

Abstract
The immunosuppressive agents cyclosporin A (CsA) and FK-506 have previously been shown to exhibit neurotrophic and neuroprotective properties in vivo. Given that significant clinical expertise exists for both drugs, they represent an attractive starting point for treatment of acute neural injuries. One putative mechanism for neuroprotection by these drugs relates to inhibition of calcineurin activity. However each drug–immunophilin complex can potentially influence additional signal transduction pathways. Furthermore, several non-immunosuppressive immunophilin ligands have been described as possessing neuroprotective properties, suggesting that neuroprotection may be separable from calcineurin inhibition. In the present study, we examined the mechanism of this neuroprotection in facial motor neurons following axotomy-induced injury. Similar to previous studies in rats, CsA and FK-506 enhanced motor neuron survival in mice following acute injury. To examine the mechanism responsible for neuroprotection by these agents, pharmacologic inhibitors of several potential alternate signalling pathways (17-(allylamino)-17-demethoxygeldanamycin, rapamycin, cypermethrin) were evaluated with respect to neuroprotection. Of these, only cypermethrin, a direct calcineurin inhibitor not previously associated with neuronal survival properties, was observed to significantly enhance motor neuron survival following injury. The results demonstrate for the first time that direct inhibition of calcineurin is neuroprotective in vivo. These data support a model in which calcineurin inhibition promotes neuronal survival, distinct from effects upon neurite outgrowth.
Keywords: programmed cell death, apoptosis, neuronal survival, immunophilin ligands, facial nerve, mice
Introduction
Immunosuppressants such as cyclosporin A (CsA) and FK-506 (Tarcolimus) are currently used clinically to treat graft-versus-host rejection following organ transplantations [1, 2]. The cellular receptors for these molecules are collectively termed immunophilins. CsA binds to proteins of the cyclophilin family such as cyclophilin A and D, whereas FK-506 interacts with FK-506 binding proteins (FKBPs) [3]. A number of studies have demonstrated that the immunosuppressive actions of CsA and FK-506 are mediated through a gain-of-function action induced by the drug–immunophilin complex; resulting in the inhibition of calcineurin phosphatase activity [4]. Notably, studies have demonstrated that expression of some immunophilins is 50 times greater in the central nervous system (CNS) than immune cells [5]. In addition, in vivo applications of CsA or FK-506 have both been shown to exert neuroprotective and neurotrophic effects in specific neuronal populations [6, 7]. Administration of FK-506 in rats has been shown to increase neuronal survival and improve functional recovery following facial and sciatic nerve axotomy, photo-thrombotic spinal cord injury and transection of the medial forebrain bundle [8–10]. FK-506 has also been shown to enhance nerve growth factor (NGF)-dependent neuritic outgrowth in PC-12 cells and primary cultures of dorsal root ganglion (sensory) neurons [11]. Similarly, CsA has been shown to protect dopaminergic neurons from 6-hydroxydopamine toxicity, and to reduce cerebral infarct volumes in experimental models of stroke [12–14]. These studies demonstrate that both CsA and FK-506 can reduce the level of neuronal cell loss under a variety of injury states. However, questions remain regarding the mechanism by which these agents promote neuronal survival following injury.
Some evidence suggests that CsA exerts its survival promoting effects through inhibition of cyclophilin D, which comprises part of the mitochondrial permeability transition pore (MPTP) [13]. Opening of the pore results in loss of mitochondrial membrane potential (DC) and mitochondrial swelling, which ultimately manifests in rupture of the mitochondrial outer membrane. Formation of the MPTP is linked to the release pro-apoptotic factors present in the mitochondrial intermembranous space such as holo-cytochrome c, apoptosis-inducing factor (AIF) and second mitochondria-derived activator of caspase/direct IAP binding protein with low pI (Smac/DIABLO), into the cytoplasm where they are involved in downstream programmed cell death (PCD) pathways [15]. Although CsA has been shown to reduce infarct size following middle cerebral artery occlusion [13], FK-506, a drug which lacks effect on the MPTP, also exhibits similar survival promoting properties [16]. Although it is possible that these agents may exert their effects through unrelated mechanisms, their commonality with respect to immune function (inhibition of calcineurin signalling) suggests a potential mechanism [4].
Interestingly, immunophilin and calcineurin expression are strongly correlated within the CNS, suggesting a functional connection [5, 17]. A linkage between calcineurin inhibition and neuronal survival is suggested from studies which shown that calcineurin mediates dephosphorylation of Bcl-2 associated death promoter (Bad); a pro-apoptotic Bcl-2 family protein [18–20]. Bad has previously been shown to influence the release of cytochrome c and other apoptogenic proteins from the mitochondrial intermembraneous space following stimulation of PCD [18–20]. The phosphorylation status of Bad has been implicated as the primary regulatory mechanism governing this BH3-only protein, because phosphorylation of serine residues S112, S136 and S155 enhance the interaction of Bad with 14-3-3, which prevents it from translocating to the mitochondria (S112 and S136), or disrupts its inhibition of anti-apoptotic Bcl-xL (S155) [21–24].
In the present study, we show that CsA and FK-506 enhanced neuronal survival following axotomy-induced facial motor neuron injury in mice, similar to previous work in rats [25]. We further demonstrate that a direct inhibition of calcineurin by cypermethrin (which acts independently of immunophilins) also promotes motor neuron survival following axotomy. In contrast, other signalling pathways related to immunophilin functions did not alter motor neuron survival. These data indicate that the survival promoting effects of CsA and FK-506 on motor neurons following injury are a direct consequence of their ability to inhibit the phosphatase activity of calcineurin.
Experimental procedures
Animals and surgical procedures
Postnatal day 3 or 8 129/SvImJ or ICR mice were generated from colony stocks. Calcineurin A alpha (Ppp3ca) null mice and controls were generated from intercrosses of (Ppp3ca–/–×Ppp3ca+/–, or Ppp3ca+/–×Ppp3ca+/–) stocks [26]. Pups were anesthetized by hypothermia and the left facial nerve exposed, freed of surrounding vascular and connective tissue and transected just distal to the stylomastoid foramen. A 1-mm segment of the distal nerve was then removed to prevent re-innervation of the nerve stump. Animals were sutured closed with 6.0 silk and then placed in water-heated platform at 30°C for 20 min. to allow recovery prior to returning to their dams. Pups were killed 7 days following axotomy (P10) and brainstems removed for subsequent analysis. Analyses of facial motor neuron survival, with drug or vehicle treatments were performed in both 129/SvImJ and ICR backgrounds, and demonstrated comparable results. Results from ICR mice are shown with the exception of Ppp3ca mice which were produced and bred on a 129/SvlmJ background. Results from Ppp3ca heterozygous mice were equivalent to those of wild-type Ppp3ca+/+, and thus only Ppp3ca heterozygous data are presented as control. All of the procedures were in accordance with the Canadian Council on Animal Care and approved by the University of Toronto Animal Care Committee (UACC).
