have already shown preliminary encouraging results. For example, patients with PMF greatly benefited from INCB18424 treatment (e.g. reduction of spleen size, inflammatory cytokine reduction). Interestingly, the allele load of V617F was not significantly reduced [5, 13, 120–126]. However, combination therapy with TG101209 and panobinostat, an HDAC inhibitor or AUY922, an HSP90 inhibitor, attenuates Jak2V617F levels [127]. Another HDAC inhibitor, ITF2357, showed selective activity against Jak2-V617F-positive cells in comparison to Jak2-WT expressing cells [128, 129]. Thus, combining epigenetic therapy (HDAC inhibitors, DNA methyltransferase inhibitors) with Jak inhibitors might be generally effective in treatment of V617F-positive disease (reviewed in [130]). Interestingly, also long-term IFN-α treatment has been described to lead to complete and sustained molecular remission in patients with PV [131–133]. Because IFN-α signals via Jak1 and Tyk2, a specific Jak2 inhibitor might potentially be combined with IFN-α treatment to also achieve a reduction of the allele load.
Table 2 summarizes characteristics of Jak inhibitors described to date which show ≤200 nanomolar activity in kinase assays or have ≤500 nanomolar activity in cellular assays. Some of these inhibitors are selective pan-Jak inhibitors, others have a preference for one or several family members. The list does not represent the whole activity in the field of nanomolar Jak inhibitors. For instance, some publications described the development of nanomolar compounds for Jak2 and Jak3, but some of these were not further characterized [134–137]. The chemical structures of some of these nanomolar Jak inhibitors are shown in Figure S2. There is a whole range of structural scaffolds (pyrrolopyrimidines, pyridones, bisubstituted pyrimidines, nicotinonitrils, oxindoles, staurosporine analogues, pyrazole-benzimidazoles, aminoindazoles…) that can be used to inhibit Jaks.
Table 2.
Jak targeting inhibitors
| Inhibitor | Primary targets IC50 (nM) | Other targets IC50 (nM) | PY-STAT IC50 (nM) | Cell growth IC50 (nM) *Patient cells | Literature |
|---|---|---|---|---|---|
| CP-690,550 | Jak3 (0.7) (2.2) (1§) | DCAMKL3 (4.5) | ≥100 | 11–100 | [100, 139, 236–238] |
| Jak1 (3) (112§) | Tyk2 (250) | ||||
| Jak2 (2) (5) (20§) | |||||
| PF 956980 | Jak3 (4) | Other Jaks (n.d.) | 23–188 | [140] | |
| TG101209 | Jak2 (6) | Jak3 (169); Flt3; RET | 300–600 | 16–200;* 300–600 | [127, 239] |
| TG101348 | Jak2 (3) | Jak1 (105); Tyk2 (405) | 300;* 300 | [30, 240, 241] | |
| Jak3 (1040); Flt3;RET | |||||
| INCB018424 | Jak1 (2.7); Jak2 (4.5) | Jak3 (322); Tyk2 (19) | 100–300 | 81–300;* 67 | [120, 123, 125, 126, 242] |
| INCB16562 | Jak1 (9); Jak2 (2) | Jak3 (1895); Tyk2 (28) | 50–128 | 133;* 110 | [243] |
| AZD1480 | Jak2 (0.4) | Jak1 (1.3); Jak3 (3.9) | 46 | 60 | [244] |
| Pyridone 1 | Jak1 (1); Jak2 (2,1) | 85 | 500 | [245] | |
| Jak3 (11); Tyk2 (7) | |||||
| Pyridone 6/Jak inhib.I | Jak1 (15); Jak2: (1) | 67 | 50–100 | [246] | |
| Jak3 (5); Tyk2 (1) | |||||
| XL019 | Jak2 (2) | Jak1(130); Jak3 (250) | 60 | [124, 247, 248] | |
| Tyk2 (340) | |||||
| AT9283 | Jak2 (1.2); Jak3 (1.1) | Aurora A/B; AblT315I | 100–300 | 88 | [249, 250] |
| CEP-701 | FLT3; Jak2 (1) | 10–30 | 30–100;*≤100 | [251, 252] | |
| AZ960 | Jak2 (3); Jak3 (9) | TrkA; Ark5; Aurora-A | 15–22 | 25–33 | [253] |
| R348 | Jak1/Jak3 pathways | Syk | 260 | 180 | [254] |
| R723 | Jak2 (2) | Jak1(∼1000); Jak3(∼20) | 130–200 | [255] | |
| CYT387 | Jak2 (18); Jak1 (11) | Jak3 (155); CDK1;TBK1 | 400 | ≥200;*≥500 | [256–258] |
| SGI1252 | Jak2 (2–19); Jak1 (15); Tyk2 (8) | Jak3 (1302) | 76 | 63–472;* 100 | [259] |
| ONX0803/SB1518 | Jak2 (19–22) | 81 | [260] | ||
| PS-020613 | Jak3 (3.4) | Jak2 (105) | 64 | [261] | |
| Jak3 inhib. VI | Jak3 (27) | Jak2 (600) | ∼1000 | 250–750 | [262] |
| Pyrimid. 26 | Jak3 (45) | Jak2 (124) | 90 | [134] | |
| Gö6976 | PKC | Jak2 (130); Jak3 (370) | 500 | ≥500;*≥500 | [263] |
| MK-0457/VX680 | Aurora (18–36) | Jak2 (190) | 295 | [100, 264–266] | |
If several values between parentheses are given these represent alternative measurements from different publications. PY-STAT: phosphorylation of signal transducer and activator of transcription (STAT); §: Initial values given in the first description paper of CP-690,550; ∼: approximation.
DCAMKL3: Doublecortin-like and CAM kinase-like 3; Aurora: Aurora kinase; PKC: Protein kinase C; CDK1: Cyclin dependant kinase 1; TBK1: Tank binding kinase 1; TrkA: Tropomyosin Related Kinase A; Ark5: AMPK-related protein kinase 5; FLT3: fms-related tyrosine kinase 3; AblT315I: Ableson kinase T315I; RET: Rearranged during transfection. *Data were generated using patient derived cells.
Quite a number of compounds that were described to have micromolar activity in kinase and cellular assays are not mentioned here since many nanomolar inhibitors are now known. These compounds are more likely to have off-target effects since it was estimated that off-kinase-target effects in cellular assays are low below 1 μM, borderline between 1 and 10 μM and high above 10 μM [138]. For instance, largely used micromolar Jak inhibitors (WHI-P131/Janex1/Jak3-inhibitor-I, WHI-P154/Jak3-inhibitor-II and AG490 and others) were recently described to have micromolar activity on Jak in in vitro kinase activity and to have an effect on Jak mediated cell growth above a concentration of low- to mid-micromolar ranges [139–141]. WHI-P131 and WHI-P154 were shown to be EGFR inhibitors with IC50 values in the low nanomolar range [140]. The two inhibitors also had activity against 5, respectively 8 other kinases out of 30 tested. They showed no activity below 10 μM in cellular assays in comparison to the nanomolar activity of CP-690550 and PF-956980.
Analogue-sensitive kinases and possible applications to Jaks
Chemical genetics to characterize kinases
Small molecule kinase inhibitors have been very useful to better understand the pathways activated by Janus kinases in different physiological and disease settings. However, most compounds do not only inhibit one Jak but also inhibit other kinases, in particular other Janus kinases. Thus, it is not possible to directly attribute the biochemical effects of an inhibitor only to its effects on the kinase in question. Off-target effects of drugs often contribute to toxicity although they may also have beneficial clinical effects which can be exploited (e.g. gleevec also inhibits c-Kit) [142]. A better understanding of the function of a given kinase as well as the effects of its specific inhibition would be very helpful in drug development.
Different approaches are available to investigate the importance of a given kinase. ‘Knockout’ approaches have provided important insights into the roles of specific proteins. However, knockout mice may not be viable at all, or ‘secondary’ adaptations may occur as surviving cells (and organisms) have been selected for survival. si-RNA-based suppression of a certain protein may be incomplete and could evoke double-stranded RNA-mediated side effects. ‘Knockout’ or si-RNA approaches might also have effects which cannot be attributed to the enzymatic activity of the targeted kinase but, e.g., to a potential structural role of the kinase.
A chemical genetic approach has been established recently to address the importance of a given kinase: A genetically modified kinase and a chemically modified ATP or inhibitor are the key ingredients in this approach which confers specificity to a bulky ATP/inhibitor by engineering the kinase of choice so that it can accommodate the modified ATP/inhibitor. The so-called ‘ATP’- or ‘inhibitor-analogue’ is thus specifically inhibiting the so-called ‘analogue-sensitive kinase’ (as-kinase). To achieve this, the ‘gatekeeper’ residue in the ATP binding pocket of a kinase is mutated to a smaller residue (e.g. glycine) (Fig. 2). Thereby, the space of the ATP binding pocket is increased and is now accessible to a ‘bulky’ derivative of a kinase inhibitor (or of ATP) which does not fit into the ATP binding pocket of the wild-type kinase of interest, nor into the one of other kinases present in the cell [143, 144]. The chemical genetic approach is applicable to the whole kinome, and presents some advantages over other systems. Table 3 provides an overview of tyrosine kinases that were successfully investigated in this way.
Fig 2.
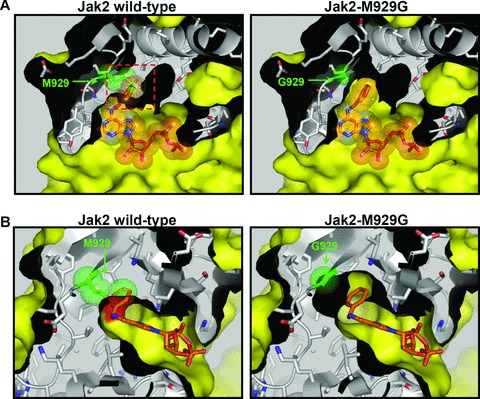
Principle of the specific inhibition of analogue-sensitive kinases. (A) Top view and (B) side view of the catalytic cleft of the Jak2 kinase domain with the bulky ATP-analogue N6-benzyl-ADP (highlighted in orange). The sterical clash between the benzyl group of the bulky inhibitor and the gatekeeper residue M929 in the wild-type Jak2 kinase domain is highlighted as a red frame (A) or surface (B). Mutation of the gatekeeper residue to glycine (Jak2-M929G) extends the catalytic pocket and allows the accommodation of the bulky compound. The representation was generated using the solved crystal structure of the Jak2 kinase domain (PDB entry code 2B7A).
Table 3.
