

Abstract
Plasma high-density lipoproteins (HDLs) protect endothelial cells against apoptosis induced by oxidized low-density lipoprotein (oxLDL). The specific component(s) of HDLs implicated in such cytoprotection remain(s) to be identified. Human microvascular endothelial cells (HMEC-1) were incubated with mildly oxLDL in the presence or absence of each of five physicochemically distinct HDL subpopulations fractionated from normolipidemic human plasma (n= 7) by isopycnic density gradient ultracentrifugation. All HDL subfractions protected HMEC-1 against oxLDL-induced primary apoptosis as revealed by nucleic acid staining, annexin V binding, quantitative DNA fragmentation, inhibition of caspase-3 activity and reduction of cytoplasmic release of cytochrome c and apoptosis-inducing factor. Small, dense HDL 3c displayed twofold superior intrinsic cytoprotective activity (as determined by mitochondrial dehydrogenase activity) relative to large, light HDL 2b on a per particle basis (P < 0.05). Equally, all HDL subfractions attenuated intracellular generation of reactive oxygen species (ROS); such anti-oxidative activity diminished from HDL 3c to HDL 2b. The HDL protein moiety, in which apolipoprotein A-I (apoA-I) predominated, accounted for ∼70% of HDL anti-apoptotic activity. Furthermore, HDL reconstituted with apoA-I, cholesterol and phospholipid potently protected HMEC-1 from apoptosis. By contrast, modification of the content of sphingosine-1-phosphate in HDL did not significantly alter cytoprotection. We conclude that small, dense, lipid-poor HDL 3 potently protects endothelial cells from primary apoptosis and intracellular ROS generation induced by mildly oxLDL, and that apoA-I is pivotal to such protection.
Keywords: HDL particle heterogeneity, reactive oxygen species, anti-apoptotic activity, anti-oxidative activity, sphingosine-1-phosphate
Introduction
Apoptosis is a key feature of the progression of atherosclerotic plaques [1]. Indeed, apoptosis of endothelial cells, vascular smooth muscle cells and macrophages contributes to plaque growth, lipid core development, plaque rupture and thrombosis. The process of apoptosis in atherosclerotic lesions is triggered by chronic inflammation and oxidative stress [2]. Indeed, oxidative stress is now recognized as a major emerging risk factor for premature cardiovascular disease [3].
During atherogenesis, pro-inflammatory processes in the arterial wall elicit production of a spectrum of one- and two-electron oxidants which act on lipid and protein components of low-density lipoproteins (LDL) to form oxidized LDL (oxLDL) [4]. OxLDL possess multiple pro-atherogenic and pro-inflammatory properties which include induction of apoptosis and necrosis in endothelial cells and macrophages of the arterial wall [5].
In contrast to LDL, high-density lipoproteins (HDLs) exert vasculoprotective actions; in addition to cellular cholesterol efflux and anti-oxidative/anti-inflammatory properties, HDLs protect endothelial cells from the cytotoxicity of oxLDL [6]. Moreover, HDLs decrease endothelial cell apoptosis induced by mildly oxLDL [7], tumour necrosis factor-α (TNF-α) [8] and growth factor deprivation [9]. HDLs equally protect endothelial cells from necrosis [10, 11].
Both apolipoprotein and lipid components appear to contribute to the cytoprotective properties of HDL; these primarily include apolipoprotein A-I (apoA-I), the major HDL apolipoprotein [7, 8], and sphingosine-1-phosphate (S1P), a minor bioactive lipid [10–12]. Gene transfer of human apoA-I to rats has been recently shown to decrease apoptosis in cardiomyocytes [13]. Plasma membrane receptors and transporters involved in cellular cholesterol efflux, including scavenger receptor class B type I (SR-BI) and ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1), may potentially mediate the cytoprotective effects of HDL via their interaction with apoA-I and ensuing cellular efflux of toxic oxidized lipids [14, 15].
S1P circulates at high nanomolar concentrations primarily associated with small, dense HDL 3 [16–18], and is likely derived from red blood cells and/or platelets [19, 20]. Equally, S1P can function as a ligand for G protein-coupled S1P receptors on endothelial and smooth muscle cells, thereby enhancing cell growth and survival [21]. Consistent with these findings, intracellular signal transduction induced by HDL associated lysosphingolipids, primarily S1P, may in part account for the cytoprotective effects of HDL [22].
HDL particles are highly heterogeneous in their structure, physicochemical properties, intravascular metabolism and anti-atherogenic and vasculoprotective activities. Small, dense, protein-rich HDL 3 display elevated capacities to accept cellular cholesterol [23], to inhibit expression of adhesion molecules [24] and to protect LDL from oxidation [25] as compared to large, light, lipid-rich HDL 2 [26]. Our recent data suggest that small, dense HDL 3 enriched in S1P [18] exert potent protection of endothelial cells from primary apoptosis induced by mild oxLDL [27]; however, HDL components involved in such cytoprotection remain indeterminate. We suggested that apoA-I and/or S1P constitute major factors in the anti-apoptotic activity of small HDL. We present evidence that apoA-I is pivotal to the potent protection of endothelial cells against primary apoptosis induced by mild oxLDL; by contrast, S1P failed to enhance HDL and/or apoA-I-mediated cytoprotection.
Methods
To investigate cytoprotective effects of HDL subfractions, we employed human microvascular endothelial cells (HMEC-1) which are widely used for in vitro studies of multiple biological aspects of atherosclerosis, including inflammation and apoptosis [28–30], and thus represent an appropriate model to study biological properties of oxLDL and protective effects of HDL. Indeed, mechanisms of apoptotic signalling evoked by oxLDL in HMEC-1 and cytoprotection induced by HDL are similar in HMEC-1 and other cultured vascular cells, such as bovine aortic endothelial cells and smooth muscle cells [5–7, 31, 32].
Cellular apoptotic and necrotic morphology was determined microscopically using two vital fluorescent dyes, a permeant intercalating green probe SYTO-13 and a non-permeant intercalating orange probe propidium iodide (PI) [33]. Apoptosis and necrosis were equally quantified using the annexin V-flourescein isothiocyanate (FITC)/PI kit for flow cytometry (Beckman Coulter, Roissy, France).
Lipoproteins were preparatively fractionated by isopycnic density gradient ultracentrifugation from normolipidemic human serum or ethylenediaminetetraacetic acid plasma as previously described [34, 35]. Five major subfractions of HDL were isolated, i.e. large, light HDL 2b (density 1.063–1.087 g/ml) and 2a (density 1.088–1.110 g/ml), and small, dense HDL 3a (density 1.110–1.129 g/ml), 3b (density 1.129–1.154 g/ml) and 3c (density 1.154–1.170 g/ml). LDL (200 mg apolipoprotein B/dl) was oxidized by ultraviolet (UV) irradiation in the presence of Cu2+ (5 μM). This approach produces mildly oxLDL that contain low levels of lipid peroxidation products (4–13 mol of conjugated dienes/mol LDL corresponding to the end of the lag-phase of LDL oxidation by Cu2+[36]) and selectively induces apoptosis in endothelial cells [31], in contrast to heavily oxLDL that contain high levels of products of lipid and protein oxidation (e.g. >200 mol of conjugated dienes/mol LDL corresponding to the end of the propagation phase of LDL oxidation by Cu2+) and largely induce cellular necrosis.