Drug preparation and procedures
Sterile CsA (Sandimmune) was purchased from Novartis Pharmaceuticals (Dorval, Canada), and FK-506 (Tacrolimus – Prograft) was obtained from Fujisawa Pharmaceutical Co., Ltd. (Osaka, Japan). Drugs were removed from sealed glass ampules and diluted in 0.9% sodium chloride immediately prior to use. Cypermethrin and 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) were purchased from LC Laboratories (Woburn, MA, USA) and was dissolved in 100% ethanol at the initial concentration of 15 mg/ml. Rapamycin (Sirolimus – Rapamune) was obtained from Wyeth Pharmaceuticals (Montreal, Canada). Drugs were diluted in a vehicle consisting of ethanol (final concentration 33%), PEG-60 (hydrogenated castor oil, 17%) diluted in 100 mM phosphate, 0.9% NaCl (PBS), pH 7.4. CsA (20 mg/kg), FK-506 (3 mg/kg), cypermethrin (10 mg/kg) and rapamycin (3 mg/kg) were administered once per day via subcutaneous injections. 17-AAG was administered subcutaneously twice daily at 5 mg/kg for a total dose of 10 mg/kg per day (P3 axotomy). The dosages of various pharmacologic inhibitors utilized in this study are in accordance with doses previously demonstrated to exert neural effects [25, 27–30].
Tissue preparation and determination of neuronal count
Mice were anesthetized with 2.5% Avertin, killed and the brains carefully removed. Brainstems were then isolated and the tissues immersion-fixed in 4% paraformaldehyde/100 mM phosphate, 0.9% NaCl, pH 7.4 (PBS) at 4°C overnight. Total motor neuron counts, counts of facial motor neurons expressing activated caspase-3 and immunofluorescent labelling of activated astrocytes and microglia were performed with tissues from distinct sets of animals. Following fixation, samples were processed for either cryostat or paraffin sections. Upon processing, each sample was given a coded identification number so that sections derived from each block could be analysed in a blinded manner. Paraffin embedded specimens were serially sectioned at a thickness of 7 μm through the full extent of the facial nucleus. Sections were stained with 0.1% thionin and motor neuron counts obtained for every ninth section. Facial motor neurons were counted if they contained a clear nucleus and nucleoli within the facial nucleus; counts were not corrected for split nucleoli. Motor neuron number was determined by the method of physical dissector [31] (frame interval = 63 μm). For frozen sections, samples were first cryoprotected in 30% sucrose, 100 mM PBS overnight at 4°C, then frozen in OCT compound (Fisher Scientific Co., Ottawa, Canada) and sections obtained at a thickness of 30 μm at –22°C. For serially sectioned cryostat blocks, sections were collected at 90-μm intervals (every fourth section collected).
Histochemical and immunohistochemical analyses
Samples for peroxidase-based immunohistochemistry were first placed in a solution of 3% H2O2/methanol for 30 min. at room temperature to destroy endogenous peroxidase activity, followed by several washes of PBS. Samples were blocked in 5% goat serum, 0.2% Tween-20 in PBS for 1 hr at room temperature, followed by incubation in primary antibodies overnight at 4°C. Epitope recognition reagents consisted of the following: activated caspase-3 (1:200, Cell Signaling Technology, Inc., Danvers, MA, USA #9661), biotinylated tomato lectin (Lycopersicon esculentum agglutinin – 2.5 μg/ml, Sigma-Aldrich Co., St. Louis, MO, USA #L0651), glial fibrillary acidic protein (GFAP; 1:800, DAKO Canada, Inc., Mississauga, Canada #Z0334). Following primary incubation and washing, sections were incubated with fluorescent or biotinylated secondary antibody (1:200, Bio-Rad Laboratories, Hercules, CA, USA #172-1019) for 2 hrs. Biotinylated samples were further incubated with HRP-conjugated streptavidin (1:100, Vector Laboratories, Inc., Burlingame, CA, USA #PK-6100) for 1 hr and visualized using 3,3-diaminobenzidine. Immunofluorescent secondary antibodies were anti-rabbit AlexaFluor-488 (Invitrogen Corp., Carlsbad, CA, USA #A11008) or Texas Red Avidin D (Vector Laboratories, Inc. #A-2006).
Levels of Bad serine 112 phosphorylation were determined through immunofluorescence using phosphorylated Bad S112 antibody (1:200, Cell Signaling Technology, Inc. #5284) in conjunction with anti-rabbit AlexFlour-488. Signal intensities for individual facial motor neurons (≥ 100 per facial nuclei) were determined for the injured and uninjured facial nuclei of all animals in each treatment group (vehicle, cypermethin, FK-506). A minimum of two independent replicates of the experimental series was performed for each group. For all motor neuron imaging, image collection parameters were held constant. Inclusion parameters used for motor neuron imaging were identical to those used to perform stereoscopic counts (complete cell, nuclear and nucleolar profile). Collected images were analysed in terms of 256 grey levels using ImageJ. Total Bad S112 signal intensity within the non-nuclear component of each motor neuron was then averaged with respect to motor neuron area to obtain individual values. For each injured/uninjured facial nuclear pair, signal intensity was normalized with respect to background signal. For each group, secondary antibody alone resulted in no detectable signal, and correlation analyses of motor neurons area among treatment groups demonstrated no significant differences. Results are presented for injured facial motor neurons in each group as a percentage relative to that observed in uninjured facial motor neurons.
Statistical methods
Statistical analyses were performed with Graphpad PRISM. Assessment of differences between means was determined by unpaired, two-tailed parametric Student’s t test with assumption of unequal variance. Null hypotheses were rejected at the 0.05 level. Results are expressed as mean ± S.E.M. and determined to be significant if P < 0.05.
Image acquisition and manipulation
Fluorescent and bright field images were captured using a Nikon Eclipse E-1000 motorized microscope equipped for epifluorescence at 10×, 20× and 40×, using Hamamatsu ORCA-285 and Nikon DS-Fi1 cameras for fluorescent and bright field imaging, respectively. Image capture software were Simple PCI (Compix, Inc., Imaging Systems, Sewickley, PA, USA) and NIS Elements F (Nikon Corp., Mississauga, Canada). Images were captured and exported as TIFF files, and figures created using in Adobe Photoshop 10 and Illustrator 13. No manipulations other than contrast and brightness adjustments were performed on captured images.
