

Abstract
Autism spectrum disorders (ASD) comprise a complex and heterogeneous group of conditions of unknown aetiology, characterized by significant disturbances in social, communicative and behavioural functioning. Recent studies suggested a possible implication of the high-density lipoprotein associated esterase/lactonase paraoxonase 1 (PON1) in ASD. In the present study, we aimed at investigating the PON1 status in a group of 50 children with ASD as compared to healthy age and sex matched control participants. We evaluated PON1 bioavailability (i.e. arylesterase activity) and catalytic activity (i.e. paraoxonase activity) in plasma using spectrophotometric methods and the two common polymorphisms in the PON1 coding region (Q192R, L55M) by employing Light Cycler real-time PCR. We found that both PON1 arylesterase and PON1 paraoxonase activities were decreased in autistic patients (respectively, P < 0.001, P < 0.05), but no association with less active variants of the PON1 gene was found. The PON1 phenotype, inferred from the two-dimensional enzyme analysis, had a similar distribution in the ASD group and the control group. In conclusion, both the bioavailability and the catalytic activity of PON1 are impaired in ASD, despite no association with the Q192R and L55M polymorphisms in the PON1 gene and a normal distribution of the PON1 phenotype.
Keywords: autism, arylesterase, paraoxonase, polymorphisms
Introduction
Autism spectrum disorders (ASD), which include the prototypic autistic disorder, Asperger’s syndrome and pervasive developmental disorders not otherwise specified (PDD-NOS), are a category of neurobehavioural conditions falling under the umbrella term of PDD. This severe and sustained group of disorders is characterized by qualitative impairments in reciprocal social interaction, impairments in verbal and nonverbal communication and a pattern of repetitive, stereotypical behaviours and interests [1]. Although the neurobiological underpinnings of ASD are currently undeciphered, it is becoming increasingly clear that it is a systemic genetically influenced disorder with significant epigenetic and environmental contributors [2–4]. There is growing evidence suggesting that low-level chronic exposure to organophosphate (OP) compounds (e.g. insecticides and nerve agents) may affect neurodevelopment [5–7], and a recent study demonstrated that both prenatal and postnatal dialkylphosphate metabolites were associated with an increased risk for PDD at 24 months of age [8].
Human serum paraoxonase 1 (PON1), encoded by the PON1 gene on chromosome 7q21.3, is a high-density lipoprotein (HDL)-associated esterase/lactonase that catalyses the hydrolysis of arylesters, toxic OP compounds, carbamates and lactones [9]. Although the physiological role is still uncertain, PON1 plays a role in protection against oxidative modification of low-density lipoproteins (LDL)[10, 11], homocysteine-thiolactone [12, 13] and bacterial endotoxins [14]. There is an impressive interindividual variation in PON1 activity and concentration [15]. Two common polymorphisms in the PON1 coding region, Gln→Arg (Q192R) and Leu→Met (L55M), have been described to contribute to this variability [16–18]. While phenylacetate hydrolytic activity (ARE.ase) is a reliable surrogate for serum PON1 bioavailability, PON1 catalytic efficiency can be evaluated by measuring the paraoxon rate of hydrolysis (PO.ase). The PO.ase activity is determined in part by the Q192R polymorphism: the Q form of PON1 is more efficient at hydrolyzing sarin and soman, whereas the R form more efficiently hydrolyzes paraoxon [19]. The second coding region polymorphism, PON1 L55M, does not affect catalytic activity, but may affect PON1 protein stability [20], and has been associated with plasma PON1 protein levels via an interaction with the C-108T promoter polymorphism [21–23]. Several recent studies have underlined the importance of determining both PON1 activities and functional alloform phenotypes as opposed to analysing any number of PON1 single nucleotide polymorphisms (SNPs), when inferring associations between PON1 and disease [15, 24, 25].
Recent evidence pointed to a possible implication of PON1 in ASD. D’Amelio and collaborators demonstrated that Caucasian-American, but not Italian families, display a significant association between autism and less active PON1 gene variants [26], and Paşca et al. reported low ARE.ase activity in a cohort of children with autism [27]. The aim of the present study was to evaluate whether the measurement of arylesterase (ARE.ase) and NaCl stimulated paraoxonase (ssPO.ase) enzymatic activities of PON1, together with the assessment of PON1 Q192R and L55M polymorphisms might yield more information than either genotype or activity alone in a cohort of patients with ASD.