Details of reagents and chemicals, blood samples, fractionation and physicochemical characterization of serum lipoproteins [25], endothelial cell culture [37], characterization of apoptosis [37], S1P assays [18] and statistical analysis are available as an online supplement.
Results
HDL mediated protection of HMEC-1 from oxLDL-induced cytotoxicity
Progressive reduction in HDL hydrated density and size across HDL particle subfractions from HDL 2b to HDL 3c was associated with progressive elevation in protein content and diminution in lipid content (Table S1). Consistent with earlier data [25], the lipid core of small HDL featured enrichment in cholesteryl ester (CE) relative to triglyceride (TG), with almost twofold increment in the CE/TG weight ratio from large HDL 2b (6.3 ± 1.2) to small HDL 3c (11.8 ± 3.8).
The protective effect of plasma HDL (d 1.063–1.21 g/ml) against endothelial cell death mediated by oxLDL does not require direct contact between HDL and oxLDL, but rather depends on the duration of the cellular contact of HDL, which should exceed 12 hrs [7]. We therefore preincubated HMEC-1 with HDL subfractions for 24 hrs before incubation with oxLDL. Incubation of cells with oxLDL alone induced marked cytotoxicity as documented by diminished mitochondrial function with significant decrease in 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction (–68%, P < 0.01). All HDL subfractions protected endothelial cells from the cytotoxic action of mild oxLDL (Fig. 1A).
Fig 1.
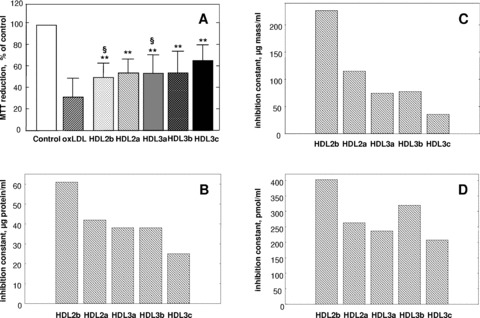
HDL subfractions protect HMEC-1 from oxLDL-induced cytotoxicity. The cytoprotective action of individual HDL subfractions was evaluated by MTT reduction at a fixed concentration of total HDL protein (25 μg/ml; A). Data from 10 experiments performed on 8 separate HDL preparations are shown as a percentage of MTT reduction in controls; **P < 0.01 versus oxLDL; §P < 0.05 versus HDL 3c. Calculated values of inhibition constants for each HDL subfraction expressed as micrograms protein/ml (B), μg mass/ml (C) or pmol/ml (D) are equally shown. To calculate inhibition constants, cytoprotective effects of HDL subfractions were measured as a function of total HDL protein concentration, total HDL mass or the number of HDL particles and curve fitting for each data set was performed with a model of non-competitive inhibition. HMEC-1 (40,000 cells/well) were plated in 24-well plates and incubated with HDL subfractions for 24 hrs and subsequently with oxLDL for 18 hrs.
At equal protein concentration (25 μg/ml), however, the dense HDL 3c subfraction displayed significantly elevated cytoprotective activity (51% protection, P < 0.01; MTT assay) as compared to larger, lighter HDL 2b and -3a subspecies (28% and 33%, P < 0.05 versus HDL 3c; Fig. 1A). The cytoprotective effects of HDL subfractions were dose dependent and increased in parallel with increment in HDL concentration over the range 5–100 μg protein/ml; again, the densest HDL 3c subclass exerted the most potent cellular protection at all concentrations studied (data not shown).
To further compare the intrinsic anti-apoptotic activities of HDL subfractions, cytoprotective effects of HDL subfractions were evaluated as a function of total HDL protein concentration, total HDL mass or the number of HDL particles; curve fitting was performed with a model of non-competitive inhibition on each data set. We observed that small, dense HDL 3c displayed lower values of inhibition constants (i.e. superior cytoprotective activity) relative to other HDL subfractions on the basis of total HDL protein (Fig. 1B), total HDL mass (Fig. 1C) or the number of HDL particles (Fig. 1D), thereby indicating greater intrinsic cytoprotective activity of HDL 3c independently of the HDL concentration basis. For example, preincubation of HMEC-1 in the presence of small HDL 3c (250 nM) provided protection of 50 ± 19%, whereas protection of HMEC-1 was attenuated (37 ± 23%; n= 3) at the same concentration of HDL 2b particles.
Pooling of results from all experiments revealed that cytoprotection afforded by HDL subfractions positively correlated with HDL density (r= 0.35, P= 0.002), dense HDL 3 subspecies exhibiting greatest potency. By contrast, HDL mediated cellular protection from oxLDL toxicity did not parallel the capacity of HDL subfractions to efflux cellular cholesterol. Indeed, all five HDL subfractions did not differ significantly in their capacity to efflux cholesterol from HMEC-1 cells (2.3 ± 0.4, 2.8 ± 0.4, 2.5 ± 0.6, 3.6 ± 0.4 and 2.8 ± 0.3% for HDL 2b, 2a, 3a, 3b and 3c, respectively, n= 3) when compared at equivalent protein concentration.
HDL mediated protection of HMEC-1 from oxLDL-induced apoptosis
Death of HMEC-1 endothelial cells treated with mildly oxLDL (200 μg apoB-100/ml; 4–13 mol of conjugated dienes/mol LDL) occurred mainly through an apoptotic process, as indicated by the number of cells exhibiting characteristic morphological nuclear changes, such as chromatin condensation and nuclear fragmentation clearly visualized by SYTO13/IP labelling (Fig. 2A and B). It is of note that oxLDL induced the appearance of cells exhibiting fragmented nuclei, which were permeable to PI, a vital DNA marker excluded from intact nuclei and indicative of a late stage of apoptosis (post-apoptotic necrotic patterns). By contrast, the level of primary necrosis was very low (<5%) as shown by low levels of lactate dehydrogenase (LDH) release and by small numbers of PI-stained cells with the morphological features of primary necrosis (data not shown). All HDL subfractions protected HMEC-1 from apoptosis induced by oxLDL, attenuating condensed chromatin staining by SYTO-13 and post-apoptotic nuclear staining by PI; again however, small dense HDL 3c revealed superior cytoprotection relative to less dense HDL subfractions (Fig. 2C–G).
Fig 2.

HDL subfractions protect HMEC-1 from oxLDL-induced apoptosis. Apoptotic and necrotic morphology was evaluated using fluorescent microscopy either in the absence of added lipoproteins (A), or in the presence of oxLDL added after preincubation in the absence (B) or in the presence of HDL 2b (C), 2a (D), 3a (E), 3b (F) and 3c (G) subfractions. Normal nuclei, loose chromatin staining by SYTO-13 (green); apoptotic nuclei, condensed chromatin staining by SYTO-13 (green) and/or chromatin fragmentation; post-apoptotic necrosis, nuclear staining by PI (orange). HMEC-1 (20,000–30,000 cells/well) were plated in 12-well plates and incubated in the absence (control) or in the presence of each HDL subfraction (25 μg total protein/ml) for 24 hrs. Subsequently, oxLDL was added and cells were incubated for 18 to 24 hrs. Results are representative of three independent experiments performed on two independent HDL preparations.