Results
Motor neuron survival is enhanced by CsA and FK-506 treatment following facial axotomy
We first sought to determine whether the survival promoting effects previously observed in rat, could be reproduced in murine strains. For both rats and mice, neonatal axotomy of the facial nerve results in a well-characterized pattern of motor neuron loss through the process of PCD [32, 33]. Although strain variations exist, this lesion paradigm typically results in a loss of >80% of facial motor neurons by 1 week after axotomy [33]. In the current study, mice were subjected to unilateral facial nerve axotomy on postnatal day 3 (P3), and given either CsA or FK-506 once daily for 7 days following injury until the time of killing. As shown in Fig. 1, an enhancement of motor neuron survival was observed in mice following administration of either CsA (20 mg/kg) (Fig. 1C and D) or FK-506 (3 mg/kg) (Fig. 1E and F) compared to vehicle-treated controls (Fig. 1A and B) within injured facial nuclei (dotted regions). Stereological serial counts of surviving motor neurons were performed through the full extent of the facial nucleus for nuclei on both contralateral (uninjured) and ipsilateral (injured) to the axotomized facial nerve, in order to quantify the effect of drug treatment on axotomized motor neurons. As shown in Fig. 1G (left panel), CsA treatment improved motor neuron survival from 15 ± 1% (vehicle) to 27 ± 3%, whereas FK-506 administration (Fig. 1G, centre panel) resulted in 40 ± 3% motor neuron survival compared to control (16 ± 2%). To determine whether the survival promoting effects of CsA and FK-506 acted through similar or disparate mechanisms, mice were treated with both agents following facial nerve axotomy. Stereological analysis of facial nuclei from these animals demonstrated levels of motor neuron survival comparable to that observed using FK-506 alone (Fig. 1G, right panel), indicating that the effects of these agents are not additive with respect to enhancement in neuronal survival, thus suggesting they act through similar targets.
Fig 1.
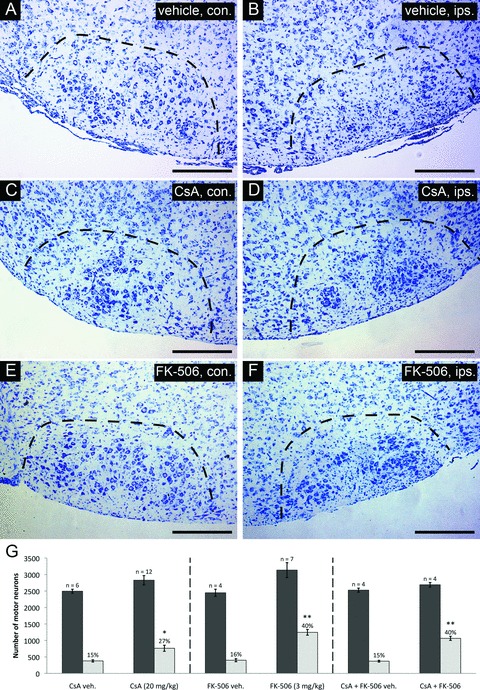
Survival characteristics of injured facial motor neurons following CsA or FK-506 treatment. Thionin-stained cross-sections taken through the facial nucleus at 7 days following axotomy are shown for each treatment. Sections shown are through comparable levels of the facial nucleus from representative individuals of each treatment group. (A), (C), (E) depict coronal cross-sections through uninjured facial nuclei (contralateral nucleus – con.). (B), (D), (F) show coronal cross-sections taken through injured nuclei (ipsilateral nucleus – ips.). Scale bars represent a distance of 250 μm. (G) Summary of stereological counts of facial motor neurons in axotomized and uninjured facial nuclei following cyclosporin A or FK-506 treatment. Administration of CsA and FK-506 under the conditions indicated resulted in the survival of 27 ± 3% and 40 ± 3% total facial motor neurons, respectively, compared to 15 ± 1% and 16 ± 2% for respective vehicle-treated controls. (* indicates statistical significance between treatment group [i.e. CsA and controls] at P < 0.05; ** indicates statistical significance between treatment groups [i.e. FK-506 and CsA + FK-506 co-treatment versus CsA] at P < 0.01)
CsA and FK-506 protect motor neurons following injury by reducing caspase-3 activation
To better understand the mechanism by which CsA and FK-506 treatments enhance the survival of injured facial motor neurons, we investigated the downstream pattern of PCD progression by examining executioner caspase activation within the facial nucleus. Such caspases have been shown to be proteolytic activated within injured facial motor neurons following neonatal axotomy [34–36]. Inhibition of this response can therefore be taken as evidence of inhibition of PCD in facial motor neurons at a point upstream of executioner caspase activation. For these experiments, levels of caspase-3 activation were examined through the full extent of facial nuclei by stereological counts at 20 hrs following injury (a peak period of injury-induced motor neuron death). Significant elevation in levels of activated caspase-3 was observed in injured as compared to uninjured facial nuclei in vehicle-treated controls (Fig. 2A and B). Administration of CsA (Fig. 2C and D) and FK-506 (Fig. 2E and F) significantly reduced the levels of activated caspase-3 in injured facial nuclei relative to vehicle treatment. Stereological counts of motor neurons expressing activated caspase-3 throughout the facial nucleus demonstrated that both CsA and FK-506 significantly reduced caspase-3 activation (16 ± 2% and 36 ± 3% reduction compared to vehicle, respectively) (Fig. 2G). Interestingly, facial motor neurons expressing activated caspase-3 were significantly increased in uninjured facial nuclei of CsA-treated animals relative to vehicle-treated controls (95 ± 3%) (Fig. 2C and G). In contrast, FK-506 administration did not appear to alter the levels of naturally occurring PCD in uninjured facial nuclei during the same period (Fig. 2E and G).
Fig 2.