Material and methods
Participants
The participants enrolled in this study were 50 children with a diagnosis of ASD and 30 healthy children, balanced both with regard to age (respectively, 6.54 ± 0.48 years, 6.74 ± 0.50 years, P= 0.76) and sex (respectively, 78.0% males, 73.3% males, P= 0.78) (Table 1). For the genetic analysis, additional participants were recruited for the control group, with a total of 85 apparently healthy participants being genotyped. All participants in this study were Romanians. Each patient was examined by an experienced child neuropsychiatrist and assigned a diagnosis of autism (29 patients, 58%), PDD-NOS (17 patients, 34%) or Asperger syndrome (4 patients, 8%) based on the criteria defined in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition Revised (DSM-IVR) [1]. None of the patients followed any special diet (i.e. gluten free, casein free, high-dose vitamin supplementation). Comparison participants were drawn from the same geographical area as our patients, aiming to recover the same demographics for the control and patient groups. All the controls were somatically and behaviourally healthy, had no past or present history of neuropsychiatric disorders and none of them had ever taken medications for psychiatric conditions. Informed consent was obtained and the research protocol was in agreement with the Declaration of Helsinki of the World Medical Association.
Table 1.
Group composition by gender and age, PON1 enzymatic activities and PON1 polymorphisms distribution in ASD patients versus control participants
| ASD (n=50) | Controls (n=30) | P | ||
|---|---|---|---|---|
| Age (years) | 6.54 ± 0.48 | 6.74 ± 0.50 | 0.769 | |
| Sex: male, n% | 39 (78.0%) | 22 (73.3%) | 0.787 | |
| Paraoxonase 1 | ||||
| Arylesterase activity (kU/l) | 69.87 ± 2.04 | 85.05 ± 3.32 | <0.001 | |
| Paraoxonase activity (U/l) | 422.72 ± 36.18 | 610.79 ± 69.19 | 0.021 | |
| ASD (n=50) | Controls (n=85) | χ2 | P | |
| PON1 Q192R | ||||
| 26 (52.0%) | 43 (50.6%) | 0.02 | 0.98 | |
| QR | 21 (42.0%) | 37 (43.5%) | ||
| RR | 3 (6%) | 5 (5.9%) | ||
| Q/R | 0.73/0.27 | 0.72/0.28 | 0.01 | 0.90 |
| PON1 L55M | ||||
| LL | 15 (30.0%) | 31 (36.5%) | 1.13 | 0.56 |
| LM | 30 (60.0%) | 43 (50.6%) | ||
| MM | 5 (10.0%) | 11 (12.9%) | ||
| L/M | 0.60/0.40 | 0.62/0.38 | 0.08 | 0.77 |
All values are the mean ± S.E.M.
Blood samples
Blood specimens were obtained after overnight fast. Samples were withdrawn from a cubital vein into blood tubes and immediately stored on ice at 4°C. For the enzymatic determinations, plasma was separated by centrifugation at 3000 rpm for 10 min. and stored at −20°C until analysis.
Paraoxonase 1 activities
PON1 ssPO.ase and ARE.ase activities in heparinized plasma were measured spectrophotometrically according to Eckerson et al.[28], with minor modifications. Determination of ssPO.ase activity was performed by using paraoxon (O,O-diethyl-O-p-nitrophenyl phosphate, Sigma Chemical Co., Seelze, Germany). The basal assay mixture included 1.0 mM paraoxon, 2M NaCl and 1 mM CaCl2 in 50 mM glycine-NaOH buffer (pH 10.5). The reaction was initiated by the addition of the plasma sample, and the absorbance was monitored at 405 nm for 90 sec. ARE.ase activity was measured using phenyl acetate (Sigma Chemical Co., Steinheim, Germany) 1 mM in 20 mM Tris-HCl (pH 8) containing 1 mM CaCl2. The rate of phenyl acetate hydrolysis at 25°C was determined by monitoring the increase of absorbance at 270 nm over a 90 sec. period. A blank sample, containing incubation mixture without plasma, was run simultaneously to correct for spontaneous substrate breakdown, for both activity determinations. All samples were run in duplicate; the average value was used for activity calculation using a molar extinction coefficient of 18,290 M–1 cm–1 at 412 nm for p-nitrophenol, and 1310 M–1 cm–1 at 270 nm for phenol. Results are expressed as U/l for PON1 ssPO.ase activity (nanomole paraoxon hydrolyzed per minute), and kU/l for PON1 ARE.ase activity (micromole phenyl acetate hydrolyzed per minute).
Paraoxonase 1 genotyping
Genomic DNA was isolated from whole peripheral blood using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and was kept at −20°C until further determinations. Gene polymorphisms were determined using Light Cycler real-time PCR technology based on fluorescence resonance energy transfer. PON1 L55M and Q192R polymorphism analyses were performed according to the method proposed by Pocsai et al.[29], using Light Cycler™ high-speed thermal cycler (Hoffmann-LaRoche, Basel, Swizerland) and Light Cycler™ DNA Master HybProbe kit (Roche Molecular Biochemicals, Mannheim, Germany). Melting curve analysis allowed the identification of the PON1 L55M and Q192R polymorphisms according to the melting point temperatures: 61.0°C for M allele, 57°C for L allele, 57.5°C for Q allele and 51.5°C for R allele.