Apoptosis and necrosis were equally quantified by flow cytometry as cellular binding of annexin V; indeed, annexin V specifically interacts with phosphatidylserine in the extracellular membrane leaflet of apoptotic cells [38]. Preincubation with small, dense HDL 3b and -3c (–54%, P < 0.01 and –148%, P < 0.001, respectively; Fig. 3A), and to a lesser extent with large light HDL 2b (–47%, P < 0.01), significantly diminished annexin V binding to HMEC-1 induced by oxLDL. Caspase-3 (DEVDase) was activated in oxLDL-treated HMEC-1 (Fig. 3B). All HDL subfractions prevented activation of caspase-3 provoked by oxLDL (P < 0.05; Fig. 3B); again, small HDL 3c was more protective than large HDL 2b (P < 0.05) and 2a (P < 0.01). Quantitative DAPI experiments revealed fragmentation of cellular DNA in oxLDL-treated HMEC-1 (Fig. 3C); all HDL subfractions attenuated DNA fragmentation induced by mildly oxLDL, with small dense HDL 3c providing most potent protection (Fig. 3C).
Fig 3.
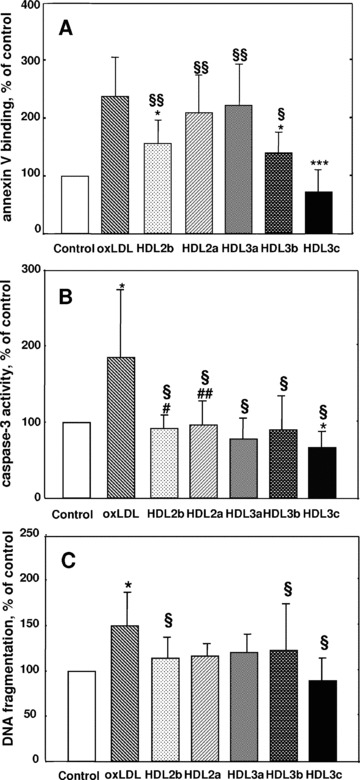
HDL subfractions protect HMEC-1 against oxLDL-induced annexin V binding (A), caspase-3 activation (B) and DNA fragmentation (C). Apoptosis and necrosis were quantified by flow cytometry as annexin V binding using the annexin V-FITC/PI kit at excitation and emission wavelengths of 492 and 520 nm, respectively, for annexin V-FITC and 550 and 680 nm, respectively, for PI (A). Data are shown as a percentage of annexin V binding in the absence of added lipoproteins (controls); *P < 0.05, ***P < 0.001 versus oxLDL; §P < 0.05, §§P < 0.01 versus HDL 3c. Caspase-3 activity was measured fluorometrically using Ac-DEVD-AMC fluorescent substrate (excitation and emission wavelengths 380 and 430 nm, respectively (B). Chromatin fragments were determined by the fluorometric DAPI procedure (C). HMEC-1 were plated in Petri dishes and incubated in the absence (control) or in the presence of each HDL subfraction (25 μg total protein/ml) for 24 hrs. OxLDL was added and cells were incubated for 16 to 24 hrs. Data are shown as a percentage of caspase-3 activation and DNA fragmentation in the absence of added lipoproteins (controls). Results of 5 (annexin V binding), 6 (caspase-3 activation) and 7 (DNA fragmentation) experiments performed on 5 (annexin V binding) or 4 (caspase-3 activation and DNA fragmentation) independent HDL preparations are shown; *P < 0.05 versus control; §P < 0.05 versus oxLDL; #P < 0.05, ##P < 0.01 versus HDL 3c.
The mechanism of apoptosis triggered by mildly oxLDL involves two calcium-dependent pathways, a caspase-dependent pathway mediated by calpain/Bid/cytochrome c/caspase-3 and a caspase-independent pathway mediated by apoptosis-inducing factor (AIF) [39].
OxLDL induced cytoplasmic release of cytochrome c and AIF from HMEC-1, and degradation of Bid (Fig. 4). Preincubation of cells with small dense HDL 3c reversed this cascade of pro-apoptotic events, whereas large, light HDL 2b was markedly less effective (Fig. 4).
Fig 4.
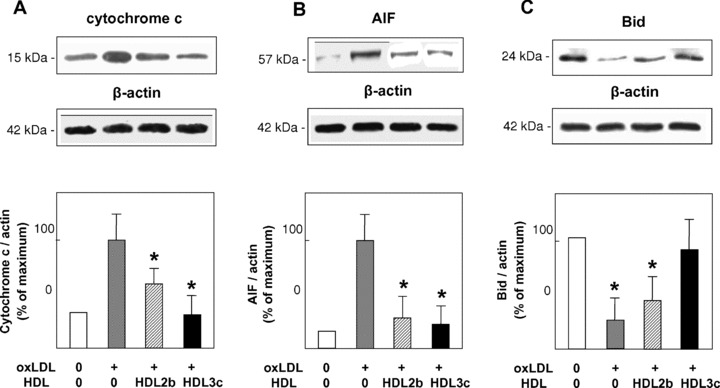
HDL subfractions protect HMEC-1 against oxLDL-induced cytoplasmic release of cytochrome c and AIF and degradation of Bid. HMEC-1 were plated in Petri dishes, pre-incubated in the absence (control) or in the presence of each HDL subfraction (25 μg total protein/ml) for 24 hrs and subsequently incubated with oxLDL for 16 hrs. Following cellular homogenisation, cytosolic proteins were immunoblotted in the mitochondria free-cytosol using anti-cytochrome c (A), anti-AIF (B) and anti-Bid (C) monoclonal antibodies, and equally using anti β-actin antibody to detect β-actin as a housekeeping protein. Data are scaled to the highest ratio of the protein of interest to β-actin. Results of three experiments performed on three independent HDL preparations are shown; *P < 0.05 versus oxLDL.
HDL mediated protection of HMEC-1 from oxLDL-induced generation of reactive oxygen species (ROS)
Preincubation of endothelial cells with each HDL subfraction diminished intracellular generation of ROS induced by oxLDL as measured using dichlorofluorescein diacetate (H2DCFDA) as a fluorescent probe (Fig. 5). Such anti-oxidative activity decreased with decrement in HDL density from HDL 3c (54% inhibition) to HDL 2b (21%; P < 0.01 versus HDL 3c). The anti-oxidative effects of HDL subfractions were dose dependent and increased with HDL concentration in the range 10–50 μg/ml; again, small, dense HDL 3c and 3b particles displayed more potent protection as compared to large HDL 2 at all concentrations studied (data not shown). The intracellular anti-oxidative activity of HDL particles was positively correlated with HDL density (r= 0.51, P= 0.01) and anti-apoptotic activity (r= 0.71, P= 0.001) expressed as inhibition of annexin V binding (Fig. 5).