Described analogue-sensitive tyrosine kinases: (a list of analogue-sensitive serine/threonine kinases as well as those of yeast and other organisms is provided in [181])
| Y-Kinase | Gatekeeper residue | Citation |
|---|---|---|
| v-Src | I338 | [144, 156, 177–179, 267, 268] |
| c-Src | T338 | [144, 180, 201] |
| fyn | T339 | [144, 179, 201, 268] |
| Abl | T325 | [144] |
| BCR-Abl | T315 | [269] |
| lck | T316, | [270] |
| v-erbB | T210 | [271–273] |
| EGFR | T790 | [201, 271] |
| TrkA | F592 | [168] |
| TrkB | F616 | [168, 171–173, 274] |
| TrkC | F617 | [168] |
| Btk | T474A | [167] |
| Zap70 | M414 | [150] |
| Syk | M442 | [151, 152] |
Chemical genetics in practice: possible pitfalls and requirements
A newly generated ‘analogue-sensitive’ mutant kinase has to be tested to see whether it fulfils certain criteria:
First, it has to be verified that the mutated kinase still performs ‘normally’, like the wild-type kinase, as it has been reported that the introduced gatekeeper mutation may impair ATP and substrate binding or catalysis. Interestingly, the significance of the gatekeeper for the structure of the active site also becomes evident by gatekeeper mutations ‘naturally’ occurring in kinases in cancer. A good example for this is the disease-relevant mutation of the small gatekeeper residue (threonine 790) in the EGF receptor to a larger methionine. This mutation was identified in lung cancer patients and confers resistance to the kinase inhibitors erlotinib and gefitinib [112, 145]. A similar mutation of the gatekeeper was also described for BCR-Abl (T315I) in CML patients [146, 147]. It is thought that the EGFR mutation T790M derepresses kinase activity by favouring the active conformation of the conserved DFG-motif within the catalytic cleft and by increasing the affinity of ATP binding [148]. Furthermore, it was postulated that this mutation to a bulkier residue may increase the stability of the ‘hydrophobic spine’, a stack of hydrophobic residues that forms upon activation of protein kinases [148, 149]. The converse argument could thus be applied for the chemical genetic approach where a large gatekeeper (notably a methionine residue in case of the Janus kinases) is mutated to a smaller residue such as glycine (Fig. 2). The effects of such a mutation cannot be precisely predicted but a reduced activity of the kinase could be a consequence. In line with this, a reduced kinase activity upon mutation of the gatekeeper residue for chemical genetic approaches has been described [150–152]. In cases for which the gatekeeper mutation led to an inactivation of the kinase, this could be overcome by exchanging the gatekeeper by another amino acid (e.g. alanine instead of glycine [144, 151]) and/or by introducing secondary mutations, either by random mutagenesis or by a structure-guided mutational approach [150–153]. However, even in cases in which the gatekeeper mutation resulted in an inactive kinase, valuable results could be obtained, e.g. for IRE1. Here, the addition of a bulky inhibitor resulted in an activated conformation of the kinase domain and the activation of the RNAse domain involved in RNA splicing of the ER-stress response transcription factor XBP1 [154, 155].
Second, it has to be verified that the kinase can indeed be inhibited by a bulky inhibitor or accepts a bulky ATP analogue as cofactor. Several of these analogue compounds are commercially available, and novel inhibitors are being developed that have even less effect on any wild-type kinase [156]. Ideally, inhibition should be selective, inducible and reversible.
Advantages and possible applications of the chemical genetics approach
Investigation of specific kinase-mediated effects
This approach circumvents problems inherent to the use of knockout scenarios which are unable to distinguish between catalytic activity-dependent and -independent kinase functions and which might be subject to potential compensation by other kinases. Many kinases are recognized to have other functions in addition to their kinase activity. Many kinases are multidomain proteins which are embedded in signalling complexes with multiple contacts and even kinase domains have been described to have additional functions [154, 157–159]. As for Jaks, they are, apart from their function as enzymes, involved in the stable surface expression of at least some cytokine receptors [63, 79–82, 160–163]. A recent review focuses on cases in which kinase inhibitors and knockouts have produced different functional outcomes [164].
Specific and flexible pharmacologic intervention allows target validation of compounds from drug screens
A clear advantage is the fact that the as-kinase can be reversibly inhibited, and the time period of inhibition can be tightly controlled which is not possible when, for example, the whole protein expression is regulated or if kinase inactive mutants are used [165, 166]. The activity of an analogue-sensitive kinase can be inhibited quickly in a time-dependent manner and can be directly compared to the actions of other small molecule inhibitors (of which the specificity profile is not clear yet) on the corresponding wild-type kinase. With regard to drug screening, this approach promises to be useful in generating gene expression profiles characteristic of inhibition of a certain kinase of the kinome, such a ‘finger print’ may be used, e.g. to identify ‘unspecific’ targets of a novel drug candidate [167]. Mutant alleles encoding as-kinases have also been successfully introduced into mice [168–170]. Administration of bulky inhibitors (by intraperitoneal injection and/or by addition into the drinking water) indeed allowed the assignment of novel in vivo functions, in particular to the BDNF receptor TrkB [168, 171–173]. Such in vivo model systems can also be very valuable for pharmacologically relevant target validation [174]. Using an inhibitor selective of the as-kinase as a reference compound in comparison with a novel drug candidate, it will be possible to clearly differentiate off-target effects from those elicited by the kinase in question. Moreover, these transgenic mice expressing an analogue-sensitive allele could be used to detect biomarkers associated with the activity and inhibition of a given kinase [175].
Identification of direct substrates
Moreover, such analogue-specific mutant kinases can be applied to unequivocally identify direct substrates of the kinases by employing a bulky ATP analogue that is no substrate to wild-type kinases [170, 176, 177]. Direct substrates can be identified by the radioactive label of the transferred gamma-phosphate group of a bulky ATP analogue [177–180]. Recently, also a non-radioactive method for substrate identification has been published. Here, the direct substrates of an analogue-sensitive kinase are thiophosphorylated (using correspondingly modified bulky ATP analogues), followed by alkylation which creates an epitope for a thiophosphate ester-specific antibody [170, 181].
Structure-based interpretation of the Jak mutations
With a rapidly growing number of Janus kinase mutations that are reported it is tempting to speculate about their impact on the molecular events that favour disease development and/or progression. An obvious way to approach this is to bring structural considerations into the game. For the Jaks, this is facilitated by the availability of solved crystal structures of the kinase domains of Jak1, Jak2 and Jak3. Furthermore, molecular modelling approaches allow to rather accurately predict the location of amino acid residues in protein domains that show a high degree of conservation. However, the prediction of the molecular effects of specific mutations requires biochemical information about the activation status of the respective mutant Janus kinase. Here we provide an overview of the Jak mutations that have been reported in the context of haematologic diseases (Table 1), with the exception of the Jak3 SCID mutations and the different Jak fusion proteins. Many of these mutations have not yet been functionally investigated. As illustrated in Table 1, mutations associated with haematologic diseases can occur in any of the four Jak protein domains. However, most of these activating or potentially activating mutations have been identified in the pseudokinase domain of the Jaks. This is not surprising because this domain is known to be involved in the negative regulation of kinase activity by interacting with the kinase domain [93, 182]. Mutations within the FERM domain and the kinase domain have a much higher probability to either interfere with receptor binding or abrogate kinase activity. Receptor binding is crucial for constitutive activity in mutated Jaks. The Jak2-V617F mutant is rendered inactive if cytokine receptor binding is abrogated, and concomitantly looses its transforming potential [77, 78]. The location of the reported mutations in haematologic neoplasm (Table 1) within the primary sequence of the Jaks is illustrated in Figure S3. The figure shows an alignment of the four human Janus kinases with the sequences of structurally resolved FERM, SH2 and kinase domains. In Figures S4-7, we included representations showing the location of all the mutations in model structures or solved crystal structures of the different Jak domains. Mutations whose activating effects were biochemically validated, i.e. those mutations leading to constitutive activation of signal transduction and promoting factor-independent growth when transfected into cells are underlined or presented in a separate figure for the pseudokinase domain (Figs. S4 and S7). For interpretations, we only considered the biochemically characterized mutations. This is due to the fact that large sequencing studies have shown that the bulk of kinase mutations in cancer are ‘passenger’-mutations. Because ‘driver’-mutations seem to appear at frequencies undistinguishable from the ‘passenger’-mutations, functional testing of candidate ‘driver’-mutations is necessary [183–185].
Mutations within the FERM and SH2 domains
Various mutations within the FERM and SH2 domains of Jak1, Jak3 and Tyk2 were described to be associated with haematologic diseases (Fig. S4 and Table 1) but only for I87T and P132T in the FERM domain of Jak3 as well as for T478S within the SH2 domain of Jak1 biochemical data have been gathered.
Due to the divergent nature of the Janus kinase FERM domain in comparison to other FERM domains, molecular models such as the one presented in Figure S4 can only provide information about the location of highly conserved and structurally important residues. As the alignment in Figure S3 shows, these mutations mostly affect residues which are not well conserved between the Jaks and the sequences of other FERM domains. Thus, the information provided by the model structure remains very hypothetical, especially concerning the location of the residues K204, R360 in Jak1 and P151R in Jak2. The accuracy of the predicted location of I62, P132 and I87 is more reliable. Our model structure of the Jak3 FERM domain suggests that P132 and I87 could be involved in the positioning of the alpha helix in the F1 subdomain by contacting residues at both ends of the helix (L49 for P132/G63 and L64 for I87). As mutation of both P132 and I87 has been shown to lead to constitutive activity of the kinase, one could speculate that the region around the F1 α-helix is involved in the regulation of kinase activity. Interestingly, a mutation within the corresponding helix in Jak1, I62V, was also reported, but unfortunately not further characterized [186].
The effect of the reported mutations within the SH2 domain of Jak1 and Jak3 is hard to predict as two of the mutations are located in loop regions (S512 in the EF-loop of Jak1 and P434 in the DE-loop of Jak3). However, in principle both S512 and P434 are located in interesting regions within the SH2 domain as the EF-loop participates in the recognition of phosphotyrosine motifs in SH2 domains and the DE-loop of the N-terminal SH2 domain of SHP2 was shown to inhibit the phosphatase activity of this enzyme [187]. The reported activating effect of the T478S mutation in Jak1 is also not that obvious. Although this residue is usually involved in the formation of a hydrogen bond network around the phosphate group of a bound phosphotyrosine residue, an exchange to serine should not impair that function. In fact, most SH2 domains contain a serine residue at this position (serine residue in ∼50% and threonine in ∼25% of SH2 domains (see also Fig. S3). Rather than directly affecting a potential interaction with a phosphotyrosine motif, the T478S mutant may partially destabilize the SH2 domain and could thereby affect interdomain interactions in a way that leads to constitutive signalling.
Mutations within the kinase-like domain
The alignment of the pseudokinase domains (Fig. S3) shows that several mutational ‘hotspots’ exist in the primary sequence where mutations in two or three of the Jak family members occur (e.g. the region preceding the β1-strand, the region between the β3- and β5-strands as well as the region between the β6 and β7-strands). Model structures of the pseudokinase domains of Jak1, Jak2 and Jak3 allow predicting the position of the different mutations. As illustrated in Figures 3, S5 and S6, the majority of the pseudokinase domain mutations affect the N-terminal lobe of the domain and modify residues involved in either the postulated interface with the kinase domain [27] or structurally important residues whose mutation can destabilize the N-lobe and thus also affect a possible interface between the pseudokinase and kinase domains. Mutations in the C-lobe of the pseudokinase domain are rare, which could suggest that the structural integrity of this region is essential for Jak function and/or that its surface does not participate in the kinase domain activity regulation. Figure 3 shows a model structure of the Jak2 pseudokinase domain with the positions of the biochemically validated activating mutations (please refer to Figure S5 for a representation of validated activating pseudokinase mutants for all the Jak family members and to Figure S6 for all identified pseudokinase mutations in the Jaks). Based on our model structures, we hypothesize that the activating exon 12 mutations (referring to Jak2) and the mutations between the β3 and β5 strands (e.g. the Jak2-V617F and most other Jak2 exon 14 mutations) belong to the same structural hotspot (structural hotspot I in Fig. 3) and exert their effects via a similar molecular mechanism. For example, the exon 12-encoded residue K539 (which seems to be a key residue in exon 12) and the amino acid V617 are in close proximity (Fig. 3) and are both exposed at the surface. Thus, the corresponding mutant K539L and V617F could well act via the same molecular mechanism, which is in line with the clinical phenotype of both mutants (MPN, myeloid leukaemia) (Table 1). Aside this first structural hotspot, a second structural hotspot for mutations is apparent at the interface between the N- and C-lobe which regroups the activating exon 16 mutants in the β-sheet of the C-terminal lobe as well as the exon 14 mutant L611S. Interestingly, in the case of Jak2, the mutations located in this structural hotspot II lead to a different clinical phenotype, namely lymphoblastic leukaemia (Table 1). Such genotype-phenotype specificity was recently proposed by Bercovich and colleagues who postulated that the residues R683 and V617F are located in different protein–protein interfaces [188]. Considering all reported activating mutations in the Jak2 pseudokinase domain, we postulate that this genotype-phenotype specificity can be extended to also incorporate the other activating exon 12, 14 and 16 mutations. Thus, mutations in the structural hotspots I and II might, in addition to activating Jak2, influence the recruitment to different signalling complexes including different cytokine receptors and lead to different signalling events. Such a genotype-phenotype specificity is not yet obvious for the corresponding mutations in Jak1 and Jak3, where the same structural hotspots are affected by mutations (please refer to Figs. S5 and S6 and to Table 1).