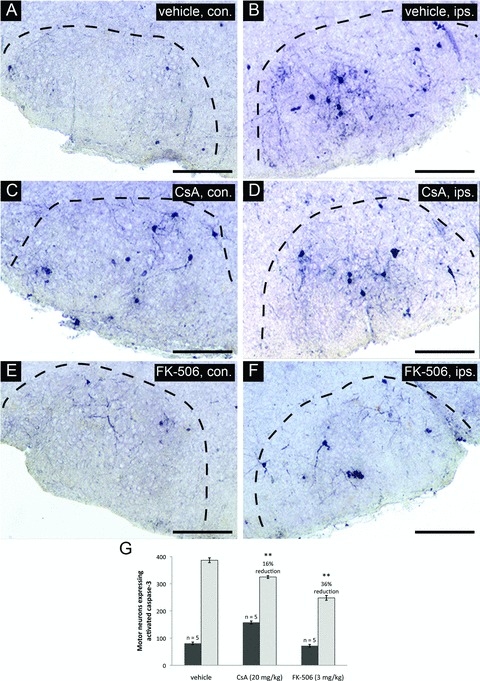
CsA or FK-506 treatment reduces levels of activated caspase-3 in facial motor neurons following axotomy. Coronal cross-sections through the facial nucleus were examined for activated caspase-3 at 20 hrs following axotomy for drug treatment groups and vehicle-treated controls. (A), (C) and (E) show sections through uninjured facial nuclei (contralateral – con.). (B), (D) and (F) show sections through injured facial nuclei (ipsilateral – ips.). Scale bars represent a distance of 250 μm. (G) Stereological counts of activated caspase-3 positive neurons through the facial nucleus show that both CsA and FK-506 treatments significantly reduced the number of motor neurons positive for activated caspase-3 compared to vehicle treatment (** indicates statistical significance at P < 0.01 between treatment groups and vehicle controls). Notably, CsA treatment was observed to increase the total number of motor neurons with activated caspase-3 in the uninjured facial nuclei compared to vehicle controls (** indicates statistical significance at P < 0.01 between CsA and vehicle treatments) whereas FK-506 did not appear to have any effects on uninjured facial motor neurons.
CsA and FK-506 do not alter levels of reactive gliosis or microglial infiltration following injury
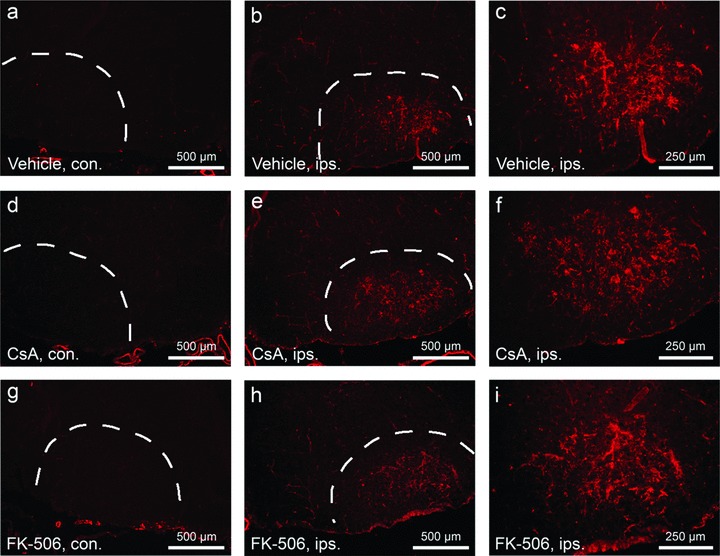
Microglial activation and reactive gliosis are frequently observed following many forms of neural injury, and have been suggested to contribute to levels of neuronal death [37, 38]. To determine the effect of CsA and FK-506 treatments on these secondary cellular responses, the degree and pattern of reactive gliosis and microglial infiltration were examined in control and treated facial nuclei at 2 (data not shown) and 4 days after injury (Figs 3 and 4). As indicated in the figure, levels of activated microglia present within injured facial nuclei were not altered by either drug treatment (Fig. 3). Similarly, numbers of reactive (GFAP+) astrocytes within injured facial nuclei were also observed to be comparable between treated and controls groups (Fig. 4). Together, these data support a model that CsA and FK-506 act in a cell autonomous manner to promote neuronal survival of injured facial motor neurons.
Fig 3.
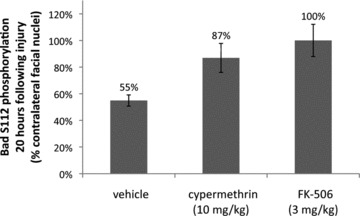
Inhibition of calcineurin-mediated Bad S112 dephosphorylation by cypermethrin and FK-506. Levels of Bad S112 phosphorylation were examined by immunofluorescence in coronal sections of injured and uninjured facial nuclei for vehicle, cypermethrin and FK-506-treated animals at 20 hrs following facial nerve axotomy. Bad S112 phosphorylation in the injured facial nuclei was significantly reduced following axotomy to 55 ± 4% compared to the uninjured facial nuclei (P < 0.01). Both cypermethrin and FK-506 treatments inhibited Bad S112 dephosphorylation by calcineurin and restored phosphorylation levels in the injured facial nuclei to 87 ± 11% (* indicates statistical significance at P < 0.05 between vehicle and cypermethrin treatment) and 100 ± 12% (** indicates statistical significance at P < 0.01 between vehicle and FK-506 treatment) of the uninjured facial nuclei, respectively.
Fig 4.

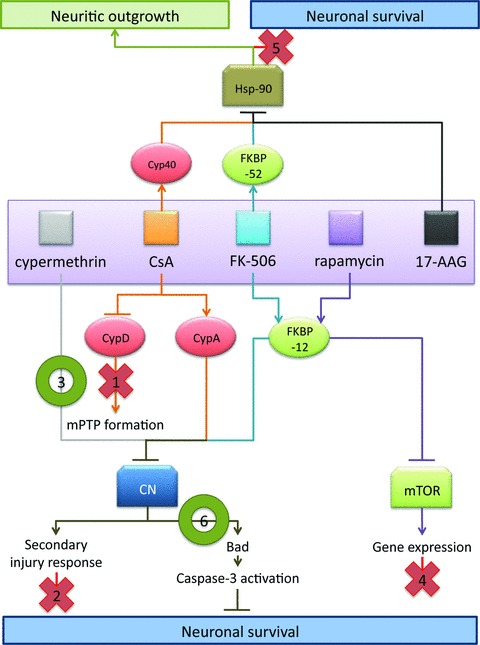
Mechanistic model of neuroprotection mediated by CsA and FK-506. Based upon results in facial motor neurons, the following model is proposed to explain the effects observed using CsA and FK-506. As indicated in this model, data from the present study suggest that the observed survival promoting and neurotrophic effects of these immunophilin ligands are mediated through distinct sets of molecular interactions. Evidence for exclusion of specific pathway is as indicated below. (1) CsA-mediated MPTP blockade is likely not the principle route of CsA-mediated neural rescue given that CsA and FK-506 treatment exhibit several similar mechanistic features, and co-administration of CsA and FK-506 did not enhance motor neuron survival over FK-506 treatment alone. (2) Administration of CsA or FK-506 did not act to reduce levels of reactive gliosis or infiltration of activated microglia; thus the enhanced survival of facial motor neurons was not due to repression of secondary injury responses. (3) Cypermethrin, an immunophilin-independent inhibitor of calcineurin, significantly enhanced motor neuron survival, hence highlighting calcineurin inhibition as a principal mechanism of regulating neuronal survival mediated by immunophilin ligands. (4) FK-506 and rapamycin both interact with FKBP-12, but only FK-506/FKBP-12 complexes inhibit calcineurin signalling. The failure of rapamycin to enhance motor neuron survival, following injury implicates calcineurin in the signalling pathway of motor neuron injury, and rules out mTOR-mediated effects. (5) Application of 17-AAG (a pharmacologic inhibitor of Hsp-90) failed to exhibit enhance motor neuron survival following injury, indicating that FK-506- and CsA-mediated enhancement in neuronal survival is distinct from the documented disruption in steroid receptor complex formation involved in enhancing neuritic outgrowth. (6) Inhibition of calcineurin-mediated Bad S112 dephosphorylation was observed at 20 hrs following axotomy for treatments with a single dose of either cypermethrin or FK-506, demonstrating a common mechanism by which cypermethrin, FK-506 and possibly CsA can enhance neuronal survival following injury.