Fig 5.
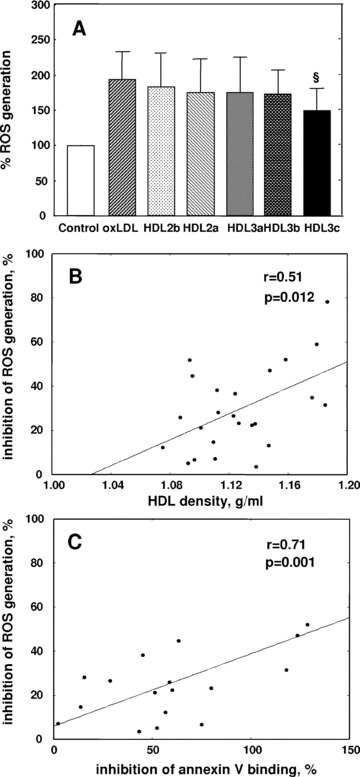
HDL subfractions protect HMEC-1 against oxLDL-induced intracellular ROS generation. (A) HMEC-1 (40,000 cells/ml; 250 μl/well) were plated in 96-well plates for 24 hrs and incubated in the absence (control) or in the presence of each HDL subfraction (25 μg total protein/ml) for 24 hrs. Cells were labelled with 2′,7′-dichlorofluorescein diacetate (H2DCFDA) fluorescent substrate and incubated with oxLDL (Δ234 nm, 17–34 mol conjugated dienes/mol LDL) for 1 hr. Intracellular ROS generation was measured fluorimetrically (excitation and emission wavelengths 485 and 530 nm, respectively). Data are presented as a percentage of ROS generation in the absence of added lipoproteins (controls). Results of 11 experiments performed on 7 independent HDL preparations are shown. (B) Correlation between HDL mediated inhibition of ROS generation in HMEC-1 and HDL particle density. (C) Correlation between HDL mediated inhibition of ROS generation in HMEC-1 and HDL mediated protection of HMEC-1 from apoptosis assessed by flow cytometry as a decrease in annexin V binding. Effects of HDL subfractions are shown as percentage protection relative to differences between incubation in the absence of added lipoproteins (controls) and in the presence of oxLDL alone; §P < 0.05 versus oxLDL.
Since H2DCFDA does not allow to identify individual ROS, we subsequently determined the source of intracellular ROS generated by oxLDL using a series of specific inhibitors for NAD(P)H oxidases (diphenylene iodonium and apocynin), xanthine oxidase (allopurinol), mitochondrial electron transport chain (ETC) at a level of complex I (rotenone) and complex III (myxothiazol), cytochrome P450 (sulfaphenazole) and metabolism of arachidonic acid (eicosatetraynoic acid for lipoxygenase and indomethacine for cyclooxygenase). We found that NAD(P)H oxidase inhibitors diphenylene iodonium and apocynin as well as the complex III inhibitor myxothiazol potently inhibited intracellular ROS increase elicited by oxLDL (Fig. 6A). ROS production was also blocked by the antioxidants Trolox and pyrolidine dithiocarbamate employed as a positive control. In striking contrast, other tested agents were without significant effect, indicating that NAD(P)H oxidase and mitochondrial ETC provided major contribution to ROS generated by HMEC-1 in the presence of oxLDL.
Fig 6.
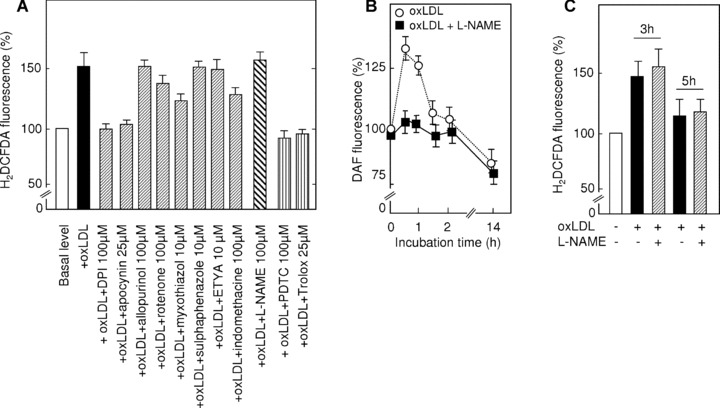
Role of different sources of oxidants for ROS generation in HMEC-1 induced by oxLDL. (A) Effect of specific inhibitors on ROS generation by HMEC-1 induced by oxLDL. HMEC-1 were incubated with oxLDL (200 μg apoB/ml) for 3 hrs and loaded with H2DCFDA. Subsequently, fluorescence resulting from the H2DCFDA oxidation by intracellular ROS was measured. Data are shown as means of 3 independent experiments. (B) Effect of L-NAME on nitric oxide generation by HMEC-1 induced by oxLDL. HMEC-1 were incubated with oxLDL (200 μg apoB/ml) for the indicated time and loaded with DAF-FM. Subsequently, fluorescence of DAF was measured. Data are shown as means of three independent experiments. (C) Effect of L-NAME on ROS generation by HMEC-1 induced by oxLDL. HMEC-1 were incubated with oxLDL (200 μg apoB/ml) for 3 or 5 hrs and loaded with H2DCFDA-AM. Subsequently, fluorescence resulting from H2DCFDA oxidation by intracellular ROS was measured. Data are shown as means of three independent experiments.
We also investigated the potential role of nitric oxide in the intracellular increase of ROS triggered by oxLDL. In these experiments, we used two different approaches involving first, the fluorescent nitric oxide specific probe 4-amino-5-methylamino-2′, 7′-diflouroflourescein (DAF-FM) which allows measurement of intracellularly generated nitric oxide and second, a specific inhibitor of endothelial nitric oxide synthase GN-nitro-L-arginine methyl ester (L-NAME). We observed that oxLDL triggered rapid, transient and slight increase in intracellular nitric oxide levels in HMEC-1, which was completely inhibited by L-NAME (Fig. 6B). Interestingly, longer incubations of HMEC-1 with oxLDL were characterized by progressive decrease in intracellular nitric oxide concentrations, consistent with previous reports on the effects of oxLDL on nitric oxide production in vascular and macrophagic cells [40]. In order to investigate whether nitric oxide contributed to the increase in intracellular ROS induced by oxLDL, we measured the oxidation of H2DCFDA in the presence of L-NAME. As shown in Fig. 6, C, L-NAME did not inhibit induction of intracellular ROS formation elicited by oxLDL, strongly suggesting that nitric oxide is not involved in the oxLDL-induced ROS generation measured by H2DCFDA.
Role of apolipoproteins in the anti-apoptotic effects of HDL subfractions
The particle content of apoA-I fell progressively with HDL size from 4.3 mol/mol in HDL 2b to 2.9 mol/mol in HDL 3c (Table S1). By contrast, maximal particle contents of apoA-II occurred in HDL 2a and 3a (1.3–1.5 mol/mol). Consistent with earlier published data [41], the molar ratio of apoA-I to apoA-II was highest in the largest and smallest HDL particles, respectively (HDL 2b, 5.7 ± 3.4; HDL 3c, 4.9 ± 1.1), thereby suggesting that lipoprotein A-I (LpA-I) particles predominate in large HDL 2b and small HDL 3c.