Statistical analysis
The data are presented as means ± S.E.M. (standard error of the mean). The normal distribution of continuous data was checked using the Kolmogorov–Smirnov test. In parameters with normal distribution comparisons between the ASD group and their age and sex matched controls were made using Student’s t-test. When normality was not present and equal variance could not be assumed, the Mann-Whitney rank-sum test was applied. The effect size (Cohen’s d based on sample size) for variables with a significant group mean difference was computed. For the polymorphisms under study, genotypic and allelic frequencies were calculated. The distribution of genotypes in all groups was tested for deviation from Hardy–Weinberg equilibrium. The Pearson’s chi-square (χ2) test and Fisher’s exact test were applied to assess differences in the genotype and allelic (respectively) distributions between groups of patients and controls. Linkage disequilibrium (LD) was assessed using the THESIAS software, version 3.1 [30]. The multiple regression analysis for the ssPO.ase activity and ARE.ase was conducted using five independent variables (age, sex, PON1 L55M, PON1 Q192R and group). The linear regression analysis (performed on the square-root transformed data) was performed in order to check for the influence of the two polymorphisms in the PON1 gene on ARE.ase and ssPO.ase. A P-value of less than 0.05 was considered statistically significant. SPPS software (version 15.0) was used for data analysis.
Results
We evaluated the activity of PON1 using as substrates both phenylacetate (ARE.ase activity) and paraoxon (ssPO.ase activity) in 50 children with ASD and 30 age and sex matched healthy individuals. As shown in Table 1, both ARE.ase (69.87 ± 2.04 kU/l versus 85.05 ± 3.32 kU/l, P < 0.001) and ssPO.ase (422.72 ± 36.18 U/L versus 610.79 ± 69.19 U/l, P < 0.05) activities were significantly lower in the patient group than in the control group; the effect size (Cohen’s d based on sample size) was 0.92 for ARE.ase and 0.57 for ssPO.ase.
We investigated the PON1 genotypes distributions as a possible explanation for our observation of decreased PON1 function. We found that the distribution of SNPs is unaffected. Allele and genotype frequencies for the ASD group and extended control group (n= 85) are summarized in Table 1. Genotype distributions were at or near Hardy–Weinberg equilibrium for ASD patients and controls (Fisher exact test, P= 0.20 and 0.80 for the L55M and 0.41 and 0.40 for the Q192R polymorphisms in patients and controls, respectively). We found that PON1 L55M and PON1 Q192R genotype and allele distribution was similar in patients and controls (Table 1). Neither were there any differences in the frequencies of combined haplotypes (QM: QL: RL: RM was 0.38: 0.35: 0.25: 0.01 in patients and 0.37: 0.35: 0.26: 0.01 in controls; χ2= 0.31, P= 0.95). LD between markers was estimated by both |D’| calculation and determination of the associated P-value. A strong LD between the R allele of the PON1 192 polymorphism and the L allele of the PON1 55 polymorphism was observed in both ASD participants (|D’| = 0.84, P < 0.001) and controls (|D’| = 0.90, P < 0.001).
Multiple regression analysis showed that ssPO.ase activity was independently affected (adjusted R2= 0.42, F5, 74= 12.69, P < 0.001) by the factors of PON1 Q192R (β= 0.57, P < 0.001) and group (β=−0.23, P= 0.008), whereas the ARE.ase activity was affected (adjusted R2= 0.20, F5, 74= 5.04, P < 0.001) by the factors of PON1 L55M (β=−0.30, P= 0.008) and group (β=−0.39, P < 0.001).
The linear regression analysis (performed on the square-root transformed data) showed that the PON1 Q192R polymorphism explained 49.9% of ssPO.ase activity variance in ASD patients (adjusted R2= 0.499, P < 0.001), and 25.9% of the variance of the ssPO.ase activity in the control group (adjusted R2= 0.259, P < 0.001). On the other hand, the PON1 L55M polymorphism did not influence the ARE.ase activity in the ASD group (adjusted R2= 0.035, P= 0.102), but it showed a borderline influence in the control group (adjusted R2= 0.085, P= 0.065).
The ratio of ssPO.ase to ARE.ase activities (double-substrate method applied on the logarithm-transformed data) was used to identify individual phenotypes according to Eckerson et al.[31]. PON1 ratio was trimodally distributed on the histograms in both groups, and the discriminative values, which separate the low-activity phenotype AA and the high-activity phenotype BB from the intermediate AB phenotype, were found as 0.7 and 1.2, respectively (Fig. 1). The AA : AB : BB phenotype frequencies obtained were similar in the ASD group and the control group (respectively, 0.480 : 0.480 : 0.040, 0.400 : 0.533 : 0.067; χ2= 0.64, P= 0.72).