Enhancement in neuronal survival is not mediated via calcineurin-independent signalling pathways
Due to the array of potential cellular interaction targets for both CsA and FK-506, several possible mechanisms of neuronal survival effects exist beyond that of calcineurin inhibition. Recently, several calcineurin-independent signalling pathways have been suggested as possible means by which CsA and FK-506 exert their neuroprotective effects using non-immunosuppressive (calcineurin-independent) immunophilin ligands such as GPI-1046 and V-10,367 [3, 14, 39–41]. In addition, an alternative calcineurin-independent model of FK-506-mediated neuritic outgrowth has recently been proposed [42, 43]. In this model, FK-506 promotes neuritic outgrowth via binding with FKBP-52 [42]. The resulting drug–immunophilin complex inhibits formation of the heat shock protein 90 (Hsp-90)/steroid receptor complex, which in turn releases p23 to activate downstream ERK pathways [43]. To determine whether such calcineurin-independent signalling pathways are implicated in survival enhancements observed in injured facial motor neurons as a result of treatments with CsA or FK-506, several additional pharmacologic inhibitors were examined for their ability to enhance motor neuron survival following axotomy. 17-AAG, a cyclophilin/FKBP/calcineurin-independent inhibitor of Hsp-90 and rapamycin, a FKBP-dependent, calcineurin-independent inhibitor of mammalian target of rapamycin (mTOR), were administered daily to animals following axotomy until the time of killing at doses previously demonstrated to produced neural effects in vivo[27, 28, 30]. As shown in Fig. 5, motor neuron counts performed on the ipsilateral (injured) and contralateral (uninjured) facial nuclei demonstrated that neither 17-AAG (10 mg/kg) (Fig. 5A and B) nor rapamycin (3 mg/kg) (Fig. 5C and D) had any effects on neuronal survival following injury compared to vehicle (17-AAG versus vehicle controls: 22 ± 1% and 25 ± 2%, respectively, 4 days after injury; rapamycin versus vehicle controls: 17 ± 2% and13 ± 1%, respectively). These results demonstrate that such calcineurin-independent pathways do not contribute to the survival enhancement of injured facial motor neurons observed using CsA or FK-506.
Fig 5.

CsA and FK-506 do not suppress levels of activated microglia following facial axotomy. Cryostat sections through facial nuclei were labeled with tomato lectin to examine numbers of activated microglia at 4 days following motor neuron injury. (A), (D) and (G) show sections of unoperated (contralateral – con.) facial nuclei. (B), (E) and (H) show sections through operated (ipsilateral – ips.) facial nuclei. Scale bars indicate a distance of 500 μm. (C), (F) and (I) show higher magnification views of that shown in (B), (E) and (H), respectively. Scale bars represent a distance of 250 μm. Note that similar levels of microglial activation were observed between vehicle and CsA/FK-506 treatment groups.
Role of calcineurin inhibition in CsA- and FK-506-mediated facial motor neuron survival
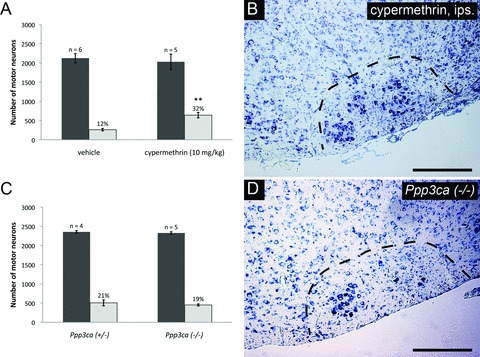
The absence of survival promoting effects observed following the administration of several calcineurin-independent agents prompted us to investigate the contribution calcineurin inhibition has with respect to neuronal survival in greater detail. To perform manner disparate to that of CsA and FK-506. Cypermethrin, is a type II synthetic pyrethroid insecticide which inhibits calcineurin through direct binding, thus functions as a cyclophilin/FKBP-independent calcineurin inhibitor [44]. To examine the potential of cypermethrin to enhance neuronal survival following injury, cypermethrin (10 mg/kg) was administered to animals on a daily basis following axotomy until the time of killing. Total counts of facial motor neurons demonstrated that cypermethrin treatment significantly enhanced motor neuron survival following axotomy compared to vehicle controls (32 ± 3% versus 12 ± 1%, respectively) (Fig. 6A and B). This represents the first demonstration that cypermethrin and, by extension, direct calcineurin inhibition enhance motor neuron survival following injury in vivo.
Fig 6.
CsA and FK-506 do not suppress levels of reactive gliosis following facial axotomy. Sections were labeled with GFAP to examine numbers of reactive astrocytes at 4 days following facial axotomy. (A), (D) and (G) show sections through unoperated facial nuclei (contralateral – con.). (B), (E) and (H) show sections through operated facial nuclei (ipsilateral – ips.). Scale bars represent 500 μm. (C), (E) and (I) show injured facial nuclei at higher magnification. Scale bars represent 250 μm. Note that similar levels of reactive gliosis were observed between vehicle and CsA/FK-506 treatment groups.
CNA isoform α is dispensable for regulating motor neuron survival following injury
To further investigate the potential role of calcineurin inhibition in enhancing motor neuron survival, neonatal facial axotomies was performed in mice lacking the CNA-α isoform. However, genetic deletion of this calcineurin isoform alone was not observed to enhance motor neuron survival (Ppp3ca−/–versus Ppp3ca+/−: 19 ± 1% and 21 ± 3%, respectively) (Fig. 6C and D). Similarly, deletion of CNA-α was not observed to reduce developmental motor neuron PCD, as indicated by the comparable numbers of facial motor neurons seen in Ppp3ca+/− and Ppp3ca−/– animals (Fig. 6C). Thus, CNA-α appears to be dispensable both for developmental and injury-mediated loss of facial motor neurons. Thus the motor neuron survival enhancement observed using CsA, FK-506 and cypermethrin appear to reflect an inhibition which extends beyond CNA-α alone, and most probably due to the inhibition of β and γ isoforms in facial motor neurons as well.