To assess the relative role of protein versus lipid components in the anti-apoptotic activity of HDL subfractions, apolipoprotein and lipid moieties of each subfraction were separated using Folch extraction and their cellular protective capacity measured in the MTT assay at concentrations corresponding to the initial level of HDL protein (25 μg/ml). Importantly, no S1P was co-isolated with the protein fraction as verified by HPLC (data not shown).
Significantly, the protein fraction accounted for the majority of the cytoprotective effect (71 ± 21%) relative to that of lipids (34 ± 2%, n= 5), thereby demonstrating that the protein moiety, with apoA-I as the major component (Table S1), was central to HDL mediated cytoprotection.
SR-BI is a major apoA-I-binding HDL receptor on human endothelial cells [42]; we therefore additionally evaluated the potential role of SR-BI in mediating the cytoprotective effects of HDL subfractions against oxLDL. Preincubation of HMEC-1 with a monoclonal antibody to human SR-BI did not influence the toxicity of oxLDL (MTT reduction of 42% and 40% of control levels in the absence and presence of the antibody; n= 2). By contrast, the antibody markedly diminished (up to –68%) the protective effects of HDL 3c (MTT reduction of 82 ± 5 and 53 ± 4% of control levels in the absence and presence of the antibody; n= 3). No effect on the cytoprotective activity of HDL 3c was observed when HMEC-1 were pre-incubated with an irrelevant antibody (data not shown).
Role of S1P in the anti-apoptotic effects of HDL subfractions
S1P was asymmetrically distributed across the HDL particle spectrum, with preferential enrichment in HDL 3 as compared to HDL 2 [18] (Fig. 7A). Indeed, S1P was 3-fold enriched in HDL 3c and 3b (approximately 1 mole per 17 HDL particles) as compared to HDL 2b (approximately 1 mole per 50 HDL particles). HDL molar content of S1P positively correlated with HDL density (r= 0.70, P < 0.001), with anti-apoptotic activity expressed as inhibition of annexin V binding (r= 0.56, P= 0.01; Fig. 7B) and with the intracellular anti-oxidative activity of HDL (r= 0.59, P < 0.01; Fig. 7C), thereby suggesting that S1P may contribute to the cytoprotective effects of HDL subfractions on HMEC-1 cells.
Fig 7.
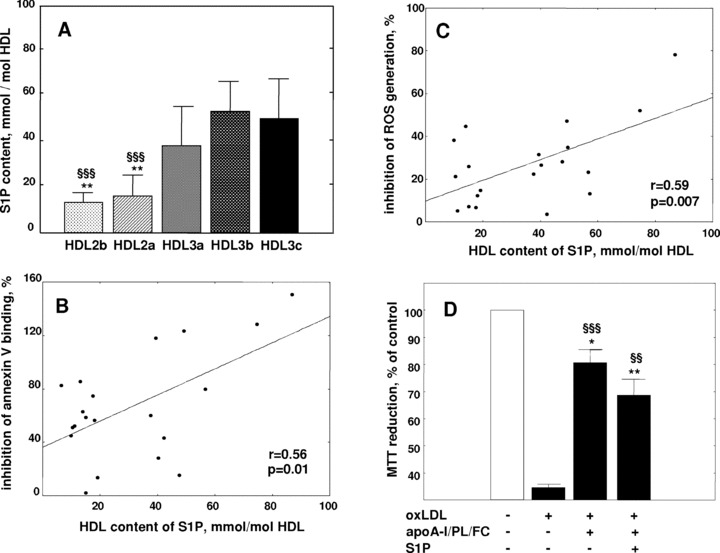
Role of S1P in the anti-apoptotic effects of HDL subfractions. (A) Molar content of S1P in HDL subfractions from normolipidemic patients (n= 5); **P < 0.01 versus HDL 3c, §§§P < 0.001 versus HDL 3b. (B) Correlation between HDL content of S1P and HDL mediated protection of HMEC-1 from apoptosis assessed by flow cytometry as an inhibition of annexin V binding. (C) Correlation between HDL content of S1P and HDL mediated inhibition of ROS generation in HMEC-1. Effects of HDL subfractions are shown as a percentage of protection relative to differences between incubations in the absence of added lipoproteins (controls) and in the presence of oxLDL alone. (D) Cytoprotective effects of S1P-free and S1P-containing rHDL prepared from the major HDL components (apoA-I, POPC, cholesterol) using cholate dialysis. Data from three independent experiments are shown as MTT reduction at a fixed concentration of apoA-I (25 μg/ml); **P < 0.01, *P < 0.05 versus control; §§§P < 0.001, §§P < 0.01 versus oxLDL.
The potential contribution of S1P to the capacity of HDL subfractions to inhibit oxLDL-induced apoptosis in HMEC-1 was then evaluated in four in vitro experimental systems, involving (i) reconstituted HDL (rHDL) discs containing S1P, (ii) S1P in the presence of bovine serum albumin (BSA), (iii) S1P enrichment of HDL subfractions and (iv) use of a pharmacological inhibitor to ligate S1P1/S1P3 receptors.
S1P-free and S1P-containing rHDL were prepared from the major HDL components (apoA-I, palmitoyloleoyl phosphatidylcholine (POPC) and cholesterol) using cholate dialysis [43].
Importantly, the S1P-containing discs were considerably (more than 10-fold) enriched in S1P as compared to normal plasma HDL on the basis of apoA-I content. rHDL that did not contain S1P (at an apoA-I/cholesterol/POPC molar ratio of 1:9:61) protected HMEC-1 from oxLDL-induced apoptosis by 70 ± 13% (P < 0.001; Fig. 7D). However, this protective effect was not enhanced by the addition of S1P to rHDL (protection of 52 ± 15%, P < 0.01 versus incubation with oxLDL alone).
Albumin readily binds S1P and transports approximately 30–40% of total plasma S1P in human beings [16, 17]. When S1P was added to HMEC-1 cells in the presence of 0.4% BSA at concentrations corresponding to those transported by HDL subfractions (1–100 nM; Fig. 7A, Table S1), no inhibition of oxLDL-induced primary apoptosis was observed. In these studies, annexin V binding to HMEC-1 preincubated with S1P plus BSA represented 286–290% relative to control cells lacking oxLDL, a level which did not significantly differ from that measured in HMEC-1 preincubated in the absence of S1P (211%; n= 3). Nor was any effect on oxLDL-induced apoptosis observed following HMEC-1 preincubation with 1–1000 nM dihydro-S1P which is devoid of anti-apoptotic activity (data not shown).