Fig 1.
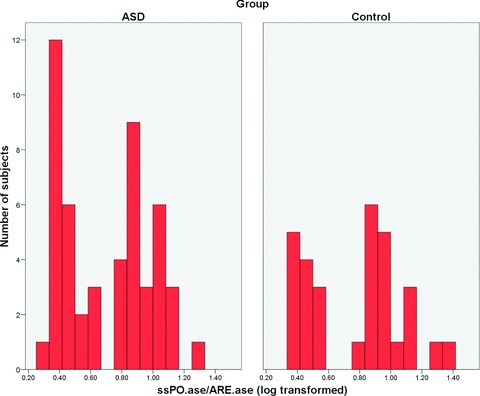
Histogram of the PON1 phenotype distribution in the ASD and control groups. The PON1 phenotype was determined by computing the ratio of ssPO.ase to ARE.ase activities (double-substrate method applied on the logarithm-transformed data). The discriminative values, which separate the low-activity phenotype AA and the high-activity phenotype BB from the intermediate AB phenotype, were found as 0.7 and 1.2, respectively. The phenotype frequencies were similar in patients versus control participants (P > 0.05).
Discussions
Recent investigations indicated a putative involvement of the HDL-associated esterase/lactonase PON1 in the pathogenesis of autism by demonstrating low ARE.ase activity in ASD [27], and by showing an association with less active PON1 gene variants in autistics from North America [26]. In the current study, we aimed at investigating, in a representative cohort of Romanian children with ASD, both PON1 catalytic activity and bioavailability (i.e. ssPO.ase and ARE.ase) and the two most common polymorphisms in the PON1 gene (Q192R and L55M).
The present report does not lend support to either the L55M or the Q192R polymorphism or combined haplotypes of the PON1 gene being associated with ASD in Romania, which is congruent with the lack of association reported in another European autistic group [26]. A previous study [32], investigating the transmission disequilibrium of the PON1 L55M polymorphism in 196 trios, did not demonstrate any association with autism. In our sample, the Q and the L alleles were predominant, and the previously described strong LD between PON1 codon 55 and PON1 codon 192 was observed [33]. Although there are no available data on the PON1 gene variants distribution in Romania, the observed frequencies are similar to those described for other European populations [34–37].
Because wide interindividual variability in plasma PON1 levels within a genetic class has been reported and the PON1 phenotype is a better predictor of disease [24], one should not be mislead by SNPs analysis alone in an epidemiological study. Indeed, we provide evidence that both PON1 enzyme activities are significantly decreased in ASD patients compared with healthy individuals, irrespective of the polymorphisms distribution. In a previous report, we observed a decreased ARE.ase activity in a cohort of autistic children [27], but ssPO.ase activity failed to reach significance; most probably because of the high variance seen in this activity and the small population sample.
Our current findings showed that the participants’ group (ASD versus control) influenced both activities, but no influence of age or sex was detected. The multiple regression analysis performed in our participants also indicated that, as previously demonstrated in a large cross-sectional investigation [38], the major determinant of the PON1 catalytic activity (PO.ase) was the Q192R genotype, whereas PON1 bioavailability (ARE.ase) was affected only by the L55M genotype. Interestingly, in the ASD group, the ssPO.ase/Q192R dependency was more pronounced than in controls. On the other hand, the dependency of the bioavailability of PON1 (ARE.ase) on the L55M polymorphism was absent in the ASD patients, whereas it reached a borderline small level in healthy participants.
The phenotype distribution, computed by graphically examining the ssPO.ase/ARE.ase ratio, was analogous in ASD and controls. The similar phenotypic distribution and the fact that overall there was no difference in the ssPO.ase/ARE.ase ratio between groups suggest that the relationship between the two activities is generally maintained in autistics, regardless of a reduction in the hydrolytic protection.
In recent years, great concern has been raised about the negative impact of chronic low OP exposure on the nervous system development. Prospective studies recorded significant decreases in birth weight, head circumference and slower reflexes in prenatally exposed infants [5, 39, 40]. Developmental disorders in children, including atypical autism, have been described among farming families who generally use OP [7]. In the same line of evidence, it has recently been reported an adverse association of OP pesticide exposure, as measured by six non-specific dialkylphosphate metabolites, with mental development and pervasive developmental problems at 24 months of age [8]. Alternatively, post-natal exposure to chlorpyrifos, a well-known neurotoxic OP, reduced the expression of nerve growth factor and reelin (RELN) in the forebrain of rats [41].