Fig 3.
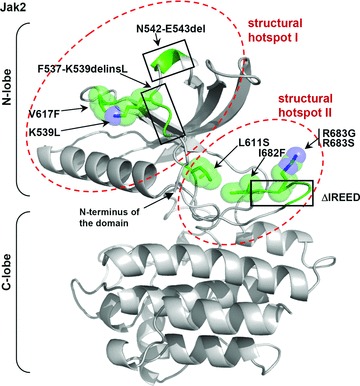
Model structure of the pseudokinase domain of Jak2 with the predicted locations of patient-derived mutations with validated activating effect. Residues for which point mutations were reported in patients are represented as green stick models with spheres indicating the Van-der-Waals radii of atoms. Regions carrying insertions and/or deletions are indicated by a green coloured backbone without stick models (black frames). The structural mutation hotspots I and II are highlighted in red. The model structures of the FERM, SH2 and pseudokinase domains of Jak1, Jak2 and Jak3 shown in this review were generated as described in the Supporting Information. For molecular modelling and graphic representation of all protein structures in this review, the programs WHAT IF [275] and Pymol [DeLano, WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA] were used.
In the context of the Jak2-V617F mutation, previously described molecular models of the full length Jak2 were used to interpret the observed biological effects. The generally accepted theory concerning the effects of the V617F (or more general pseudokinase domain mutations) postulates that the residue V617 is part of the binding interface by which the pseudokinase domain contacts the kinase domain and negatively regulates its activity [27]. Accordingly, mutation of this residue to a larger hydrophobic residue should prevent optimal contact and reduce the affinity of the inhibitory interaction. However, it was shown that a V617Y exchange does not lead to constitutive activity, indicating that the situation may be more complex [189]. Although the hypothesis concerning the interface between the pseudokinase and kinase domain makes a lot of sense and explains much of the biological data, it must be noted that the true molecular mechanism could be different and that only a solved structure encompassing at least the pseudokinase and the kinase domains would provide reliable evidence for the mechanism. As the existing Jak1 and Jak2 pseudokinase model structures (references [27, 97, 186, 188, 190, 191] and references [40, 192, 193] which use previously generated model structures of Lindauer et al. [190] and Giordanetto et al. [192]) are based on the conformation of active kinase domains or non-phosphorylated kinase domains which were co-crystallized with an ATP competitive inhibitor, they may reflect neither the real conformation of the activation loop segment of the pseudokinase domain nor the real orientation between the N- and C-lobes of the kinase. In the existing pseudokinase models, the proposed activation loop (A-loop) conformation is an ‘out’-conformation in which the activation loop is oriented away from the catalytic site. As the pseudokinase domain does not have intrinsic kinase activity, one could speculate that the conformation of the A-loop may rather be an ‘in’-conformation resembling the conformation found in the crystal structures of inactive kinase domains (for reviews: [101, 102]). In such a conformation, the A-loop would cross the catalytic cleft, the regulatory αC-helix would be rotated and the relative orientation between the two lobes of the pseudokinase domain would change. In addition, the models which were used to develop the potential inhibitory mechanism are not based on the later described crystal structures of the Jak kinase domains. It would be interesting to incorporate the solved structures of Jak1, Jak2 and Jak3 and their special features such as the Lip/JSI-region into these interaction models. However, the solved kinase domain structures are also not optimal because they reflect the activated states of the kinases with an ‘out’-conformation of the A-loop and all its consequences for the structure. As described above, there may be important differences between the inactive A-loop conformations of Jak3 compared to the other Jaks due to the alanine residue preceding the DGF-motif. On the other hand, the conformation of the activated A-loops of Jak1, Jak2 and Jak3 seen in the crystal structures are almost identical (see Fig. S7). The potential difference in the inactive state could suggest a different inhibitory interface between the kinase and pseudokinase domains in the case of Jak3. Most interestingly, such a theory would be in line with experimental data as it was reported that the Jak3 M592F mutation, which corresponds to the Jak2-V617F mutation does not activate Jak3 whereas corresponding mutations in Jak1 and Tyk2 are activating [96]. Of course, this theory remains speculative and only crystal structures of the different Jaks encompassing at least the pseudokinase and kinase domains will provide definite proof for these regulatory mechanisms.
Mutations within the kinase domain
In comparison to the kinase-like domain, far less activating mutations have been described for the kinase domain. Mutational hotspots are well known in protein kinases and they are often kinase domain mutations. One example is the V600 mutation within the A-loop in BRAF [194] which generates an active kinase. The mutation of the corresponding residue is also found in MET, FLT3 and KIT [195–200]. However, the mutations in the Jak kinase domain are rare events compared to the occurrence of mutations in the pseudokinase domain.
Most mutations are confined to a loop-region between the β2 and β3 strands of Jak2 (R867Q, D873N, T875N) (Fig. S7). Similarly, the other reported mutations (P933R in Jak2 and R879C/H/S in Jak1) affect residues which are exposed on the surface and do not affect the structure of the domain. Considering the kinase–pseudokinase interaction model by Lindauer and colleagues [190], none of the activating Jak2 mutations can be attributed to the proposed interface between the two domains.
Perspectives
Further insights into the structure of Janus kinases, ideally solved structures of domain combinations, will certainly give answers to questions currently still open: How do the domains interact with each other and with the cytokine receptor? Which structural changes are imposed on Jaks during activation of the cytokine receptor complex? How do the disease-associated mutations in Jaks translate into a gain-of-function phenotype? What is the molecular basis of the mutational hotspots associated with either MPN or leukaemia? Moreover, also proteomic analyses of the Jak-associated proteins will provide insights into the nature of the signalling complexes engaged by the different mutants and the wild-type kinases. As more and more Jak mutations are discovered in haematopoietic disorders and also in solid tumours [98], and since Jaks would be potentially interesting targets in inflammatory disorders, the interest in developing specific Jak inhibitors will surely increase.
The outlined opportunities of chemical genetics can be used to understand the consequences of the inhibition of a single given Janus kinase. This approach may thereby complement previous studies with knockout cells and mice, kinase-dead mutants and less specific inhibitors. It is applicable to both, the wild-type and mutated kinases and can be used in mouse models. Therefore, it will help to further elucidate the biology of Jaks. Moreover, analogue-sensitive kinases enable the identification of the direct substrates and possibly of a transcriptome signature of a given kinase. Recent developments of chemical genetics include, e.g. the possibility to generate analogue-sensitive kinases that can be inhibited by irreversible kinase inhibitors. These may be fluorescently labelled which should enable exact quantitation of kinase inhibition as required for systems biology approaches [201].
Further structural information will certainly be helpful in the development of inhibitors. As the bulk of the gain-of-function mutations affect the pseudokinase domain of Jaks, allosteric inhibitors targeting domain–domain interactions would seem to be useful. Inhibitors selective only for the mutated kinase would seem ideal as they would promise to have the least side effects. In the end, clinical studies have to prove what works best, highly selective kinase inhibitors or broad spectrum inhibitors, or drugs targeting different crucial pathways in combination.
Acknowledgments
We thank the numerous colleagues whose work has contributed to our current understanding of the JAK–receptor interaction mechanism and apologise to those whose work could not be cited here due to space restrictions. Our work was supported by the University of Luxembourg grant (R1F105L01).
Supporting Information
Additional supporting information may be found in the online version of this article:
Molecular modelling of the FERM, SH2 and kinase-like domainsof Jaks: For molecular modeling and graphic representation of protein structures, the programs WHAT IF [275] and Pymol [DeLano, WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA] were used. The structure of the kinase domain of Protein Tyrosine Kinase 2 Beta (PTK2B), Brookhaven data bank entry code 3CC6, was used as template for the model structure of the Jak2 pseudokinase domain (amino acids 540-810). For the modelling of the activation loop region (aa R688-R715 in Jak2) in an "in"-conformation, the conformation of the inactive activation loop of the insulin receptor (IR; aa T1145-P1172 ; PDB entry code 1IRK) was chosen as a template. The modelling of the Exon12 amino acids 533-539 of Jak2 was based on the N-terminal region of the EGFR kinase domain structure (aa 700-706; PDB code 2GS6). Energy minimizations were performed under vacuum conditions with the GROMOS program library (W. F. van Gunsteren, distributed by BIOMOS Biomolecular Software B.V., Laboratory of Physical Chemistry, University of Groningen, Netherlands). The initial alignment of the pseudokinase domain sequences of human Jak1, Jak2, Jak3 and Tyk2 with the sequences of the structurally explored kinase domains of PTK2B, Src, FGFR and IR (PDB entry codes: 3CC6, 2PTK, 1FGK and 1IRK) was performed by the use of the BLAST program. Modifications were then introduced to meet structural requirements derived from the known kinase structures. The sequential alignment of the known structures is based on the superposition of their backbone coordinates. The structures of the pseudokinase domains of Jak1 and Jak3 were generated using the Jak2 model as a template. The Swiss-Prot accession numbers for the used Jak sequences used are: NP_002218 (hJak1), NP_004963 (hJak2), P29597 (hTyk2) and AAC50950 (hJak3). The model structure of the Jak1 FERM domain was previously described [71]. The Jak3 FERM model was based on the template of the Jak1 model. The SH2 domain model of Jak1 and Jak3 are based on the crystal structure of the C-terminal SH2 domain of SHP2 (PDB entry code 2SHP).
Table S1 Four Janus kinases transmit the signals ofmany cytokines.
Fig. S1 Non-conserved residues around the ATP- and substratebinding sites. A: Non-conserved residues in the kinase domainsof Jak1, Jak2 and Jak3 that may be exploited for the design of morespecific Jak inhibitors (PDB entry codes for the structures: 3EYG,2B7A and 1yvj). An overlay of Jak1, Jak2 and Jak3 kinase domainstructures is shown and the three kinase domains are shownseparately. The Jak1 residues are highlighted in yellow, the Jak2residues in green and the Jak3 residues in turquoise. The JSIregion is highlighted by a red frame. The kinase inhibitors aredepicted as stick models. Jak1: MI1; CP-690550;3-{(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-D]pyrimidin-4-YL)-amino]-piperidin-1-YL}-3-oxopropanenitrile,Jak2: IZA; CMP6;2-tert-butyl-9-fluoro-3,6-dihydro-7H-benz[H]-imidaz[4,5-F]-isoquinoline-7-one, Jak3: 4ST; AFN941;1,2,3,4-tetrahydrogen-staurosporine. B: Table with theselected non-conserved residues in the kinase domains of Jak1,Jak2, Jak3 and Tyk2.
Fig. S2 Chemical Structures of Jak kinase inhibitors actingin the nanomolarrange. The measured or approximatedIC50 values for Jak inhibition, Phospho STAT inhibition or growth inhibition are also indicated.