Inhibition of calcineurin-mediated Bad dephosphorylation by FK-506 and cypermethrin following injury
A previous in vitro study had demonstrated an inhibition of calcineurin-mediated Bad dephosphorylation by FK-506 following glutamate stimulation was correlated with enhanced survival of the stimulated neurons, thus suggesting that inhibition of calcineurin-mediated Bad dephosphorylation is, at least partially, responsible for enhanced survival of facial motor neurons following administration of CsA, FK-506 and cypermethrin [19]. To determine whether Bad serine 112 (S112) dephosphorylation occurs within facial motor neurons following axotomy, and whether this process is regulated through calcineurin, we examined levels of S112 Bad phosphorylation by immunofluorescence at 20 hrs following facial nerve axotomy in vehicle, FK-506 and cypermethrin-treated animals. In vehicle-treated animals, Bad S112 phosphorylation was on average reduced in injured facial motor neurons to 55 ± 4% of that observed in the contralateral (uninjured) facial nuclei (Fig. 7). In contrast, FK-506 and cypermethrin-treated animals exhibited substantially reduced levels of Bad S112 dephosphorylation compared to vehicle-treated controls, demonstrating that both treatments inhibited calcineurin-mediated Bad dephosphorylation at serine 112. Although cypermethrin treatment enhanced levels of Bad S112 phosphorylation to 87 ± 11% of that seen in uninjured facial motor neurons, treatment with FK-506 appeared to have completely restored levels of Bad S112 phosphorylation to that observed in the contralateral facial nuclei (100 ± 12%) (Fig. 7). These results support the notion that regulation of Bad phosphorylation status may be a critical PCD signalling event downstream of calcineurin inhibition. The results further suggest that this can occur through either indirect inhibition of the drug–immunophilin complex, or directly through inhibition of calcineurin.
Fig 7.
17-AAG and rapamycin treatments do not enhance facial motor neuron survival following injury. (A) Histogram of stereological counts of facial motor neurons from mice treated with 17-AAG (10 mg/kg) or vehicle performed at 4 days after axotomy. Similar to rapamycin, treatment with 17-AAG did not enhance levels of motor neuron survival compared to controls (22 ± 1% versus 25 ± 2% for 17-AAG and vehicle-treated groups, respectively), indicating these respective immunophilin-related pathways are not involved in neuroprotection. (B) depicts a typical injured facial nucleus from an animal treated daily with 17-AAG following axotomy. (C) Histogram of facial motor neuron survival following rapamycin treatment. Mice receiving daily rapamycin administration (3 mg/kg) following injury until time of killing showed no significant difference in motor neuron survival compared to vehicle-treated controls (17 ± 2% versus 13 ± 1%, respectively). Shown in (D) is a typical injured facial nucleus from rapamycin-treated animals.
Discussion
In the present study, we demonstrate that three agents whose sole commonality is the inhibition of calcineurin to enhance the survival of motor neurons following acute injury. Two of these agents, CsA and FK-506, have previously been shown to enhance neuronal survival in vivo, whereas the third (cypermethrin) has not been previously described with respect to neuronal survival effects in vivo. By contrast, examination of reagents representing alternative signalling pathways (17-AAG) or a non-calcineurin inhibiting FKBP-dependent immunosuppressant (rapamycin) failed to enhance the survival of motor neurons following axotomy. With respect to neuronal survival, FK-506 and to a lesser extent CsA, at daily doses of 3 mg/kg and 20 mg/kg, respectively, significantly enhanced motor neuron survival following axotomy. We have sought to determine the effects of these ligands in mice due to several experimental advantages. Previous studies have shown that administration of CsA or FK-506 enhanced motor neuron survival following neonatal facial axotomy in rat [25]. Although rats exhibit several advantages as a mammalian model system including size and detailed physiologic knowledge, this system poses several obstacles to complete mechanistic studies including genetic heterogeneity, difficulty in performing homologous gene targeting, and incomplete genome sequencing status. By contrast, several strains of mice including 129/SvlmJ are well-characterized inbred strains well suited to gene deletion and targeting studies, and whose genomes have been fully sequenced [45]. Compared to previous findings in rat, application of CsA in mice demonstrated lower levels of motor neuron rescue following injury (rat – 34%, mouse – 27% at doses of 17.5 mg/kg and 20 mg/kg, respectively). Although administration of FK-506 resulted in comparable levels of motor neuron survival between these species (rat – 35%, mouse – 40% survival), the dosage required to obtain this level of neuronal survival was greater for mice (rat – 1 mg/kg, mouse – 3 mg/kg). These effects may reflect differences in drug metabolism between the two species, as mice exhibit higher rates of metabolism for a number of known drugs [46]. They are unlikely to be due to differential access through the blood brain barrier, as this structure is not yet fully established in rodents over the period of examination (P3-P10) [47]. In mice, elevation of the applied dose of CsA or FK-506 beyond the above levels did not enhance motor neuron survival further, suggesting that the dosages applied are at or above optimal levels. Similarly, combined application of CsA and FK-506 did not further enhance motor neuron survival following injury, suggesting that these agents work through similar signalling pathways.
Given the immunosuppressive nature of CsA and FK-506, it might be postulated that the observed enhancement in motor neuron survival mediated by these agents is related to their suppression of immunologic phenomena retarding the rate of destruction of facial motor neurons. This is unlikely for several reasons. (1) FK-506 and CsA act to protect motor neurons from cell death over a time period which is incompatible with immunosuppression [48], given that suppression of caspase-3 activity is seen in the facial nucleus by 20 hrs after injury (earliest time-point examined). (2) In addition, if CsA and FK-506-mediated motor neuron survival through suppression of inflammatory responses within the facial nucleus, one would expect cellular processes such as reactive gliosis and infiltration of activated microglia to be suppressed in the presence of these drugs; both of which were not observed in the present study. (3) Finally, if direct or immune-stimulated cell-cell interactions were a significant cause of motor neuron death within the facial nucleus, dying motor neurons would not exhibit the apoptotic morphologic and biochemical features which have previous been well characterized for this lesion paradigm [32, 36]. Rather, the data demonstrate that the neuronal survival effects observed are the result of a cell autonomous reduction in PCD with facial motor neurons.