Studies of the PON1 enzyme in newborns showed that its levels are on average three to fourfold lower than those of adults [42], suggesting that newborn children and infants will be more susceptible to OP compounds. Taking maternal PON1 status into consideration greatly increases the risk for this adverse behaviour [6]. Because the dose–response relationship for OP toxicity are steep [43], a small percentage difference in PON1 hydrolytic rates should account for large differences in toxicity [19].
The low levels of PON1 activity in our ASD cohort are congruent with the recently proposed model of gene–environment interactions in ASD [3], which involves PON1, prenatal exposure to OP and RELN, a large extracellular serine protease that orchestrates neuronal positioning during corticogenesis. Within this framework, an autistic phenotype could arise in genetically or epigenetically vulnerable individuals producing lower amounts of RELN that are exposed to OP during critical neurodevelopmental periods and in which neuronal migration would be affected to a different extent depending on the enzymatic efficacy of PON1 [44].
The implications of a low level of PON1 activity for the pathogenesis of ASD can also be translated to other factors than just the OP toxicity. PON1 displays a peroxidase-like activity [45], being involved in the protection against oxidative stress [46]. Several studies suggest increased oxidative stress in autism by showing alterations in antioxidant enzymes (e.g. superoxide dismutase, glutathione peroxidase, catalase), increased lipid peroxidation, disturbances in transsulfuration and altered glutathione levels [47–49]. In this context, a reduction in the PON1 activity level could be one of the contributing factors to the redox alterations in autism. PON1 is also able to protect cells against the toxicity of bacterial endotoxins, lipopolysaccharides (LPS), and prevent or greatly reduce the release of cytokines [14]. Recent evidence suggests that ASD may be accompanied by aberrant immune responses, and peripheral blood mononuclear cells from ASD children produce excessive proinflammatory cytokines with a sub-optimal dose of endotoxin (LPS) at high frequency [50, 51]. Therefore, low levels of PON1 activity in children with ASD might contribute to this exuberant cytokine response and to the subsequent disturbances in the cellular immunity. Previous studies have showed a link between PON1 alleles that debilitate its activity with Alzheimer’s dementia [52] and Parkinson’s disease [53], indicating an impact on the cholinergic system. Therefore, an altered PON1 status could also be translated at the level of the previously described alterations in the cholinergic system in autism [54].
Our study does not bring any direct explanation for the low PON1 activity in ASD. The mechanisms behind the observed decrease in the level of PON1 activity are yet to be investigated and here we can only mention a few hypotheses. Some inhibitors of the enzymatic activity of PON1 in children with autism or factors influencing liver PON1 gene expression could be involved. PON1 could also be inactivated under oxidative stress [55] or following pesticide exposure [56]. PON1 can also undergo S-glutathionylation with a consequent reversible inactivation [57]. The enzymatic activity of PON1 is also modulated by subtle dietary factors, as well as other polymorphisms in the PON1 gene [9]. A lessening in PON1 activities in ASD, especially in the PON1 bioavailability, could also be a consequence of a dysfunction of liver conjugation capacity or an altered synthesis and/or secretion of HDL [58]. The fact that neonatal PON1 activity is decreased in infants from mothers having a chronic viral hepatic disease [59] raises also the possibility of an influence of mother’s liver status and/or infection. PON1 has a potentially imprinted expression in mouse placenta [60], which, if confirmed in human beings, could have implications for the imprinting hypothesis for the development of autism [61].
There are a few limitations in our study. First, it is a retrospective study and more information could be gained by following the PON1 status dynamics in children who develop ASD. Second, we did not obtain more accurate clinical phenotypic descriptions in order to balance our patient group for the heterogeneity of ASD [62]. Third, we did not collect data on OP exposure in our participants. On the other hand, because many of the developmental brain abnormalities associated with ASD arise during the first two semester of the pregnancy, environmental factors are more likely to play a role in the pathophysiology of ASD via maternal factors, and consequently, it would seem essential to look, in future studies, for an effect of mother’s PON1 activity in children with ASD.
Conclusions
Both PON1 bioavailability and catalytic activity are impaired in children with ASD. There is a normal distribution of PON1 phenotype and no association with the two most common polymorphisms in the coding region of the gene.