Fig. S3 Sequence alignment of full length Jak1, Jak2 Jak3 andTyk2 with sequences of structurally explored FERM, SH2 and kinasedomains. Residues which are conserved in all the Jaks and inthree of four reference sequences are indicated in red.Residuesthat are rather conserved in only the Janus kinases areindicated in blue. Residues for which mutations havebeen identifiedin patients with haematologicdiseases are highlighted in yellow(Jak1), green (Jak2), turquoise (Jak3) and grey (Tyk2) and thecorresponding mutations are indicated below the sequences. Due tothe large number of exon 12, exon 14 and exon 16 mutationsidentified in Jak2, these mutations are not specifically named(please refer to Table 1 in the main document). Mutation which wereonly found in combination with another mutation are followed by a"+" sign. Regions which are subject to deletions and/or insertionsare underlined. An initial alignment was performed using the BLASTprogram and modifications were subsequently introduced to meet thestructural requirements derived from the known referencestructures. Accession numbers for the used Jak sequences used are:NP_002218 (hJak1), NP_004963 (hJak2), AAC50950 (hJak3) and P29597(hTyk2). A: Reference sequences and structures for the FERMdomain are from focal adhesion kinase (FAK; PDB code: 2AL6),radixin (RAD, PDB code: 1GC7), moesin (MOE, PDB code: 1EF1) andmerlin (MER; PDB code: 1H4R). The FERM subdomains F1 to F3 areindicated above the sequences. B: Reference sequences andstructures for the SH2 domains are from phospholipase Cg (PLC, PDBcode: 2PLD), the C-terminal SH2 domain of the p85 alpha subunit ofphosphoinositide 3-kinase (P85aC; PDB code: 1BFJ), the C-terminalSH2 domain of SHP2 (SHP2C; PDB code: 2SHP) and Bcr-Abl (BAbl, PDBcode: 2ABL). Secondary structure elements for SHP2C are given.Reference sequences and structures for the pseudokinase domain werefrom the following kinases: protein tyrosine kinase 2 beta (Ptk2B;PDB code: 3CC6), c-Src (SRC, PDB code: 1FMK), fibroblast growthfactor receptor (FGFR; PDB code: 1FGK) and insulin receptor (IR;PDB code: 1IR3). The 30 amino acid sequence from the epidermalgrowth factor receptor (EGFR; PBD code: 1m17) and the correspondingstructure served as template for the modellingof the N-terminalparts of the Jak pseudokinase domains (e.g. exon 12 region inJak2). Secondary structure elements are given above the sequence.C: Reference sequences and structures were the same as usedfor the pseudokinase sequence alignment. Secondary structureelements for the Jak2 kinase domain are indicated above thesequence (α: alpha helix; β: beta strand; 3: 3/10helix).
Fig. S4 Predicted location of Jak mutations within the FERMand SH2 domains of Jak1 and Jak3. The Jak1 and Jak3 residuesfor which mutants have been reported in patients are represented asyellow or turquoise stick models and Van-der-Waals radii are shownas spheres. Mutants for which an activating effect has been shownare underlined. Residues L49, G62 and L64 on both sides of theJak3-F1α-helix which can be contacted by P132 and I87 arerepresented in orange. The potential phosphotyrosine binding pocketof Jak1 is highlighted in red.
Fig. S5 Model structures of the Jak1, Jak2, and Jak3pseudokinase domains highlighting mutations with biochemicallyvalidated activating effects (see table 1). Residues for which activating point mutations were reported in patients are represented as yellow (Jak1), green (Jak2) or turquoise (Jak3) stick models with spheres indicating the Van-der-Waals radii of atoms. Regions carrying insertions and/or deletions are indicated by a coloured backbone without stick models (black frames). The proposed Jak2 structural mutation hotspots I and II are highlighted in red.
Fig. S6 Model structures of the Jak1, Jak2, and Jak3pseudokinase domains highlighting all reported mutations(biochemically validated and non-validated; table 1). Residues for which point mutations were reported in patients are represented as yellow (Jak1), green (Jak2) or turquoise (Jak3) stick models with spheres indicating the Van-der-Waals radii of atoms. Regions carrying insertions (ins) and/or deletions (del) are indicated by a coloured backbone without stick models. Please refer to table 1 for the detailed denomination of the different insertions and/or deletions in Jak2 exons 12 and 16. The proposed structural mutation hotspots I and II are highlighted in red.
Fig. S7 Crystal structures of the Jak1, Jak2, and Jak3 kinasedomains highlighting all reported mutations (PDB entry codes 3EYG,2B7A and 1yvj, respectively). An overlay of Jak1, Jak2 and Jak3 kinase domain structures is shown and the three kinase domains are shows separately. Residues for which point mutations were reported in patients are represented as yellow (Jak1) or green (Jak2) stick models with spheres indicating the Van-der-Waals radii of atoms. The activation loops of the three Jaks arehighlighted in the overlay representation by a dashed box. The phosphotyrosine residues within the activation loop of the kinases are represented as dotted spheres.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortutay C, Valiaho J, Stenberg K, et al. KinMutBase: a registry of disease-causing mutations in protein kinase domains. Hum Mutat. 2005;25:435–42. doi: 10.1002/humu.20166. [DOI] [PubMed] [Google Scholar]
- 3.Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr Opin Genet Dev. 2007;17:31–9. doi: 10.1016/j.gde.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Janne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2006;12:4416s–20s. doi: 10.1158/1078-0432.CCR-06-0555. [DOI] [PubMed] [Google Scholar]
- 5.Tefferi A. Molecular drug targets in myeloproliferative neoplasms: mutant ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1. J Cell Mol Med. 2009;13:215–37. doi: 10.1111/j.1582-4934.2008.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeh TC, Pellegrini S. The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell Mol Life Sci. 1999;55:1523–34. doi: 10.1007/s000180050392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinrich PC, Behrmann I, Haan S, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11:69–74. doi: 10.1016/s0168-9525(00)89000-9. [DOI] [PubMed] [Google Scholar]
- 9.O’Sullivan LA, Liongue C, Lewis RS, et al. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol. 2007;44:2497–506. doi: 10.1016/j.molimm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 10.Pestka S, Krause CD, Sarkar D, et al. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 11.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 12.Hintzen C, Haan C, Tuckermann JP, et al. Oncostatin M-induced and constitutive activation of the JAK2/STAT5/CIS pathway suppresses CCL1, but not CCL7 and CCL8, chemokine expression. J Immunol. 2008;181:7341–9. doi: 10.4049/jimmunol.181.10.7341. [DOI] [PubMed] [Google Scholar]
- 13.Pesu M, Laurence A, Kishore N, et al. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–42. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pesu M, Candotti F, Husa M, et al. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol Rev. 2005;203:127–42. doi: 10.1111/j.0105-2896.2005.00220.x. [DOI] [PubMed] [Google Scholar]
- 16.Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–40. [PubMed] [Google Scholar]
- 17.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 18.Reiter A, Walz C, Watmore A, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662–7. doi: 10.1158/0008-5472.CAN-04-4263. [DOI] [PubMed] [Google Scholar]
- 19.Murati A, Gelsi-Boyer V, Adelaide J, et al. PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8;9) translocation. Leukemia. 2005;19:1692–6. doi: 10.1038/sj.leu.2403879. [DOI] [PubMed] [Google Scholar]
- 20.Bousquet M, Quelen C, De Mas V, et al. The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24:7248–52. doi: 10.1038/sj.onc.1208850. [DOI] [PubMed] [Google Scholar]
- 21.Adelaide J, Perot C, Gelsi-Boyer V, et al. A t(8;9) translocation with PCM1-JAK2 fusion in a patient with T-cell lymphoma. Leukemia. 2006;20:536–7. doi: 10.1038/sj.leu.2404104. [DOI] [PubMed] [Google Scholar]
- 22.Griesinger F, Hennig H, Hillmer F, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329–33. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 23.Poitras JL, Dal Cin P, Aster JC, et al. Novel SSBP2-JAK2 fusion gene resulting from a t(5;9)(q14.1;p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer. 2008;47:884–9. doi: 10.1002/gcc.20585. [DOI] [PubMed] [Google Scholar]
- 24.Cirmena G, Aliano S, Fugazza G, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet. 2008;183:105–8. doi: 10.1016/j.cancergencyto.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 25.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 26.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 27.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 28.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 29.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Wernig G, Mercher T, Okabe R, et al. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lacout C, Pisani DF, Tulliez M, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 33.Zaleskas VM, Krause DS, Lazarides K, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS ONE. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bumm TG, Elsea C, Corbin AS, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res. 2006;66:11156–65. doi: 10.1158/0008-5472.CAN-06-2210. [DOI] [PubMed] [Google Scholar]
- 35.Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci USA. 2005;102:18962–7. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 38.Staerk J, Lacout C, Sato T, et al. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107:1864–71. doi: 10.1182/blood-2005-06-2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercher T, Wernig G, Moore SA, et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108:2770–9. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walz C, Crowley BJ, Hudon HE, et al. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–83. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 42.Plo I, Nakatake M, Malivert L, et al. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–12. doi: 10.1182/blood-2008-01-134114. [DOI] [PubMed] [Google Scholar]
- 43.Kralovics R, Teo SS, Buser AS, et al. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106:3374–6. doi: 10.1182/blood-2005-05-1889. [DOI] [PubMed] [Google Scholar]
- 44.Bock O, Hussein K, Brakensiek K, et al. The suppressor of cytokine signalling-1 (SOCS-1) gene is overexpressed in Philadelphia chromosome negative chronic myeloproliferative disorders. Leuk Res. 2007;31:799–803. doi: 10.1016/j.leukres.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 45.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 46.Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 47.Naka T, Narazaki M, Hirata M, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–9. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 48.Ungureanu D, Saharinen P, Junttila I, et al. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol. 2002;22:3316–26. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hookham MB, Elliott J, Suessmuth Y, et al. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood. 2007;109:4924–9. doi: 10.1182/blood-2006-08-039735. [DOI] [PubMed] [Google Scholar]
- 50.Haan S, Wuller S, Kaczor J, et al. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene. 2009;28:3069–80. doi: 10.1038/onc.2009.155. [DOI] [PubMed] [Google Scholar]
- 51.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 52.Shide K, Shimoda HK, Kumano T, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 53.Capello D, Deambrogi C, Rossi D, et al. Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia-negative chronic myeloproliferative disorders. Br J Haematol. 2008;141:504–11. doi: 10.1111/j.1365-2141.2008.07072.x. [DOI] [PubMed] [Google Scholar]
- 54.Teofili L, Martini M, Cenci T, et al. Epigenetic alteration of SOCS family members is a possible pathogenetic mechanism in JAK2 wild type myeloproliferative diseases. Int J Cancer. 2008;123:1586–92. doi: 10.1002/ijc.23694. [DOI] [PubMed] [Google Scholar]
- 55.Quentmeier H, Geffers R, Jost E, et al. SOCS2: inhibitor of JAK2V617F-mediated signal transduction. Leukemia. 2008;22:2169–75. doi: 10.1038/leu.2008.226. [DOI] [PubMed] [Google Scholar]