If the immunophilin ligands CsA and FK-506 act in a cell intrinsic manner to reduce PCD in injured motor neurons, what is the mechanism of these effects? Neuroprotection by CsA has long been rationalized in terms of an inhibitory effect upon MPTP formation through interaction with cyclophilin D [3, 13]. However, with respect to enhancing facial motor neuron survival, this appears unlikely to be the principal mechanism given that: (i) combined application of CsA and FK-506 does not result in elevated levels of motor neuron survival above that seen with FK-506 alone, suggesting that these agents act through similar signalling mechanisms; (ii) FK-506 does not interfere with MPTP formation in the manner that CsA does and (iii) the maximum rescue effect of FK-506 is significantly greater than that seen with CsA. These effects suggest that the survival promoting effects of CsA lie beyond an MPTP effect.
An alternative mechanism recently proposed to explain the neurotrophic/neuroprotective effects of FK-506 and CsA, highlights the ability of both of these agents to disrupt binding between components of steroid hormone receptor complexes (Hsp-90 and p23) [42, 43]. In this model, drug–immunophilin complexes (FK-506 with FKBP-52, and CsA potentially with cyclophilin-40 [49–51]) disrupts the interaction of Hsp-90 with other components of the receptor complex, thereby allowing p23 to dissociate and initiate ERK signalling pathways [42, 43]. As FKBP-52 and Cyp40 share a similar binding site on Hsp-90 [49, 51]; this pathway provides a common putative signalling mechanism for FK-506 and CsA, respectively. To examine whether the inhibition of Hsp-90 plays a role in regulating the neuronal survival observed with FK-506 and CsA treatments, Hsp-90 was pharmacologically inhibited using 17-AAG. Using several different dosing regimens at 17-AAG concentrations previously shown to inhibit Hsp-90 in mice in vivo[28, 30], no enhancement of motor neuron survival was observed following axotomy. Thus with respect to acute motor neuron injury (in contrast to neuritic outgrowth) [42], Hsp-90-mediated signalling does not appear to regulate the survival promoting effects of CsA and FK-506.
The results in facial motor neurons suggest that it is the ability of CsA and FK-506 to inhibit calcineurin phosphatase activity which is the principal cause of the observed effects. Such a possibility has previously been proposed to explain the neurotrophic/neuroprotective actions of these agents [6, 7]. However in recent years, attention has shifted away from calcineurin as a neural target as new non-immunosuppressive (i.e. calcineurin independent) FK-506 derivatives have been suggested to possess neurotrophic/neuroprotective activities in several neural injury paradigms [14, 40, 52]. To determine the role which calcineurin inhibition plays in regulating the survival promoting effects of CsA and FK-506, we examined the ability of cypermethrin, a type II synthetic pyrethroid insecticide which is a cell permeable, immunophilin-independent inhibitor of calcineurin to promote neuronal survival. The results demonstrate for the first time, the ability of cypermethrin to promote motor neuron survival following injury in vivo. Given the alternative mechanistic modes of cypermethrin, CsA and FK-506 action, these data indicate that it is the common inhibition of calcineurin activity which mediates the survival promoting effects observed in facial motor neurons. To further distinguish the lack of immunophilin binding as a requirement in mediating these effects, rapamycin (an immunophilin ligand which exerts its immunosuppressive effects via FKBP binding but independent of calcineurin inhibition [53]) was examined with respect to facial motor neuron survival, and found to be without effect. Thus, FKBP binding alone by rapamycin (or other immunophilin ligands) is insufficient for neuroprotection in the absence of calcineurin inhibition.
The concept that the inhibition of calcineurin activity can enhance motor neuron survival is attractive in that this phosphatase is highly expressed within CNS tissue, and is known to dephosphorylate pro-apoptotic Bcl-2 protein Bad at residues critical to the promotion of its pro-apoptotic activity [18–24]. In this study, we have shown that Bad S112 phosphorylation is reduced significantly in facial motor neurons following axotomy, and that the inhibition of calcineurin by FK-506 or cypermethrin can largely reverse this effect. This is the first demonstration that cypermethrin or similar agents in its class have been shown to enhance neuronal survival in vivo following injury. Together with the results seen in 17-AAG, rapamycin, CsA and FK-506-treated animals, these data clearly demonstrate the critical role which calcineurin has in regulating the survival of motor neurons following injury in vivo.
To examine the role which specific CNA isoforms might play in regulating motor neuron PCD, we have examined survival of facial motor neurons following genetic deletion of the CNA-α gene (Ppp3ca), the dominant isoform of CNA in the CNS, after facial nerve axotomy. Analysis of these mice demonstrates that inhibition of CNA-α activity is dispensable with respect to survival of injured facial motor neurons. In this regard, it is notable that two additional CNA isoforms exist within the CNS (Ppp3cb and Ppp3cc) [54, 55]; and these lower abundance isoforms may serve either a direct or compensatory role with respect to motor neuron injury. Thus, the CNA response of motor neurons may be similar to that seen in T cells. Despite the fact that calcineurin is the known physiologic target of CsA and FK-506 in T cells, T cells which lack Ppp3ca remain sensitive to these agents, thus demonstrating the functional redundancy which exists among CNA isoforms [26].
A variety of signalling mechanisms have previously been postulated to explain the neurotrophic/neuroprotective effects seen for CsA and FK-506. Using a variety of agents and approaches as summarized in Fig. 8, we demonstrate that it is the inhibition of calcineurin activity which promotes neuronal survival observed in facial motor neurons following axotomy in vivo. Given this, how does one rationalize these findings with previous studies which demonstrate an enhancement in neural regeneration using non-immunosuppressive immunophilin ligands? We believe that an explanation lies in clearly distinguishing effects upon neural regeneration – neuritic outgrowth in response to an injury stimulus distal to the neuronal cell soma, and effects upon neuronal survival. This latter form occurs when the injury stimulus is proximal to the cell soma. PCD, autophagy and necrosis represent different forms of response to this type of cellular injury. In contrast, a sizeable body of experimental data suggests that more distal forms of neuronal injury trigger a distinct set of cellular response through signalling pathways such as the c-Jun/JNK pathway [56–58], therefore the neural regenerative effects seen with calcineurin-dependent/independent inhibitors may reflect actions on disparate components of the injury response in the CNS.
Fig 8.