Incubation of normolipidemic human plasma with S1P (5 μM) resulted in the enrichment of HDL in S1P which was more pronounced in large than in small subfractions (2.9-fold in HDL 2b versus 1.8-fold in HDL 3c; n= 3). S1P-enriched HDL tended to be more potent than the corresponding native HDL subfractions in inhibiting primary apoptosis in endothelial cells. Indeed, S1P enrichment of HDL 2b, 2a and 3a decreased annexin V binding to HMEC-1 by 30%, 35% and 24%, respectively (n= 3); effects observed in S1P-enriched small, dense HDL 3b and 3c (annexin V binding was reduced by 23% and 9%, respectively) were weaker, consistent with their lower levels of enrichment; these effects were non-significant. As a result, the molar content of S1P in HDL used in the S1P enrichment experiments weakly negatively correlated with annexin V binding (r=–0.38, P= 0.008).
Ligation of S1P1/S1P3 receptors with a pharmacological inhibitor moderately attenuated the cytoprotective action of HDL 3c (–26%, P < 0.05). Indeed, after preincubation with the inhibitor, HDL 3c reduced apoptosis of HMEC-1 by 28 ± 8% as compared to that in the absence of the inhibitor (38 ± 13%; n= 5). By contrast, ligation of S1P1/S1P3 receptors by Pertussis toxin did not affect the capacity of HDL 3c to protect HMEC-1 from oxLDL-induced apoptosis (data not shown).
Discussion
As apoptosis of endothelial cells is a key feature of endothelial dysfunction and atherosclerotic plaque progression, it is highly relevant that small, dense, protein-rich HDL 3c exhibited superior cytoprotective activity towards human endothelial cells (HMEC-1) as compared to large, light, lipid-rich HDL 2 when expressed on the basis of total protein, total mass or particle number. Moreover, inhibition constants calculated for cytoprotective actions of HDL 3c were lower than those obtained for other HDL subfractions independently of the basis on which HDL concentration was expressed, thereby documenting superior intrinsic anti-apoptotic activity of small HDL 3c. Indeed, small dense HDLs efficiently inhibited both caspase-dependent (mediated by calpain/Bid/cytochrome c/caspase-3) and caspase-independent (mediated by AIF) apoptotic pathways.
The present studies identify the protein moiety of HDL subfractions as the major component in their cytoprotective activities. Furthermore, rHDL discs constituted of apoA-I-, phosphatidylcholine and cholesterol potently protected HMEC-1 from oxLDL-induced apoptosis (–70%). ApoA-I predominates in the HDL protein moiety (up to 70% of total HDL protein [44]) that was pivotal to the anti-apoptotic activity of total human HDL [7]. Significantly, small, protein-rich HDL 3c, which exert potent anti-apoptotic activity, are preferentially enriched in apoA-I relative to apoA-II (molar ratio of 4.9). Consistent with these results, apoA-I potently inhibited TNF-α-induced apoptosis in endothelial cells when complexed with phosphatidylcholine and cholesterol, whereas replacement of apoA-I with apoA-II markedly attenuated the protective effect [8]. These data provide direct evidence that apoA-I is central to the anti-apoptotic activity of HDL particles.
Mechanistically, initial interaction of HDL with the cellular plasma membrane with ensuing intracellular protein synthesis represent prerequisites for the cytoprotective action of both HDL and of its apolipoprotein moiety [7]. Preincubation of HMEC-1 with a monoclonal antibody to human SR-BI, but not with an irrelevant antibody, markedly diminished the protective effects of HDL 3c. These data are consistent with the hypothesis that cell tethering of HDL particles is in part mediated by SR-BI, a major apoA-I-binding HDL receptor on human endothelial cells [45]. Amphipathic α-helices located in the central domain of apoA-I are involved in its binding to SR-BI [45]. The association rate constants of monoclonal antibodies, whose apoA-I binding sites are located in the central domain, increase with HDL size [46]. A conformation-dependent mechanism involving the accessibility of apoA-I might therefore underlie the elevated anti-apoptotic activity of small, dense HDL 3c; indeed, structural integrity of apoA-I is essential for HDL mediated cytoprotection [7].
Mild oxLDL induces apoptosis primarily via its elevated content of lipid hydroperoxides and to a lesser degree oxysterols and other oxidized lipids [37]. Such diversity of biologically active components translates into a complex pattern of apoptotic mechanisms triggered by oxLDL [5]. Indeed, oxLDL can trigger both (i) the extrinsic apoptotic pathway mediated by the FLICE inhibitory protein and Fas/Fas ligand [47] that activates caspase-8, which in turn directly activates caspase-3 and (ii) calcium/calpain pathway that activates Bid and the mitochondrial apoptotic pathway, thereby indirectly activating caspase-3 [39]. The anti-apoptotic activity of HDL may therefore be mechanistically related to accelerated inactivation of pro-apoptotic oxidized lipids and/or ROS, to cellular efflux of oxidized lipids, primarily oxycholesterols [14, 15], and/or to intracellular signalling which boosts survival [6].
All HDL subfractions attenuated generation of ROS in HMEC incubated with oxLDL; the potential of HDL subfractions to attenuate intracellular ROS increased progressively however with increment in HDL density from HDL 2b to 3c. Experiments with specific inhibitors for cellular enzymes revealed that NAD(P)H oxidase and mitochondrial ETC provided major contribution to ROS early generated by HMEC-1 in the presence of oxLDL; by contrast, the role of nitric oxide (and thus peroxynitrite) appears to be minor. Superoxide anion O2.−and hydrogen peroxide H2O2 are two major ROS produced by NAD(P)H oxidase and mitochondrial ETC, thereby implicating these oxidants in the pro-oxidant effects of oxLDL. Superoxide anion O2.− generated by NAD(P)H oxidase in this system appears to be rapidly dismutated to hydrogen peroxide, which participates in the oxLDL-induced signalling as previously reported [48].
The highly significant correlation observed between cellular anti-apoptotic and anti-oxidative activities of HDL subfractions strongly suggests that these two biological properties are mechanistically related. Indeed, consistent with its capacity to inactivate lipid hydroperoxides [49], apoA-I might play a direct role in cellular protection from oxidative stress [50]. In clear contrast, no correlation between the capacities of HDL subfractions to inhibit annexin V binding and intracellular ROS generation on the one hand, and their potential to delay LDL oxidation in an in vitro assay [25] on the other, was observed [27], thereby suggesting that cellular effects of HDL subfractions may be largely unrelated to their capacity to delay LDL oxidation. Consistent with this proposal, the cytoprotective action of both HDL and apoA-I did not necessitate direct contact between HDL and oxLDL, but rather was dependent on preincubation of HDL with cells and ensuing protein synthesis [7]. Efflux of oxidized lipids has been proposed to contribute to HDL mediated protection of macrophages from apoptosis [14, 15]; it appears however unlikely that cholesterol efflux could account for the distinct anti-apoptotic activity of small HDL 3c in our studies as HDL subspecies did not differ in their capacity to efflux cholesterol from HMEC-1.
Survival-boosting intracellular signalling mediated by HDL represents an intriguing possibility. S1P, a bioactive sphingolipid metabolite involved in signalling processes, has been proposed to account for the key cytoprotective properties of HDL including protection from apoptosis, induction of nitric oxide dependent vasorelaxation and stimulation of the expression of anti-inflammatory transforming growth factor β[22]. Remarkably, S1P was enriched in HDL 3 subfractions; furthermore, S1P content was correlated with the anti-apoptotic and anti-oxidative activities of HDL.