- 56.Wilks AF, Harpur AG, Kurban RR, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Girault JA, Labesse G, Mornon JP, et al. Janus kinases and focal adhesion kinases play in the 4.1 band: a superfamily of band 4.1 domains important for cell structure and signal transduction. Mol Med. 1998;4:751–69. [PMC free article] [PubMed] [Google Scholar]
- 58.Richter MF, Dumenil G, Uze G, et al. Specific contribution of Tyk2 JH regions to the binding and the expression of the interferon alpha/beta receptor component IFNAR1. J Biol Chem. 1998;273:24723–9. doi: 10.1074/jbc.273.38.24723. [DOI] [PubMed] [Google Scholar]
- 59.Zhao Y, Wagner F, Frank SJ, et al. The amino-terminal portion of the JAK2 protein kinase is necessary for binding and phosphorylation of the granulocyte-macrophage colony-stimulating factor receptor beta c chain. J Biol Chem. 1995;270:13814–8. doi: 10.1074/jbc.270.23.13814. [DOI] [PubMed] [Google Scholar]
- 60.Chen M, Cheng A, Chen YQ, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci USA. 1997;94:6910–5. doi: 10.1073/pnas.94.13.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cacalano NA, Migone TS, Bazan F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. EMBO J. 1999;18:1549–58. doi: 10.1093/emboj/18.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kohlhuber F, Rogers NC, Watling D, et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol Cell Biol. 1997;17:695–706. doi: 10.1128/mcb.17.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haan C, Kreis S, Margue C, et al. Jaks and cytokine receptors–an intimate relationship. Biochem Pharmacol. 2006;72:1538–46. doi: 10.1016/j.bcp.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 64.Pearson MA, Reczek D, Bretscher A, et al. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101:259–70. doi: 10.1016/s0092-8674(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 65.Hamada K, Shimizu T, Matsui T, et al. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 2000;19:4449–62. doi: 10.1093/emboj/19.17.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamada K, Shimizu T, Yonemura S, et al. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. EMBO J. 2003;22:502–14. doi: 10.1093/emboj/cdg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han BG, Nunomura W, Takakuwa Y, et al. Protein 4.1R core domain structure and insights into regulation of cytoskeletal organization. Nat Struct Biol. 2000;7:871–5. doi: 10.1038/82819. [DOI] [PubMed] [Google Scholar]
- 68.Ceccarelli DF, Song HK, Poy F, et al. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281:252–9. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- 69.Hilkens CM, Is’harc H, Lillemeier BF, et al. A region encompassing the FERM domain of Jak1 is necessary for binding to the cytokine receptor gp130. FEBS Lett. 2001;505:87–91. doi: 10.1016/s0014-5793(01)02783-1. [DOI] [PubMed] [Google Scholar]
- 70.Haan C, Is’harc H, Hermanns HM, et al. Mapping of a region within the N terminus of Jak1 involved in cytokine receptor interaction. J Biol Chem. 2001;276:37451–8. doi: 10.1074/jbc.M106135200. [DOI] [PubMed] [Google Scholar]
- 71.Haan S, Margue C, Engrand A, et al. Dual role of the Jak1 FERM and kinase domains in cytokine receptor binding and in stimulation-dependent Jak activation. J Immunol. 2008;180:998–1007. doi: 10.4049/jimmunol.180.2.998. [DOI] [PubMed] [Google Scholar]
- 72.Constantinescu SN, Huang LJ, Nam H, et al. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7:377–85. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
- 73.Greiser JS, Stross C, Heinrich PC, et al. Orientational constraints of the gp130 intracellular juxtamembrane domain for signaling. J Biol Chem. 2002;277:26959–65. doi: 10.1074/jbc.M204113200. [DOI] [PubMed] [Google Scholar]
- 74.Watowich SS, Liu KD, Xie X, et al. Oligomerization and scaffolding functions of the erythropoietin receptor cytoplasmic tail. J Biol Chem. 1999;274:5415–21. doi: 10.1074/jbc.274.9.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haan C, Heinrich PC, Behrmann I. Structural requirements of the interleukin-6 signal transducer gp130 for its interaction with Janus kinase 1: the receptor is crucial for kinase activation. Biochem J. 2002;361:105–11. doi: 10.1042/0264-6021:3610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou YJ, Chen M, Cusack NA, et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8:959–69. doi: 10.1016/s1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 77.Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008;283:5258–66. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 78.Wernig G, Gonneville JR, Crowley BJ, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood. 2008;111:3751–9. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Radtke S, Haan S, Jorissen A, et al. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J Biol Chem. 2005;280:25760–8. doi: 10.1074/jbc.M500822200. [DOI] [PubMed] [Google Scholar]
- 80.Ragimbeau J, Dondi E, Alcover A, et al. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 2003;22:537–47. doi: 10.1093/emboj/cdg038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8:1327–38. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 82.Zhao L, Dong H, Zhang CC, et al. A JAK2 Interdomain linker relays epo receptor engagement signals to kinase activation. J Biol Chem. 2009;284:26988–98. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shakespeare WC, Bohacek RS, Azimioara MD, et al. Structure-based design of novel bicyclic nonpeptide inhibitors for the src SH2 domain. J Med Chem. 2000;43:3815–9. doi: 10.1021/jm0003337. [DOI] [PubMed] [Google Scholar]
- 84.Giorgetti-Peraldi S, Ottinger E, Wolf G, et al. Cellular effects of phosphotyrosine-binding domain inhibitors on insulin receptor signaling and trafficking. Mol Cell Biol. 1997;17:1180–8. doi: 10.1128/mcb.17.3.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oishi S, Karki RG, Kang SU, et al. Design and synthesis of conformationally constrained Grb2 SH2 domain binding peptides employing alpha-methylphenylalanyl based phosphotyrosyl mimetics. J Med Chem. 2005;48:764–72. doi: 10.1021/jm0492709. [DOI] [PubMed] [Google Scholar]
- 86.Manning BD, Cantley LC. Hitting the target: emerging technologies in the search for kinase substrates. Sci STKE. 2002;2002:PE49. doi: 10.1126/stke.2002.162.pe49. [DOI] [PubMed] [Google Scholar]
- 87.Scheeff ED, Eswaran J, Bunkoczi G, et al. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure. 2009;17:128–38. doi: 10.1016/j.str.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boudeau J, Miranda-Saavedra D, Barton GJ, et al. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–52. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 89.Zhang X, Gureasko J, Shen K, et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–49. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 90.Baas AF, Boudeau J, Sapkota GP, et al. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–72. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luo H, Rose P, Barber D, et al. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. 1997;17:1562–71. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen M, Cheng A, Candotti F, et al. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain: evidence for interactions between the kinase and pseudokinase domains. Mol Cell Biol. 2000;20:947–56. doi: 10.1128/mcb.20.3.947-956.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20:3387–95. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yeh TC, Dondi E, Uze G, et al. A dual role for the kinase-like domain of the tyrosine kinase Tyk2 in interferon-alpha signaling. Proc Natl Acad Sci USA. 2000;97:8991–6. doi: 10.1073/pnas.160130297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wek RC, Ramirez M, Jackson BM, et al. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol Cell Biol. 1990;10:2820–31. doi: 10.1128/mcb.10.6.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Staerk J, Kallin A, Demoulin JB, et al. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280:41893–9. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 97.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106:9414–8. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jeong EG, Kim MS, Nam HK, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14:3716–21. doi: 10.1158/1078-0432.CCR-07-4839. [DOI] [PubMed] [Google Scholar]
- 99.Dong B, Silverman RH. Alternative function of a protein kinase homology domain in 2’, 5’-oligoadenylate dependent RNase L. Nucleic Acids Res. 1999;27:439–45. doi: 10.1093/nar/27.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 101.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 102.Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. J Biol Chem. 1998;273:11987–90. doi: 10.1074/jbc.273.20.11987. [DOI] [PubMed] [Google Scholar]
- 103.Haan S. Struktur-/Funktionsuntersuchungen zu den SH2-Domänen der Signalmoleküle STAT3 und SOCS3. Dissertation RWTH-Aachen. 2000 [Google Scholar]
- 104.Haan C, Kroy DC, Wuller S, et al. An unusual insertion in jak2 is crucial for kinase activity and differentially affects cytokine responses. J Immunol. 2009;182:2969–77. doi: 10.4049/jimmunol.0800572. [DOI] [PubMed] [Google Scholar]
- 105.Lucet IS, Fantino E, Styles M, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–83. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 106.Boggon TJ, Li Y, Manley PW, et al. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106:996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Williams NK, Bamert RS, Patel O, et al. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387:219–32. doi: 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 108.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 109.Cohen MS, Zhang C, Shokat KM, et al. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–21. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–65. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 112.Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096–101. doi: 10.1158/0008-5472.CAN-05-1346. [DOI] [PubMed] [Google Scholar]
- 113.Heymach JV, Nilsson M, Blumenschein G, et al. Epidermal growth factor receptor inhibitors in development for the treatment of non-small cell lung cancer. Clin Cancer Res. 2006;12:4441s–5s. doi: 10.1158/1078-0432.CCR-06-0286. [DOI] [PubMed] [Google Scholar]
- 114.Felip E, Santarpia M, Rosell R. Emerging drugs for non-small-cell lung cancer. Expert Opin Emerg Drugs. 2007;12:449–60. doi: 10.1517/14728214.12.3.449. [DOI] [PubMed] [Google Scholar]
- 115.Rishton GM. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discov Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 116.Ohren JF, Chen H, Pavlovsky A, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 117.Gauzzi MC, Barbieri G, Richter MF, et al. The amino-terminal region of Tyk2 sustains the level of interferon alpha receptor 1, a component of the interferon alpha/beta receptor. Proc Natl Acad Sci USA. 1997;94:11839–44. doi: 10.1073/pnas.94.22.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shaw MH, Boyartchuk V, Wong S, et al. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc Natl Acad Sci USA. 2003;100:11594–9. doi: 10.1073/pnas.1930781100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sigurdsson S, Nordmark G, Göring HH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Verstovsek S, Kantarjian H, Pardanani AD, et al. The Jak inhibitor, INCB018424, demonstrates durable and marked clinical responses in Primary Myelofibrosis (PMF) and Post-Polycythemia/Essential Thrombocythemia Myelofibrosis (Post PV/ETMF) Blood (ASH Annual Meeting Abstracts) 2008;112:1762. [Google Scholar]
- 121.Rambaldi A, Barbui T, Barosi G. From palliation to epigenetic therapy in myelofibrosis. Hematol Am Soc Hematol Educ Program. 2008:83–91. doi: 10.1182/asheducation-2008.1.83. [DOI] [PubMed] [Google Scholar]
- 122.Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2008;22:23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- 123.Mesa RA, Verstovsek S, Kantarjian H, et al. INCB018424, a selective Jak1/2 inhibitor, significantly improves the compromised nutritional status and Frank Cachexia in Patients with Myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2008;112:1760. [Google Scholar]
- 124.Shah NP, Olszynski P, Sokol L, et al. A Phase I study of XL019, a selective Jak2 inhibitor, in patients with Primary Myelofibrosis, Post-Polycythemia Vera, or Post Essential Thrombocythemia Myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2008;112:8. [Google Scholar]