Acknowledgments
This work has been supported by a CEEX grant (83/2006) received from the Romanian Academy of Medical Sciences.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual-text revision. 4th ed. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 2.Herbert MR, Russo JP, Yang S, et al. Autism and environmental genomics. Neurotoxicology. 2006;27:671–84. doi: 10.1016/j.neuro.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 3.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–58. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15:R138–50. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- 5.Young JG, Eskenazi B, Gladstone EA, et al. Association between in utero organophosphate pesticide exposure and abnormal reflexes in neonates. Neurotoxicology. 2005;26:199–209. doi: 10.1016/j.neuro.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Engel SM, Berkowitz GS, Barr DB, et al. Prenatal organophosphate metabolite and organochlorine levels and performance on the Brazelton Neonatal Behavioral Assessment Scale in a multiethnic pregnancy cohort. Am J Epidemiol. 2007;165:1397–404. doi: 10.1093/aje/kwm029. [DOI] [PubMed] [Google Scholar]
- 7.Worth J. Paraoxonase polymorphisms and organophosphates. Lancet. 2002;360:802–3. doi: 10.1016/S0140-6736(02)09914-2. [DOI] [PubMed] [Google Scholar]
- 8.Eskenazi B, Marks AR, Bradman A, et al. Organophosphate pesticide exposure and neurodevelopment in young Mexican-American children. Environ Health Perspect. 2007;115:792–8. doi: 10.1289/ehp.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa LG, Vitalone A, Cole TB, et al. Modulation of paraoxonase (PON1) activity. Biochem Pharmacol. 2005;69:541–50. doi: 10.1016/j.bcp.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Aviram M, Rosenblat M, Bisgaier CL, et al. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest. 1998;101:1581–90. doi: 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackness MI, Durrington PN, Mackness B. The role of paraoxonase 1 activity in cardiovascular disease: potential for therapeutic intervention. Am J Cardiovasc Drugs. 2004;4:211–7. doi: 10.2165/00129784-200404040-00002. [DOI] [PubMed] [Google Scholar]
- 12.Billecke S, Draganov D, Counsell R, et al. Human serum paraoxonase (PON1) isozymes Q and R hydrolyze lactones and cyclic carbonate esters. Drug Metab Dispos. 2000;28:1335–42. [PubMed] [Google Scholar]
- 13.Jakubowski H. Calcium-dependent human serum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation. J Biol Chem. 2000;275:3957–62. doi: 10.1074/jbc.275.6.3957. [DOI] [PubMed] [Google Scholar]
- 14.La Du BN, Aviram M, Billecke S, et al. On the physiological role(s) of the paraoxonases. Chem Biol Interact. 1999;119–120:379–88. doi: 10.1016/s0009-2797(99)00049-6. [DOI] [PubMed] [Google Scholar]
- 15.Richter RJ, Furlong CE. Determination of paraoxonase (PON1) status requires more than genotyping. Pharmacogenetics. 1999;9:745–53. [PubMed] [Google Scholar]
- 16.Humbert R, Adler DA, Disteche CM, et al. The molecular basis of the human serum paraoxonase activity polymorphism. Nat Genet. 1993;3:73–6. doi: 10.1038/ng0193-73. [DOI] [PubMed] [Google Scholar]
- 17.Adkins S, Gan KN, Mody M, et al. Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes. Am J Hum Genet. 1993;52:598–608. [PMC free article] [PubMed] [Google Scholar]
- 18.Garin MC, James RW, Dussoix P, et al. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concentrations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes. J Clin Invest. 1997;99:62–6. doi: 10.1172/JCI119134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies HG, Richter RJ, Keifer M, et al. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet. 1996;14:334–6. doi: 10.1038/ng1196-334. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Kumar M, Chan W, et al. Increased influence of genetic variation on PON1 activity in neonates. Environ Health Perspect. 2003;111:1403–9. doi: 10.1289/ehp.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blatter MC, James RW, Messmer S, et al. Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem. 1993;211:871–9. doi: 10.1111/j.1432-1033.1993.tb17620.x. [DOI] [PubMed] [Google Scholar]
- 22.Mackness B, Mackness MI, Arrol S, et al. Effect of the human serum paraoxonase 55 and 192 genetic polymorphisms on the protection by high density lipoprotein against low density lipoprotein oxidative modification. FEBS Lett. 1998;423:57–60. doi: 10.1016/s0014-5793(98)00064-7. [DOI] [PubMed] [Google Scholar]