Our data are not however consistent with a major role of S1P in the anti-apoptotic activity of small HDL. Indeed, rHDL composed of apoA-I, cholesterol and phosphatidyl choline provided potent protection of endothelial cells from apoptosis in the absence of S1P; furthermore, adding S1P to rHDL did not enhance their cytoprotective properties. In addition, neither HDL enrichment in S1P in vitro nor albumin-bound S1P enhanced HMEC-1 survival at physiological (nanomolar) concentrations of S1P. It has been suggested that S1P receptors, primarily S1P1 and S1P3, are implicated in the protection of endothelial cells mediated by HDL and S1P [11]. However, ligation of S1P1/S1P3 receptors with a pharmacological inhibitor led to only minor reduction (–26%) in the anti-apoptotic activity of HDL 3c in our studies, indicative of the minor importance of this pathway. Importantly, cytoprotective properties of S1P and other lysosphingolipids demonstrated in earlier studies were observed at micromolar concentrations [9] markedly exceeding physiological levels [18, 51]. Earlier studies which employed HPLC with UV detection
[9] (less specific as compared to fluorescent detection used in later studies [18, 51]) markedly overestimated HDL content of lysosphingolipids, thereby leading to incorrect interpretation of data (see [11] for discussion). Next, earlier studies were performed under conditions which did not distinguish between apoptosis and necrosis and globally assessed cell survival in the presence of heavily oxLDL [10, 11]. By contrast, we selectively induced primary apoptosis using mildly oxLDL containing low levels of oxidized lipids. We therefore conclude that the capacity of small, dense HDL to inhibit primary apoptosis induced in endothelial cells by mildly oxLDL may not involve interaction between HDL associated S1P and cellular S1P receptors as a major pathway. Intriguingly, our present data together with those reported previously [7] suggest that the interaction of HDL apoA-I with SR-BI and/or other HDL receptors can decrease oxidative stress induced by oxLDL and promote survival-boosting intracellular signalling – an interesting alternative which remains to be explored.
In summary, our data reveal that small dense HDL 3c particles provide potent protection of endothelial cells from oxLDL-induced primary apoptosis, which involves reduced intracellular generation of ROS. Moreover, apoA-I is pivotal to these distinct cellular anti-apoptotic and anti-oxidative activities, which may be relevant to HDL mediated vasculoprotection. These data have implications for therapeutic HDL raising approaches involving rHDL as they suggest that, consistent with in vivo data [52], simple complexes of apoA-I, phospholipids and cholesterol may be sufficient to afford significant protection of endothelial cells from apoptosis.
Acknowledgments
These studies were supported by National Institute for Health and Medical Research (INSERM), ARLA and ANR (COD 2005 Lisa). J.A.S. was supported by the CAPES – Ministry of Education (Brazil) and by the Nouvelle Societé Française d’Atherosclérose (France). A.K. was supported by an International HDL Research Award from Pfizer (USA). M.J.C. and A.K. gratefully acknowledge the award of a Contrat d’Interface from Assistance Publique – Hôpitaux de Paris/INSERM (France).
Supporting Information
Additional supporting information may be found in the online version of this article:
Table S1. Plasma concentrations, chemical composition andapolipoprotein composition of HDL particle subfractions fromnormolipidemic subjects (n = 7).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Littlewood TD, Bennett MR. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. 2003;14:469–75. doi: 10.1097/00041433-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 3.Heistad DD. Oxidative stress and vascular disease: 2005 Duff lecture. Arterioscler Thromb Vasc Biol. 2006;26:689–95. doi: 10.1161/01.ATV.0000203525.62147.28. [DOI] [PubMed] [Google Scholar]
- 4.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 5.Salvayre R, Auge N, Benoist H, et al. Oxidized low-density lipoprotein-induced apoptosis. Biochim Biophys Acta. 2002;1585:213–21. doi: 10.1016/s1388-1981(02)00343-8. [DOI] [PubMed] [Google Scholar]
- 6.Negre-Salvayre A, Dousset N, Ferretti G, et al. Antioxidant and cytoprotective properties of high-density lipoproteins in vascular cells. Free Radic Biol Med. 2006;41:1031–40. doi: 10.1016/j.freeradbiomed.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Suc I, Escargueil-Blanc I, Troly M, et al. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol. 1997;17:2158–66. doi: 10.1161/01.atv.17.10.2158. [DOI] [PubMed] [Google Scholar]
- 8.Sugano M, Tsuchida K, Makino N. High-density lipoproteins protect endothelial cells from tumor necrosis factor-[alpha]-induced apoptosis. Biochem Biophys Res Commun. 2000;272:872–6. doi: 10.1006/bbrc.2000.2877. [DOI] [PubMed] [Google Scholar]
- 9.Nofer JR, Levkau B, Wolinska I, et al. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL associated lysosphingolipids. J Biol Chem. 2001;276:34480–5. doi: 10.1074/jbc.M103782200. [DOI] [PubMed] [Google Scholar]
- 10.Kimura T, Sato K, Kuwabara A, et al. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276:31780–5. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- 11.Kimura T, Sato K, Malchinkhuu E, et al. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23:1283–8. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- 12.Theilmeier G, Schmidt C, Herrmann J, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–9. doi: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- 13.Van Linthout S, Spillmann F, Riad A, et al. Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation. 2008;117:1563–73. doi: 10.1161/CIRCULATIONAHA.107.710830. [DOI] [PubMed] [Google Scholar]
- 14.Jiang P, Yan PK, Chen JX, et al. High density lipoprotein 3 inhibits oxidized low density lipoprotein-induced apoptosis via promoting cholesterol efflux in RAW264.7 cells. Acta Pharmacol Sin. 2006;27:151–7. doi: 10.1111/j.1745-7254.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 15.Terasaka N, Wang N, Yvan-Charvet L, et al. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc Natl Acad Sci USA. 2007;104:15093–8. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murata N, Sato K, Kon J, et al. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352(Pt 3):809–15. [PMC free article] [PubMed] [Google Scholar]
- 17.Okajima F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator? Biochim Biophys Acta. 2002;1582:132–7. doi: 10.1016/s1388-1981(02)00147-6. [DOI] [PubMed] [Google Scholar]
- 18.Kontush A, Therond P, Zerrad A, et al. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol. 2007;27:1843–9. doi: 10.1161/ATVBAHA.107.145672. [DOI] [PubMed] [Google Scholar]
- 19.Yatomi Y, Ruan F, Hakomori S, et al. Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood. 1995;86:193–202. [PubMed] [Google Scholar]
- 20.Pappu R, Schwab SR, Cornelissen I, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–8. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 21.Waeber C, Blondeau N, Salomone S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 2004;17:365–82. doi: 10.1358/dnp.2004.17.6.829028. [DOI] [PubMed] [Google Scholar]