- 125.Verstovsek S, Kantarjian H, Pardanani AD, et al. Characterisation of allele burden in advanced Myelofibrosis (MF) patients: no change in V617F:WT Jak2 ratio in patients with high allele burdens despite profound clinical improvement following treatment with the Jak inhibitor INCB018424. Blood (ASH Annual Meeting Abstracts) 2008;112:2802. [Google Scholar]
- 126.Tefferi A, Kantarjian H, Pardanani AD, et al. The clinical phenotype of myelofibrosis encompasses a chronic inflammatory state that is favourably altered by INCB018424, a selective inhibitor of Jak1/2. Blood (ASH Annual Meeting Abstracts) 2008;112:2804. [Google Scholar]
- 127.Wang Y, Fiskus W, Natarajan K, et al. Co-treatment with JAK2 inhibitor TG101209 and panobinostat or hsp90 inhibitor AUY922 attenuates mutant JAK2-V617F levels and activity in human myeloproliferative disorder cells. Blood (ASH Annual Meeting Abstracts) 2008;112:3739. [Google Scholar]
- 128.Guerini V, Barbui V, Spinelli O, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F) Leukemia. 2008;22:740–7. doi: 10.1038/sj.leu.2405049. [DOI] [PubMed] [Google Scholar]
- 129.Rambaldi A, Dellacasa CM, Salmoiraghi S, et al. A phase 2A study of the histone-deacetylase inhibitor ITF2357 in patients with Jak2V617F positive chronic myeloproliferative neoplasms. Blood (ASH Annual Meeting Abstracts) 2008;112:100. [Google Scholar]
- 130.Vannucchi AM, Guglielmelli P, Rambaldi A, et al. Epigenetic therapy in myeloproliferative neoplasms: evidence and perspectives. J Cell Mol Med. 2009;13:1437–50. doi: 10.1111/j.1582-4934.2009.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065–72. doi: 10.1182/blood-2008-03-143537. [DOI] [PubMed] [Google Scholar]
- 132.Hasselbalch HC. Myelofibrosis with myeloid metaplasia: the advanced phase of an untreated disseminated hematological cancer. Time to change our therapeutic attitude with early upfront treatment. Leuk Res. 2009;33:11–8. doi: 10.1016/j.leukres.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 133.Quintas-Cardama A, Kantarjian HM, Garcia-Manero G, et al. Pegylated interferon-ALFA-2A (PEG-IFN–2A; PEGASYS) therapy renders high clinical and molecular response rates in patients with essential thrombocythemia (ET) and polycythemia VERA (PV) Blood (ASH Annual Meeting Abstracts) 2008;112:658. [Google Scholar]
- 134.Chen JJ, Thakur KD, Clark MP, et al. Development of pyrimidine-based inhibitors of Janus tyrosine kinase 3. Bioorg Med Chem Lett. 2006;16:5633–8. doi: 10.1016/j.bmcl.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 135.Clark MP, George KM, Bookland RG, et al. Development of new pyrrolopyrimidine-based inhibitors of Janus kinase 3 (JAK3) Bioorg Med Chem Lett. 2007;17:1250–3. doi: 10.1016/j.bmcl.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 136.Yang SM, Malaviya R, Wilson LJ, et al. Simplified staurosporine analogs as potent JAK3 inhibitors. Bioorg Med Chem Lett. 2007;17:326–31. doi: 10.1016/j.bmcl.2006.10.062. [DOI] [PubMed] [Google Scholar]
- 137.Antonysamy S, Hirst G, Park F, et al. Fragment-based discovery of JAK-2 inhibitors. Bioorg Med Chem Lett. 2009;19:279–82. doi: 10.1016/j.bmcl.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 138.Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–37. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 139.Changelian PS, Flanagan ME, Ball DJ, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–8. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 140.Changelian PS, Moshinsky D, Kuhn CF, et al. The specificity of JAK3 kinase inhibitors. Blood. 2008;111:2155–7. doi: 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]
- 141.Luo C, Laaja P. Inhibitors of JAKs/STATs and the kinases: a possible new cluster of drugs. Drug Discov Today. 2004;9:268–75. doi: 10.1016/S1359-6446(03)03014-9. [DOI] [PubMed] [Google Scholar]
- 142.Ghoreschi K, Laurence A, O’Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be. Nat Immunol. 2009;10:356–60. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bishop AC, Buzko O, Shokat KM. Magic bullets for protein kinases. Trends Cell Biol. 2001;11:167–72. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 144.Bishop AC, Ubersax JA, Petsch DT, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 145.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 147.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–8. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 148.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kornev AP, Haste NM, Taylor SS, et al. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006;103:17783–8. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Levin SE, Zhang C, Kadlecek TA, et al. Inhibition of ZAP-70 kinase activity via an analog-sensitive allele blocks T cell receptor and CD28 superagonist signaling. J Biol Chem. 2008;283:15419–30. doi: 10.1074/jbc.M709000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oh H, Ozkirimli E, Shah K, et al. Generation of an analog-sensitive Syk tyrosine kinase for the study of signaling dynamics from the B cell antigen receptor. J Biol Chem. 2007;282:33760–8. doi: 10.1074/jbc.M704846200. [DOI] [PubMed] [Google Scholar]
- 152.Miller AL, Zhang C, Shokat KM, et al. Generation of a novel system for studying spleen tyrosine kinase function in macrophages and B cells. J Immunol. 2009;182:988–98. doi: 10.4049/jimmunol.182.2.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang C, Kenski DM, Paulson JL, et al. A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat Methods. 2005;2:435–41. doi: 10.1038/nmeth764. [DOI] [PubMed] [Google Scholar]
- 154.Papa FR, Zhang C, Shokat K, et al. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–7. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- 155.Korennykh AV, Egea PF, Korostelev AA, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–93. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang C, Shokat K. Enhanced selectivity for inhibition of analog-sensitive protein kinases through scaffold optimization. Tetrahedron. 2007;63:5832–8. [Google Scholar]
- 157.Chong YP, Mulhern TD, Zhu HJ, et al. A novel non-catalytic mechanism employed by the C-terminal Src-homologous kinase to inhibit Src-family kinase activity. J Biol Chem. 2004;279:20752–66. doi: 10.1074/jbc.M309865200. [DOI] [PubMed] [Google Scholar]
- 158.Patrucco E, Notte A, Barberis L, et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–87. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 159.Yu VP, Baskerville C, Grunenfelder B, et al. A kinase-independent function of Cks1 and Cdk1 in regulation of transcription. Mol Cell. 2005;17:145–51. doi: 10.1016/j.molcel.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 160.Radtke S, Hermanns HM, Haan C, et al. Novel role of Janus kinase 1 in the regulation of oncostatin M receptor surface expression. J Biol Chem. 2002;277:11297–305. doi: 10.1074/jbc.M100822200. [DOI] [PubMed] [Google Scholar]
- 161.Radtke S, Jorissen A, De Leur HS, et al. Three dileucine-like motifs within the interbox1/2 region of the human oncostatin M receptor prevent efficient surface expression in the absence of an associated Janus kinase. J Biol Chem. 2006;281:4024–34. doi: 10.1074/jbc.M511779200. [DOI] [PubMed] [Google Scholar]
- 162.Royer Y, Staerk J, Costuleanu M, et al. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. 2005;280:27251–61. doi: 10.1074/jbc.M501376200. [DOI] [PubMed] [Google Scholar]
- 163.He K, Loesch K, Cowan JW, et al. Janus kinase 2 enhances the stability of the mature growth hormone receptor. Endocrinology. 2005;146:4755–65. doi: 10.1210/en.2005-0514. [DOI] [PubMed] [Google Scholar]
- 164.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–30. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 165.Ventura JJ, Hubner A, Zhang C, et al. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–10. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 166.Kim JS, Lilley BN, Zhang C, et al. A chemical-genetic strategy reveals distinct temporal requirements for SAD-1 kinase in neuronal polarization and synapse formation. Neural Dev. 2008;3:23. doi: 10.1186/1749-8104-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jiang S, DiPaolo J, Currie K, et al. Chemical genetic transcriptional fingerprinting for selectivity profiling of kinase inhibitors. Assay Drug Dev Technol. 2007;5:49–64. doi: 10.1089/adt.2006.032. [DOI] [PubMed] [Google Scholar]
- 168.Chen X, Ye H, Kuruvilla R, et al. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 169.Jaeschke A, Karasarides M, Ventura JJ, et al. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 170.Allen JJ, Li M, Brinkworth CS, et al. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4:511–6. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kaneko M, Hanover JL, England PM, et al. TrkB kinase is required for recovery, but not loss, of cortical responses following monocular deprivation. Nat Neurosci. 2008;11:497–504. doi: 10.1038/nn2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnson AW, Chen X, Crombag HS, et al. The brain-derived neurotrophic factor receptor TrkB is critical for the acquisition but not expression of conditioned incentive value. Eur J Neurosci. 2008;28:997–1002. doi: 10.1111/j.1460-9568.2008.06383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bariohay B, Roux J, Tardivel C, et al. Brain-derived neurotrophic factor/tropomyosin-related kinase receptor type B signaling is a downstream effector of the brainstem melanocortin system in food intake control. Endocrinology. 2009;150:2646–53. doi: 10.1210/en.2008-1184. [DOI] [PubMed] [Google Scholar]
- 174.Heck S, Qian X, Velleca M. Genetically engineered mouse models for drug discovery: new chemical genetic approaches. Curr Drug Discov Technol. 2004;1:13–26. doi: 10.2174/1570163043484806. [DOI] [PubMed] [Google Scholar]
- 175.Shokat K, Velleca M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. Drug Discov Today. 2002;7:872–9. doi: 10.1016/s1359-6446(02)02391-7. [DOI] [PubMed] [Google Scholar]
- 176.Habelhah H, Shah K, Huang L, et al. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J Biol Chem. 2001;276:18090–5. doi: 10.1074/jbc.M011396200. [DOI] [PubMed] [Google Scholar]
- 177.Shah K, Shokat KM. A chemical genetic screen for direct v-Src substrates reveals ordered assembly of a retrograde signaling pathway. Chem Biol. 2002;9:35–47. doi: 10.1016/s1074-5521(02)00086-8. [DOI] [PubMed] [Google Scholar]
- 178.Shah K, Liu Y, Deirmengian C, et al. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc Natl Acad Sci USA. 1997;94:3565–70. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ulrich SM, Kenski DM, Shokat KM. Engineering a “methionine clamp” into Src family kinases enhances specificity toward unnatural ATP analogues. Biochemistry. 2003;42:7915–21. doi: 10.1021/bi030042a. [DOI] [PubMed] [Google Scholar]
- 180.Witucki LA, Huang X, Shah K, et al. Mutant tyrosine kinases with unnatural nucleotide specificity retain the structure and phospho-acceptor specificity of the wild-type enzyme. Chem Biol. 2002;9:25–33. doi: 10.1016/s1074-5521(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 181.Allen JJ, Lazerwith SE, Shokat KM. Bio-orthogonal affinity purification of direct kinase substrates. J Am Chem Soc. 2005;127:5288–9. doi: 10.1021/ja050727t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277:47954–63. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 183.Loriaux MM, Levine RL, Tyner JW, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood. 2008;111:4788–96. doi: 10.1182/blood-2007-07-101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tyner JW, Walters DK, Willis SG, et al. RNAi screening of the tyrosine kinome identifies therapeutic targets in acute myeloid leukemia. Blood. 2008;111:2238–45. doi: 10.1182/blood-2007-06-097253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Frohling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12:501–13. doi: 10.1016/j.ccr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 186.Flex E, Petrangeli V, Stella L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–8. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hof P, Pluskey S, Dhepaganon S, et al. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 188.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 189.Dusa A, Staerk J, Elliott J, et al. Substitution of pseudokinase domain residue Val-617 by large non-polar amino acids causes activation of JAK2. J Biol Chem. 2008;283:12941–8. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- 190.Lindauer K, Loerting T, Liedl KR, et al. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14:27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 191.Walters DK, Mercher T, Gu TL, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10:65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 192.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng. 2002;15:727–37. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 193.Lee TS, Ma W, Zhang X, et al. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009;115:1692–700. doi: 10.1002/cncr.24183. [DOI] [PubMed] [Google Scholar]