- 23.Brophy VH, Jampsa RL, Clendenning JB, et al. Effects of 5’ regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. Am J Hum Genet. 2001;68:1428–36. doi: 10.1086/320600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarvik GP, Rozek LS, Brophy VH, et al. Paraoxonase (PON1) phenotype is a better predictor of vascular disease than is PON1(192) or PON1(55) genotype. Arterioscler Thromb Vasc Biol. 2000;20:2441–7. doi: 10.1161/01.atv.20.11.2441. [DOI] [PubMed] [Google Scholar]
- 25.Brophy VH, Jarvik GP, Richter RJ, et al. Analysis of paraoxonase (PON1) L55M status requires both genotype and phenotype. Pharmacogenetics. 2000;10:453–60. doi: 10.1097/00008571-200007000-00008. [DOI] [PubMed] [Google Scholar]
- 26.D’Amelio M, Ricci I, Sacco R, et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: possible regional specificity in gene-environment interactions. Mol Psychiatry. 2005;10:1006–16. doi: 10.1038/sj.mp.4001714. [DOI] [PubMed] [Google Scholar]
- 27.Paşca SP, Nemes B, Vlase L, et al. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006;78:2244–8. doi: 10.1016/j.lfs.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 28.Eckerson HW, Romson J, Wyte C, et al. The human serum paraoxonase polymorphism: identification of phenotypes by their response to salts. Am J Hum Genet. 1983;35:214–27. [PMC free article] [PubMed] [Google Scholar]
- 29.Pocsai Z, Toth Z, Paragh G, et al. Rapid genotyping of paraoxonase 55 and 192 mutations by melting point analysis using real time PCR technology. Clin Chim Acta. 2003;332:31–6. doi: 10.1016/s0009-8981(03)00083-4. [DOI] [PubMed] [Google Scholar]
- 30.Lewontin RC. On measures of gametic disequilibrium. Genetics. 1988;120:849–52. doi: 10.1093/genetics/120.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckerson HW, Wyte CM, La Du BN. The human serum paraoxonase/arylesterase polymorphism. Am J Hum Genet. 1983;35:1126–38. [PMC free article] [PubMed] [Google Scholar]
- 32.Serajee FJ, Nabi R, Zhong H, et al. Polymorphisms in xenobiotic metabolism genes and autism. J Child Neurol. 2004;19:413–7. doi: 10.1177/088307380401900603. [DOI] [PubMed] [Google Scholar]
- 33.Wheeler JG, Keavney BD, Watkins H, et al. Four paraoxonase gene polymorphisms in 11212 cases of coronary heart disease and 12786 controls: meta-analysis of 43 studies. Lancet. 2004;363:689–95. doi: 10.1016/S0140-6736(04)15642-0. [DOI] [PubMed] [Google Scholar]
- 34.Arca M, Ombres D, Montali A, et al. PON1 L55M polymorphism is not a predictor of coronary atherosclerosis either alone or in combination with Q192R polymorphism in an Italian population. Eur J Clin Invest. 2002;32:9–15. doi: 10.1046/j.1365-2362.2002.00935.x. [DOI] [PubMed] [Google Scholar]
- 35.Deakin S, Leviev I, Nicaud V, et al. Paraoxonase-1 L55M polymorphism is associated with an abnormal oral glucose tolerance test and differentiates high risk coronary disease families. J Clin Endocrinol Metab. 2002;87:1268–73. doi: 10.1210/jcem.87.3.8335. [DOI] [PubMed] [Google Scholar]
- 36.Gardemann A, Philipp M, Hess K, et al. The paraoxonase Leu-Met54 and Gln-Arg191 gene polymorphisms are not associated with the risk of coronary heart disease. Atherosclerosis. 2000;152:421–31. doi: 10.1016/s0021-9150(99)00489-x. [DOI] [PubMed] [Google Scholar]
- 37.Catano HC, Cueva JL, Cardenas AM, et al. Distribution of paraoxonase-1 gene polymorphisms and enzyme activity in a Peruvian population. Environ Mol Mutagen. 2006;47:699–706. doi: 10.1002/em.20259. [DOI] [PubMed] [Google Scholar]
- 38.Roest M, Van Himbergen TM, Barendrecht AB, et al. Genetic and environmental determinants of the PON-1 phenotype. Eur J Clin Invest. 2007;37:187–96. doi: 10.1111/j.1365-2362.2007.01769.x. [DOI] [PubMed] [Google Scholar]
- 39.Berkowitz GS, Wetmur JG, Birman-Deych E, et al. In utero pesticide exposure, maternal paraoxonase activity, and head circumference. Environ Health Perspect. 2004;112:388–91. doi: 10.1289/ehp.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whyatt RM, Rauh V, Barr DB, et al. Prenatal insecticide exposures and birth weight and length among an urban minority cohort. Environ Health Perspect. 2004;112:1125–32. doi: 10.1289/ehp.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Betancourt AM, Burgess SC, Carr RL. Effect of developmental exposure to chlorpyrifos on the expression of neurotrophin growth factors and cell-specific markers in neonatal rat brain. Toxicol Sci. 2006;92:500–6. doi: 10.1093/toxsci/kfl004. [DOI] [PubMed] [Google Scholar]