- 22.Nofer J-R, Assmann G. Atheroprotective effects of high-density lipoprotein-associated lysosphingolipids. Trends Cardiovasc Med. 2005;15:265–71. doi: 10.1016/j.tcm.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Ohta T, Saku K, Takata K, et al. Different effects of subclasses of HDL containing apoA-I but not apoA-II (LpA-I) on cholesterol esterification in plasma and net cholesterol efflux from foam cells. Arterioscler Thromb Vasc Biol. 1995;15:956–62. doi: 10.1161/01.atv.15.7.956. [DOI] [PubMed] [Google Scholar]
- 24.Ashby DT, Rye KA, Clay MA, et al. Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:1450–5. doi: 10.1161/01.atv.18.9.1450. [DOI] [PubMed] [Google Scholar]
- 25.Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol. 2003;23:1881–8. doi: 10.1161/01.ATV.0000091338.93223.E8. [DOI] [PubMed] [Google Scholar]
- 26.Kontush A, Chapman MJ. Antiatherogenic small, dense HDL – guardian angel of the arterial wall? Nat Clin Pract Cardiovasc Med. 2006;3:144–53. doi: 10.1038/ncpcardio0500. [DOI] [PubMed] [Google Scholar]
- 27.De Souza JA, Vindis C, Hansel B, et al. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis. 2008;197:84–94. doi: 10.1016/j.atherosclerosis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Porn-Ares MI, Saido TC, Andersson T, et al. Oxidized low-density lipoprotein induces calpain-dependent cell death and ubiquitination of caspase 3 in HMEC-1 endothelial cells. Biochem J. 2003;374:403–11. doi: 10.1042/BJ20021955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volanti C, Gloire G, Vanderplasschen A, et al. Downregulation of ICAM-1 and VCAM-1 expression in endothelial cells treated by photodynamic therapy. Oncogene. 2004;23:8649–58. doi: 10.1038/sj.onc.1207871. [DOI] [PubMed] [Google Scholar]
- 30.Selemidis S, Dusting GJ, Peshavariya H, et al. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. 2007;75:349–58. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 31.Escargueil-Blanc I, Meilhac O, Pieraggi MT, et al. Oxidized LDLs induce massive apoptosis of cultured human endothelial cells through a calcium-dependent pathway. Prevention by aurintricarboxylic acid. Arterioscler Thromb Vasc Biol. 1997;17:331–9. doi: 10.1161/01.atv.17.2.331. [DOI] [PubMed] [Google Scholar]
- 32.Auge N, Garcia V, Maupas-Schwalm F, et al. Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler Thromb Vasc Biol. 2002;22:1990–5. doi: 10.1161/01.atv.0000043453.21629.3b. [DOI] [PubMed] [Google Scholar]
- 33.Meilhac O, Escargueil-Blanc I, Thiers JC, et al. Bcl-2 alters the balance between apoptosis and necrosis, but does not prevent cell death induced by oxidized low density lipoproteins. FASEB J. 1999;13:485–94. doi: 10.1096/fasebj.13.3.485. [DOI] [PubMed] [Google Scholar]
- 34.Chapman MJ, Goldstein S, Lagrange D, et al. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J Lipid Res. 1981;22:339–58. [PubMed] [Google Scholar]
- 35.Guerin M, Egger P, Le Goff W, et al. Atorvastatin reduces postprandial accumulation and cholesteryl ester transfer protein-mediated remodeling of triglyceride-rich lipoprotein subspecies in type IIb hyperlipidemia. J Clin Endocrinol Metab. 2002;87:4991–5000. doi: 10.1210/jc.2002-020298. [DOI] [PubMed] [Google Scholar]
- 36.Kontush A, Hubner C, Finckh B, et al. Antioxidative activity of ubiquinol-10 at physiologic concentrations in human low density lipoprotein. Biochim Biophys Acta. 1995;1258:177–87. doi: 10.1016/0005-2760(95)00115-s. [DOI] [PubMed] [Google Scholar]
- 37.Kontush A, Chancharme L, Escargueil-Blanc I, et al. Mildly oxidized LDL particle subspecies are distinct in their capacity to induce apoptosis in endothelial cells: role of lipid hydroperoxides. FASEB J. 2003;17:88–90. doi: 10.1096/fj.02-0293fje. [DOI] [PubMed] [Google Scholar]
- 38.Boersma HH, Kietselaer BL, Stolk LM, et al. Past, present, and future of annexin A5: from protein discovery to clinical applications. J Nucl Med. 2005;46:2035–50. [PubMed] [Google Scholar]
- 39.Vindis C, Elbaz M, Escargueil-Blanc I, et al. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. Arterioscler Thromb Vasc Biol. 2005;25:639–45. doi: 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- 40.Nuszkowski A, Grabner R, Marsche G, et al. Hypochlorite-modified low density lipoprotein inhibits nitric oxide synthesis in endothelial cells via an intracellular dislocalization of endothelial nitric-oxide synthase. J Biol Chem. 2001;276:14212–21. doi: 10.1074/jbc.M007659200. [DOI] [PubMed] [Google Scholar]
- 41.Cheung MC, Albers JJ. Distribution of cholesterol and apolipoprotein A-I and A-II in human high density lipoprotein subfractions separated by CsCl equilibrium gradient centrifugation: evidence for HDL subpopulations with differing A-I/A-II molar ratios. J Lipid Res. 1979;20:200–7. [PubMed] [Google Scholar]
- 42.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–94. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 43.Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J Biol Chem. 1997;272:3953–60. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- 44.Barter PJ, Clay MA, Rye KA. High density lipoproteins: the anti-atherogenic fraction. In: Barter PJ, Rye KA, editors. Plasma lipids and their role in disease. Amsterdam: Harwood Academic Publishers; 1999. pp. 85–107. [Google Scholar]
- 45.Thuahnai ST, Lund-Katz S, Anantharamaiah GM, et al. A quantitative analysis of apolipoprotein binding to SR-BI: multiple binding sites for lipid-free and lipid-associated apolipoproteins. J Lipid Res. 2003;44:1132–42. doi: 10.1194/jlr.M200429-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Curtiss LK, Bonnet DJ, Rye KA. The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: a surface plasmon resonance study. Biochemistry. 2000;39:5712–21. doi: 10.1021/bi992902m. [DOI] [PubMed] [Google Scholar]
- 47.Sata M, Walsh K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J Clin Invest. 1998;102:1682–9. doi: 10.1172/JCI3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Escargueil-Blanc I, Salvayre R, Vacaresse N, et al. Mildly oxidized LDL induces activation of platelet-derived growth factor beta-receptor pathway. Circulation. 2001;104:1814–21. doi: 10.1161/hc4001.097179. [DOI] [PubMed] [Google Scholar]
- 49.Garner B, Waldeck AR, Witting PK, et al. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem. 1998;273:6088–95. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 50.Shao B, Bergt C, Fu X, et al. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J Biol Chem. 2005;280:5983–93. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 51.Nofer JR, Van Der Giet M, Tolle M, et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest. 2004;113:569–81. doi: 10.1172/JCI18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bisoendial RJ, Hovingh GK, Levels JH, et al. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation. 2003;107:2944–8. doi: 10.1161/01.CIR.0000070934.69310.1A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Plasma concentrations, chemical composition andapolipoprotein composition of HDL particle subfractions fromnormolipidemic subjects (n = 7).