- 194.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 195.Chiara F, Michieli P, Pugliese L, et al. Mutations in the met oncogene unveil a “dual switch” mechanism controlling tyrosine kinase activity. J Biol Chem. 2003;278:29352–8. doi: 10.1074/jbc.M302404200. [DOI] [PubMed] [Google Scholar]
- 196.Lorenzato A, Olivero M, Patane S, et al. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62:7025–30. [PubMed] [Google Scholar]
- 197.Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–9. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 198.Abu-Duhier FM, Goodeve AC, Wilson GA, et al. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113:983–8. doi: 10.1046/j.1365-2141.2001.02850.x. [DOI] [PubMed] [Google Scholar]
- 199.Ferrao PT, Gonda TJ, Ashman LK. Constitutively active mutant D816VKit induces megakayocyte and mast cell differentiation of early haemopoietic cells from murine foetal liver. Leuk Res. 2003;27:547–55. doi: 10.1016/s0145-2126(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 200.Tan A, Westerman D, McArthur GA, et al. Sensitive detection of KIT D816V in patients with mastocytosis. Clin Chem. 2006;52:2250–7. doi: 10.1373/clinchem.2006.068205. [DOI] [PubMed] [Google Scholar]
- 201.Blair JA, Rauh D, Kung C, et al. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol. 2007;3:229–38. doi: 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- 202.Malinge S, Ragu C, Della-Valle V, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood. 2008;112:4220–6. doi: 10.1182/blood-2008-01-136366. [DOI] [PubMed] [Google Scholar]
- 203.Sayyah J, Magis A, Ostrov DA, et al. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol Cancer Ther. 2008;7:2308–18. doi: 10.1158/1535-7163.MCT-08-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Percy MJ, Scott LM, Erber WN, et al. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607–14. doi: 10.3324/haematol.11643. [DOI] [PubMed] [Google Scholar]
- 205.Schnittger S, Bacher U, Haferlach C, et al. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica. 2009;94:414–8. doi: 10.3324/haematol.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007;21:1960–3. doi: 10.1038/sj.leu.2404810. [DOI] [PubMed] [Google Scholar]
- 207.Pietra D, Li S, Brisci A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–9. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 208.Williams DM, Kim AH, Rogers O, et al. Phenotypic variations and new mutations in JAK2 V617F-negative polycythemia vera, erythrocytosis, and idiopathic myelofibrosis. Exp Hematol. 2007;35:1641–6. doi: 10.1016/j.exphem.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Butcher CM, Hahn U, To LB, et al. Two novel JAK2 exon 12 mutations in JAK2V617F-negative polycythaemia vera patients. Leukemia. 2008;22:870–3. doi: 10.1038/sj.leu.2404971. [DOI] [PubMed] [Google Scholar]
- 210.Ormazabal C, Hurtado C, Aranaz P, et al. Low frequency of JAK2 exon 12 mutations in classic and atypical CMPDs. Leuk Res. 2008;32:1485–7. doi: 10.1016/j.leukres.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 211.Lee JW, Kim YG, Soung YH, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25:1434–6. doi: 10.1038/sj.onc.1209163. [DOI] [PubMed] [Google Scholar]
- 212.Kratz CP, Boll S, Kontny U, et al. Mutational screen reveals a novel JAK2 mutation, L611S, in a child with acute lymphoblastic leukemia. Leukemia. 2006;20:381–3. doi: 10.1038/sj.leu.2404060. [DOI] [PubMed] [Google Scholar]
- 213.Funakoshi-Tago M, Tago K, Sumi K, et al. The acute lymphoblastic leukemia-associated Jak2 L611S mutant induces tumorigenesis in nude mice. J Biol Chem. 2009;284:12680–90. doi: 10.1074/jbc.M808879200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang SJ, Li JY, Li WD, et al. The investigation of JAK2 mutation in Chinese myeloproliferative diseases-identification of a novel C616Y point mutation in a PV patient. Int J Lab Hematol. 2007;29:71–2. doi: 10.1111/j.1365-2257.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 215.Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–8. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 216.Steensma DP. JAK2 V617F in myeloid disorders: molecular diagnostic techniques and their clinical utility: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2006;8:397–411. doi: 10.2353/jmoldx.2006.060007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Steensma DP, Tefferi A. JAK2 V617F and ringed sideroblasts: not necessarily RARS-T. Blood. 2008;111:1748. doi: 10.1182/blood-2007-11-121608. [DOI] [PubMed] [Google Scholar]
- 218.Tono C, Xu G, Toki T, et al. JAK2 Val617Phe activating tyrosine kinase mutation in juvenile myelomonocytic leukemia. Leukemia. 2005;19:1843–4. doi: 10.1038/sj.leu.2403903. [DOI] [PubMed] [Google Scholar]
- 219.Remacha AF, Nomdedeu JF, Puget G, et al. Occurrence of the JAK2 V617F mutation in the WHO provisional entity: myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2006;91:719–20. [PubMed] [Google Scholar]
- 220.Szpurka H, Tiu R, Murugesan G, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108:2173–81. doi: 10.1182/blood-2006-02-005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Renneville A, Quesnel B, Charpentier A, et al. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia. 2006;20:2067–70. doi: 10.1038/sj.leu.2404405. [DOI] [PubMed] [Google Scholar]
- 222.Karow A, Waller C, Reimann C, et al. JAK2 mutations other than V617F: a novel mutation and mini review. Leuk Res. 2008;32:365–6. doi: 10.1016/j.leukres.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 223.Yoo JH, Park TS, Maeng HY, et al. JAK2 V617F/C618R mutation in a patient with polycythemia vera: a case study and review of the literature. Cancer Genet Cytogenet. 2009;189:43–7. doi: 10.1016/j.cancergencyto.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 224.Grunebach F, Bross-Bach U, Kanz L, et al. Detection of a new JAK2 D620E mutation in addition to V617F in a patient with polycythemia vera. Leukemia. 2006;20:2210–1. doi: 10.1038/sj.leu.2404419. [DOI] [PubMed] [Google Scholar]
- 225.Schnittger S, Bacher U, Kern W, et al. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia. 2006;20:2195–7. doi: 10.1038/sj.leu.2404325. [DOI] [PubMed] [Google Scholar]
- 226.Ohnishi H, Hosoi K, Yoshino H, et al. A novel JAK2 splicing mutation in neonatal myeloproliferative disorder accompanying congenital anomalies. Br J Haematol. 2009;145:676–8. doi: 10.1111/j.1365-2141.2009.07672.x. [DOI] [PubMed] [Google Scholar]
- 227.Gaikwad A, Rye CL, Devidas M, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144:930–2. doi: 10.1111/j.1365-2141.2008.07552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kearney L, Gonzalez De Castro D, Yeung J, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113:646–8. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 229.Malinge S, Ben-Abdelali R, Settegrana C, et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007;109:2202–4. doi: 10.1182/blood-2006-09-045963. [DOI] [PubMed] [Google Scholar]
- 230.Xiang Z, Zhao Y, Mitaksov V, et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood. 2008;111:4809–12. doi: 10.1182/blood-2007-05-090308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tomasson MH, Xiang Z, Walgren R, et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797–808. doi: 10.1182/blood-2007-09-113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sato T, Toki T, Kanezaki R, et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. Br J Haematol. 2008;141:681–8. doi: 10.1111/j.1365-2141.2008.07081.x. [DOI] [PubMed] [Google Scholar]
- 233.Kiyoi H, Yamaji S, Kojima S, et al. JAK3 mutations occur in acute megakaryoblastic leukemia both in Down syndrome children and non-Down syndrome adults. Leukemia. 2007;21:574–6. doi: 10.1038/sj.leu.2404527. [DOI] [PubMed] [Google Scholar]
- 234.De Vita S, Mulligan C, McElwaine S, et al. Loss-of-function JAK3 mutations in TMD and AMKL of Down syndrome. Br J Haematol. 2007;137:337–41. doi: 10.1111/j.1365-2141.2007.06574.x. [DOI] [PubMed] [Google Scholar]
- 235.Klusmann JH, Reinhardt D, Hasle H, et al. Janus kinase mutations in the development of acute megakaryoblastic leukemia in children with and without Down’s syndrome. Leukemia. 2007;21:1584–7. doi: 10.1038/sj.leu.2404694. [DOI] [PubMed] [Google Scholar]
- 236.Jiang JK, Ghoreschi K, Deflorian F, et al. Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperi din-1-yl)-3-oxopropanenitrile (CP-690,550) J Med Chem. 2008;51:8012–8. doi: 10.1021/jm801142b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kudlacz E, Conklyn M, Andresen C, et al. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008;582:154–61. doi: 10.1016/j.ejphar.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 238.Kudlacz E, Perry B, Sawyer P, et al. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am J Transplant. 2004;4:51–7. doi: 10.1046/j.1600-6143.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- 239.Pardanani A, Hood J, Lasho T, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21:1658–68. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 240.Geron I, Abrahamsson AE, Barroga CF, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13:321–30. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 241.Lasho TL, Tefferi A, Hood JD, et al. TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia. 2008;22:1790–2. doi: 10.1038/leu.2008.56. [DOI] [PubMed] [Google Scholar]
- 242.Fridman J, Nussenzveig R, Liu P, et al. Discovery and preclinical characterization of INCB018424, a selective JAK2 inhibitor for the treatment of myeloproliferative disorders. Blood (ASH Annual Meeting Abstracts) 2007;110:3538. [Google Scholar]
- 243.Liu PCC, Caulder E, Li J, et al. Combined inhibition of Janus Kinase 1 / 2 for the treatment of Jak2V617F-driven neoplasms: selective effects on mutant cells and improvements in measures of disease severity. Clin Cancer Res. 2009;15:6891–900. doi: 10.1158/1078-0432.CCR-09-1298. [DOI] [PubMed] [Google Scholar]
- 244.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mathur A, Mo JR, Kraus M, et al. An inhibitor of Janus kinase 2 prevents polycythemia in mice. Biochem Pharmacol. 2009;78:382–9. doi: 10.1016/j.bcp.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 246.Thompson JE, Cubbon RM, Cummings RT, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12:1219–23. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 247.Verstovsek S, Pardanani AD, Shah NP, et al. A phase I study of XL019, a selective JAK2 inhibitor, in patients with primary myelofibrosis and post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2007;110:553. [Google Scholar]
- 248.Paquette R, Sokol L, Shah NP, et al. A phase I study of XL019, a selective JAK2 inhibitor, in patients with polycythemia vera. Blood (ASH Annual Meeting Abstracts) 2008;112:2810. [Google Scholar]
- 249.Lyons J, Curry J, Mallet K, et al. JAK2 and T315I Abl activity of clinical candidate, AT9283. AACR Meeting Abstracts. 2007:5747. [Google Scholar]
- 250.Squires M, Curry J, Dawson M, et al. AT9283, a potent inhibitor of JAK2, is active in JAK2 V617F myeloproliferative disease models. Blood (ASH Annual Meeting Abstracts) 2007;110:3537. [Google Scholar]
- 251.Dobrzanski P, Hexner E, Serdikoff C, et al. CEP-701 is a JAK2 inhibitor which attenuates JAK2/STAT5 signaling pathway and the proliferation of primary cells from patients with myeloproliferative disorders. Blood (ASH Annual Meeting Abstracts) 2006;108:3594. [Google Scholar]
- 252.Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–71. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gozgit JM, Bebernitz G, Patil P, et al. Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. J Biol Chem. 2008;283:32334–43. doi: 10.1074/jbc.M803813200. [DOI] [PubMed] [Google Scholar]
- 254.Deuse T, Velotta JB, Hoyt G, et al. Novel immunosuppression: R348, a JAK3- and Syk-inhibitor attenuates acute cardiac allograft rejection. Transplantation. 2008;85:885–92. doi: 10.1097/TP.0b013e318166acc4. [DOI] [PubMed] [Google Scholar]
- 255.Markovtsov V, Tonkin E, Fang S, et al. In vitro and in vivo inhibition of JAK2 signaling by potent and selective JAK2 inhibitor. Blood (ASH Annual Meeting Abstracts) 2008;112:3721. [Google Scholar]