- 42.Cole TB, Jampsa RL, Walter BJ, et al. Expression of human paraoxonase (PON1) during development. Pharmacogenetics. 2003;13:357–64. doi: 10.1097/00008571-200306000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Costa LG, McDonald BE, Murphy SD, et al. Serum paraoxonase and its influence on paraoxon and chlorpyrifos-oxon toxicity in rats. Toxicol Appl Pharmacol. 1990;103:66–76. doi: 10.1016/0041-008x(90)90263-t. [DOI] [PubMed] [Google Scholar]
- 44.Keller F, Persico AM. The neurobiological context of autism. Mol Neurobiol. 2003;28:1–22. doi: 10.1385/MN:28:1:1. [DOI] [PubMed] [Google Scholar]
- 45.Aviram M, Hardak E, Vaya J, et al. Human serum paraoxonases (PON1) Q and R selectively decrease lipid peroxides in human coronary and carotid atherosclerotic lesions: PON1 esterase and peroxidase-like activities. Circulation. 2000;101:2510–7. doi: 10.1161/01.cir.101.21.2510. [DOI] [PubMed] [Google Scholar]
- 46.Li HL, Liu DP, Liang CC. Paraoxonase gene polymorphisms, oxidative stress, and diseases. J Mol Med. 2003;81:766–79. doi: 10.1007/s00109-003-0481-4. [DOI] [PubMed] [Google Scholar]
- 47.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–81. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Deth R, Muratore C, Benzecry J, et al. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology. 2008;29:190–201. doi: 10.1016/j.neuro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 49.Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev. 2006;9:485–99. doi: 10.1080/10937400600882079. [DOI] [PubMed] [Google Scholar]
- 50.Jyonouchi H, Sun S, Itokazu N. Innate immunity associated with inflammatory responses and cytokine production against common dietary proteins in patients with autism spectrum disorder. Neuropsychobiology. 2002;46:76–84. doi: 10.1159/000065416. [DOI] [PubMed] [Google Scholar]
- 51.Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol. 2001;120:170–9. doi: 10.1016/s0165-5728(01)00421-0. [DOI] [PubMed] [Google Scholar]
- 52.Leduc V, Poirier J. Polymorphisms at the paraoxonase 1 L55M and Q192R loci affect the pathophysiology of Alzheimer’s disease: emphasis on the cholinergic system and beta-amyloid levels. Neurodegener Dis. 2008;5:225–7. doi: 10.1159/000113709. [DOI] [PubMed] [Google Scholar]
- 53.Benmoyal-Segal L, Vander T, Shifman S, et al. Acetylcholinesterase/paraoxonase interactions increase the risk of insecticide-induced Parkinson’s disease. FASEB J. 2005;19:452–4. doi: 10.1096/fj.04-2106fje. [DOI] [PubMed] [Google Scholar]
- 54.Perry EK, Lee ML, Martin-Ruiz CM, et al. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry. 2001;158:1058–66. doi: 10.1176/appi.ajp.158.7.1058. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen SD, Sok DE. Oxidative inactivation of paraoxonase1, an antioxidant protein and its effect on antioxidant action. Free Radic Res. 2003;37:1319–30. [PubMed] [Google Scholar]
- 56.Hernandez A, Gomez MA, Pena G, et al. Effect of long-term exposure to pesticides on plasma esterases from plastic greenhouse workers. J Toxicol Environ Health A. 2004;67:1095–108. doi: 10.1080/15287390490452371. [DOI] [PubMed] [Google Scholar]
- 57.Rozenberg O, Aviram M. S-Glutathionylation regulates HDL-associated paraoxonase 1 (PON1) activity. Biochem Biophys Res Commun. 2006;351:492–8. doi: 10.1016/j.bbrc.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 58.Horvath K, Perman JA. Autistic disorder and gastrointestinal disease. Curr Opin Pediatr. 2002;14:583–7. doi: 10.1097/00008480-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 59.Schulpis KH, Barzeliotou A, Papadakis M, et al. Maternal chronic hepatitis B virus is implicated with low neonatal paraoxonase/arylesterase activities. Clin Biochem. 2008;41:282–7. doi: 10.1016/j.clinbiochem.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Okita C, Meguro M, Hoshiya H, et al. A new imprinted cluster on the human chromosome 7q21-q31, identified by human-mouse monochromosomal hybrids. Genomics. 2003;81:556–9. doi: 10.1016/s0888-7543(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 61.Badcock C, Crespi B. Imbalanced genomic imprinting in brain development: an evolutionary basis for the aetiology of autism. J Evol Biol. 2006;19:1007–32. doi: 10.1111/j.1420-9101.2006.01091.x. [DOI] [PubMed] [Google Scholar]
- 62.Ronald A, Happe F, Bolton P, et al. Genetic heterogeneity between the three components of the autism spectrum: a twin study. J Am Acad Child Adolesc Psychiatry. 2006;45:691–9. doi: 10.1097/01.chi.0000215325.13058.9d. [DOI] [PubMed] [Google Scholar]