- 256.Pardanani A, Lasho T, Smith G, et al. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23:1441–5. doi: 10.1038/leu.2009.50. [DOI] [PubMed] [Google Scholar]
- 257.Bumm T, Tyner J, Deininger J, et al. Effects of CYT387, a potent novel JAK2 inhibitor on JAK2-V617F induced MPD. Blood (ASH Annual Meeting Abstracts) 2008;112:856. [Google Scholar]
- 258.Wilks A, Burns C, Smith G, et al. A novel Jak2 inhibitor for the treatment of myeloproliferative disorders. AACR Meeting Abstracts. 2008:4880. [Google Scholar]
- 259.Ahmed K, Nussenzveig R, Chen A, et al. Preclinical characterization of the JAK-2 inhibitor, SGI-1252. Blood (ASH Annual Meeting Abstracts) 2008;112:2629. [Google Scholar]
- 260.Goh K, Ong W, Hu C, et al. SB1518: A potent and orally active JAK2 inhibitor for the treatment of myeloproliferative disorders. Blood (ASH Annual Meeting Abstracts) 2007;110:538. [Google Scholar]
- 261.Sills M, Appell K, Bohnstedt A, et al. In vitro and in vivo characterization of a novel Jak3 tyrosine kinase inhibitor. 14th IRA Conference – Abstracts issue. 2006:A155. [Google Scholar]
- 262.Adams C, Aldous DJ, Amendola S, et al. Mapping the kinase domain of Janus Kinase 3. Bioorg Med Chem Lett. 2003;13:3105–10. doi: 10.1016/s0960-894x(03)00657-7. [DOI] [PubMed] [Google Scholar]
- 263.Grandage VL, Everington T, Linch DC, et al. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol. 2006;135:303–16. doi: 10.1111/j.1365-2141.2006.06291.x. [DOI] [PubMed] [Google Scholar]
- 264.Giles F, Fridman S, Xiao A, et al. MK-0457, a novel multikinase inhibitor, has activity in refractory AML, including transformed JAK2 positive myeloproliferative disease (MPD), and in philadelphia-positive ALL. Blood (ASH Annual Meeting Abstracts) 2006 [Google Scholar]
- 265.Giles F, Bergstrom D, Garcia-Manero G, et al. MK-0457 Is a novel aurora kinase and janus kinase 2 (JAK2) inhibitor with activity in transformed JAK2-positive myeloproliferative disease (MPD) Blood (ASH Annual Meeting Abstracts) 2006:4893. [Google Scholar]
- 266.Tyler RK, Shpiro N, Marquez R, et al. VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell Cycle. 2007;6:2846–54. doi: 10.4161/cc.6.22.4940. [DOI] [PubMed] [Google Scholar]
- 267.Liu Y, Shah K, Yang F, et al. Engineering Src family protein kinases with unnatural nucleotide specificity. Chem Biol. 1998;5:91–101. doi: 10.1016/s1074-5521(98)90143-0. [DOI] [PubMed] [Google Scholar]
- 268.Bishop AC, Shah K, Liu Y, et al. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8:257–66. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- 269.Wong S, McLaughlin J, Cheng D, et al. Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci USA. 2004;101:17456–61. doi: 10.1073/pnas.0407061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Denzel A, Hare KJ, Zhang C, et al. Cutting edge: a chemical genetic system for the analysis of kinases regulating T cell development. J Immunol. 2003;171:519–23. doi: 10.4049/jimmunol.171.2.519. [DOI] [PubMed] [Google Scholar]
- 271.Fan QW, Specht KM, Zhang C, et al. Combinatorial efficacy achieved through two-point blockade within a signaling pathway – a chemical genetic approach. Cancer Res. 2003;63:8930–8. [PubMed] [Google Scholar]
- 272.Fan QW, Zhang C, Shokat KM, et al. Chemical genetic blockade of transformation reveals dependence on aberrant oncogenic signaling. Curr Biol. 2002;12:1386–94. doi: 10.1016/s0960-9822(02)01070-9. [DOI] [PubMed] [Google Scholar]
- 273.Fan QW, Weiss WA. Chemical genetic approaches to the development of cancer therapeutics. Curr Opin Genet Dev. 2006;16:85–91. doi: 10.1016/j.gde.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 274.Wang X, Ratnam J, Zou B, et al. TrkB signaling is required for both the induction and maintenance of tissue and nerve injury-induced persistent pain. J Neurosci. 2009;29:5508–15. doi: 10.1523/JNEUROSCI.4288-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:29–6. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular modelling of the FERM, SH2 and kinase-like domainsof Jaks: For molecular modeling and graphic representation of protein structures, the programs WHAT IF [275] and Pymol [DeLano, WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA] were used. The structure of the kinase domain of Protein Tyrosine Kinase 2 Beta (PTK2B), Brookhaven data bank entry code 3CC6, was used as template for the model structure of the Jak2 pseudokinase domain (amino acids 540-810). For the modelling of the activation loop region (aa R688-R715 in Jak2) in an "in"-conformation, the conformation of the inactive activation loop of the insulin receptor (IR; aa T1145-P1172 ; PDB entry code 1IRK) was chosen as a template. The modelling of the Exon12 amino acids 533-539 of Jak2 was based on the N-terminal region of the EGFR kinase domain structure (aa 700-706; PDB code 2GS6). Energy minimizations were performed under vacuum conditions with the GROMOS program library (W. F. van Gunsteren, distributed by BIOMOS Biomolecular Software B.V., Laboratory of Physical Chemistry, University of Groningen, Netherlands). The initial alignment of the pseudokinase domain sequences of human Jak1, Jak2, Jak3 and Tyk2 with the sequences of the structurally explored kinase domains of PTK2B, Src, FGFR and IR (PDB entry codes: 3CC6, 2PTK, 1FGK and 1IRK) was performed by the use of the BLAST program. Modifications were then introduced to meet structural requirements derived from the known kinase structures. The sequential alignment of the known structures is based on the superposition of their backbone coordinates. The structures of the pseudokinase domains of Jak1 and Jak3 were generated using the Jak2 model as a template. The Swiss-Prot accession numbers for the used Jak sequences used are: NP_002218 (hJak1), NP_004963 (hJak2), P29597 (hTyk2) and AAC50950 (hJak3). The model structure of the Jak1 FERM domain was previously described [71]. The Jak3 FERM model was based on the template of the Jak1 model. The SH2 domain model of Jak1 and Jak3 are based on the crystal structure of the C-terminal SH2 domain of SHP2 (PDB entry code 2SHP).
Table S1 Four Janus kinases transmit the signals ofmany cytokines.
Fig. S1 Non-conserved residues around the ATP- and substratebinding sites. A: Non-conserved residues in the kinase domainsof Jak1, Jak2 and Jak3 that may be exploited for the design of morespecific Jak inhibitors (PDB entry codes for the structures: 3EYG,2B7A and 1yvj). An overlay of Jak1, Jak2 and Jak3 kinase domainstructures is shown and the three kinase domains are shownseparately. The Jak1 residues are highlighted in yellow, the Jak2residues in green and the Jak3 residues in turquoise. The JSIregion is highlighted by a red frame. The kinase inhibitors aredepicted as stick models. Jak1: MI1; CP-690550;3-{(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-D]pyrimidin-4-YL)-amino]-piperidin-1-YL}-3-oxopropanenitrile,Jak2: IZA; CMP6;2-tert-butyl-9-fluoro-3,6-dihydro-7H-benz[H]-imidaz[4,5-F]-isoquinoline-7-one, Jak3: 4ST; AFN941;1,2,3,4-tetrahydrogen-staurosporine. B: Table with theselected non-conserved residues in the kinase domains of Jak1,Jak2, Jak3 and Tyk2.
Fig. S2 Chemical Structures of Jak kinase inhibitors actingin the nanomolarrange. The measured or approximatedIC50 values for Jak inhibition, Phospho STAT inhibition or growth inhibition are also indicated.
Fig. S3 Sequence alignment of full length Jak1, Jak2 Jak3 andTyk2 with sequences of structurally explored FERM, SH2 and kinasedomains. Residues which are conserved in all the Jaks and inthree of four reference sequences are indicated in red.Residuesthat are rather conserved in only the Janus kinases areindicated in blue. Residues for which mutations havebeen identifiedin patients with haematologicdiseases are highlighted in yellow(Jak1), green (Jak2), turquoise (Jak3) and grey (Tyk2) and thecorresponding mutations are indicated below the sequences. Due tothe large number of exon 12, exon 14 and exon 16 mutationsidentified in Jak2, these mutations are not specifically named(please refer to Table 1 in the main document). Mutation which wereonly found in combination with another mutation are followed by a"+" sign. Regions which are subject to deletions and/or insertionsare underlined. An initial alignment was performed using the BLASTprogram and modifications were subsequently introduced to meet thestructural requirements derived from the known referencestructures. Accession numbers for the used Jak sequences used are:NP_002218 (hJak1), NP_004963 (hJak2), AAC50950 (hJak3) and P29597(hTyk2). A: Reference sequences and structures for the FERMdomain are from focal adhesion kinase (FAK; PDB code: 2AL6),radixin (RAD, PDB code: 1GC7), moesin (MOE, PDB code: 1EF1) andmerlin (MER; PDB code: 1H4R). The FERM subdomains F1 to F3 areindicated above the sequences. B: Reference sequences andstructures for the SH2 domains are from phospholipase Cg (PLC, PDBcode: 2PLD), the C-terminal SH2 domain of the p85 alpha subunit ofphosphoinositide 3-kinase (P85aC; PDB code: 1BFJ), the C-terminalSH2 domain of SHP2 (SHP2C; PDB code: 2SHP) and Bcr-Abl (BAbl, PDBcode: 2ABL). Secondary structure elements for SHP2C are given.Reference sequences and structures for the pseudokinase domain werefrom the following kinases: protein tyrosine kinase 2 beta (Ptk2B;PDB code: 3CC6), c-Src (SRC, PDB code: 1FMK), fibroblast growthfactor receptor (FGFR; PDB code: 1FGK) and insulin receptor (IR;PDB code: 1IR3). The 30 amino acid sequence from the epidermalgrowth factor receptor (EGFR; PBD code: 1m17) and the correspondingstructure served as template for the modellingof the N-terminalparts of the Jak pseudokinase domains (e.g. exon 12 region inJak2). Secondary structure elements are given above the sequence.C: Reference sequences and structures were the same as usedfor the pseudokinase sequence alignment. Secondary structureelements for the Jak2 kinase domain are indicated above thesequence (α: alpha helix; β: beta strand; 3: 3/10helix).
Fig. S4 Predicted location of Jak mutations within the FERMand SH2 domains of Jak1 and Jak3. The Jak1 and Jak3 residuesfor which mutants have been reported in patients are represented asyellow or turquoise stick models and Van-der-Waals radii are shownas spheres. Mutants for which an activating effect has been shownare underlined. Residues L49, G62 and L64 on both sides of theJak3-F1α-helix which can be contacted by P132 and I87 arerepresented in orange. The potential phosphotyrosine binding pocketof Jak1 is highlighted in red.
Fig. S5 Model structures of the Jak1, Jak2, and Jak3pseudokinase domains highlighting mutations with biochemicallyvalidated activating effects (see table 1). Residues for which activating point mutations were reported in patients are represented as yellow (Jak1), green (Jak2) or turquoise (Jak3) stick models with spheres indicating the Van-der-Waals radii of atoms. Regions carrying insertions and/or deletions are indicated by a coloured backbone without stick models (black frames). The proposed Jak2 structural mutation hotspots I and II are highlighted in red.
Fig. S6 Model structures of the Jak1, Jak2, and Jak3pseudokinase domains highlighting all reported mutations(biochemically validated and non-validated; table 1). Residues for which point mutations were reported in patients are represented as yellow (Jak1), green (Jak2) or turquoise (Jak3) stick models with spheres indicating the Van-der-Waals radii of atoms. Regions carrying insertions (ins) and/or deletions (del) are indicated by a coloured backbone without stick models. Please refer to table 1 for the detailed denomination of the different insertions and/or deletions in Jak2 exons 12 and 16. The proposed structural mutation hotspots I and II are highlighted in red.
Fig. S7 Crystal structures of the Jak1, Jak2, and Jak3 kinasedomains highlighting all reported mutations (PDB entry codes 3EYG,2B7A and 1yvj, respectively). An overlay of Jak1, Jak2 and Jak3 kinase domain structures is shown and the three kinase domains are shows separately. Residues for which point mutations were reported in patients are represented as yellow (Jak1) or green (Jak2) stick models with spheres indicating the Van-der-Waals radii of atoms. The activation loops of the three Jaks arehighlighted in the overlay representation by a dashed box. The phosphotyrosine residues within the activation loop of the kinases are represented as dotted spheres.