Direct calcineurin inhibition results in enhanced facial motor neuron survival following axotomy, but CNA-α plays a dispensable role in regulating neuronal survival. (A) Daily administration of cypermethrin (10 mg/kg) demonstrated enhanced facial motor neuron survival from 12 ± 1% in vehicle-treated animals to 32 ± 3% in cypermethrin-treated mice, thus suggesting a prominent role for calcineurin inhibition in promoting neuronal survival (** indicates statistical significance between cypermethrin treatment and controls at P < 0.01). (B) shows an increased number of motor neurons in whole facial nuclei of cypermethrin-treated animals compared to controls. To further examine the role of calcineurin inhibition in enhancing neuronal survival following injury, axotomy-induced injury was performed in Ppp3ca null mice and heterozygous controls. (C) Histogram of facial motor neuron survival in Ppp3ca null mice. Mice homozygous for a targeted deletion of the dominant isoform of CNA in the CNS did not exhibit enhanced motor neuron survival compared to heterozygous littermates (19 ± 1% versus 21 ± 3%, respectively). (D) A typical injured facial nucleus from Ppp3ca null animals following axotomy, with no observable enhancement in motor neuron survival compared to heterozygous controls.
In contrast to previous studies utilizing CsA, FK-506 and related non-immunosuppressive immunophilin ligands to enhance neuronal survival following CNS injury, our data support a model in which it is the inhibition of calcineurin activity which enhances neuronal survival. As such, calcineurin inhibition is an attractive therapeutic target with significant clinical potential with respect to acute motor neuron injury for several reasons. Prior studies have demonstrated that neurotrophic factors such as GDNF, BDNF and CNTF are particularly effective in promoting neuronal survival following injury. The principal mechanism by which these agents promote neuroprotection is through the influence of Bcl-2 family proteins at the mitochondrial outer membrane; a similar mechanism to that which we now demonstrate for the calcineurin inhibitors, FK-506, CsA and cypermethrin. With respect to injury-induced motor neuron PCD, an advantage of therapeutic interventions aimed at the level of Bcl-2 family proteins is that they exhibit a significant post-injury treatment window [18, 59]. Consistent with effects seen using neurotrophic factors, we observe that neuroprotection by immunophilin ligands occur even when administration is delayed by 3 hrs following injury. These effects on motor neuron PCD are unlikely to be primarily the result of effects on calcium entry, because neuronal rescue seen using calcium chelators such as BAPTA [60], has been shown to be efficacious only when given as a pre-treatment. In contrast to neurotrophic factors however, substantial clinical experience with FK-506 and CsA demonstrates that application of these agents in human beings does not result in the serious side effects which halted human clinical trials of neurotrophic factors for motor neuron injury [61, 62]. Also, small molecule inhibitors of calcineurin circumvent other drawbacks seen with respect to clinical application of neurotrophic factors, such as their poor CNS distribution and blood-brain barrier permeability, and the difficulties associated with manufacture and certification of such agents.
Despite their long history, the mechanism by which agents such as CsA and FK-506 act to promote neuroprotection remains controversial. In the present study, we demonstrate that with respect to their ability to promote neuronal survival in vivo following acute motor neuron injury, these agents act through the inhibition of calcineurin, and exclude several other potential actions of these agents as causes for the observed effects. Although the current study had examined the effects of calcineurin inhibitors on motor neuron survival following injury, it is important to note that numerous studies have demonstrated that CsA and FK-506 are capable of protecting a variety of neuronal targets in a number of in vivo injury paradigms, ranging from stroke, to Parkinson’s disease, to methamphetamine-induced neurotoxicity [16, 40, 63, 64]. Hence the clinical potential of small molecule calcineurin inhibitors to promote neuronal survival may extend well beyond that of acute motor neuron injury.
Finally, it has been suggested that several non-immunosuppressive (calcineurin-independent) immunophilin ligands may exhibit neuroprotective effects. Although a clear picture has not yet emerged regarding the mechanism of these effects, it will be interesting to see if the distinction we observe between neuroprotective versus neuritic outgrowth pathways in motor neurons is maintained in other neural injury paradigms. Such findings will allow us to gain greater insight into the mechanism by which different neuronal populations within the mammalian CNS act to regulate the various aspects of neural injury.
Acknowledgments
K.K.W.H. was supported by CIHR/Rx&D Health Research Foundation.
References
- 1.Fung JJ, Starzl TE. FK 506 in solid organ transplantation. Transplant Proc. 1994;26:3017–20. [PubMed] [Google Scholar]
- 2.Morris RE. In vivo immunopharmacology of the macrolides FK 506 and rapamycin: toward the era of rational immunosuppressive drug discovery, development, and use. Transplant Proc. 1991;23:2722–4. [PubMed] [Google Scholar]
- 3.Snyder SH, Sabatini DM, Lai MM, et al. Neural actions of immunophilin ligands. Trends Pharmacol Sci. 1998;19:21–6. doi: 10.1016/s0165-6147(97)01146-2. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Farmer JD, Jr, Lane WS, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 5.Steiner JP, Dawson TM, Fotuhi M, et al. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature. 1992;358:584–7. doi: 10.1038/358584a0. [DOI] [PubMed] [Google Scholar]
- 6.Snyder SH, Lai MM, Burnett PE. Immunophilins in the nervous system. Neuron. 1998;21:283–94. doi: 10.1016/s0896-6273(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 7.Dawson TM, Steiner JP, Dawson VL, et al. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proc Natl Acad Sci USA. 1993;90:9808–12. doi: 10.1073/pnas.90.21.9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gold BG, Katoh K, Storm-Dickerson T. The immunosuppressant FK506 increases the rate of axonal regeneration in rat sciatic nerve. J Neurosci. 1995;15:7509–16. doi: 10.1523/JNEUROSCI.15-11-07509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madsen JR, MacDonald P, Irwin N, et al. Tacrolimus (FK506) increases neuronal expression of GAP-43 and improves functional recovery after spinal cord injury in rats. Exp Neurol. 1998;154:673–83. doi: 10.1006/exnr.1998.6974. [DOI] [PubMed] [Google Scholar]
- 10.Winter C, Schenkel J, Burger E, et al. The immunophilin ligand FK506, but not GPI-1046, protects against neuronal death and inhibits c-Jun expression in the substantia nigra pars compacta following transection of the rat medial forebrain bundle. Neuroscience. 2000;95:753–62. doi: 10.1016/s0306-4522(99)00486-8. [DOI] [PubMed] [Google Scholar]
- 11.Lyons WE, George EB, Dawson TM, et al. Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proc Natl Acad Sci USA. 1994;91:3191–5. doi: 10.1073/pnas.91.8.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuroda S, Janelidze S, Siesjo BK. The immunosuppressants cyclosporin A and FK506 equally ameliorate brain damage due to 30-min middle cerebral artery occlusion in hyperglycemic rats. Brain Res. 1999;835:148–53. doi: 10.1016/s0006-8993(99)01535-8. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto S, Friberg H, Ferrand-Drake M, et al. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1999;19:736–41. doi: 10.1097/00004647-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Steiner JP, Hamilton GS, Ross DT, et al. Neurotrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proc Natl Acad Sci USA. 1997;94:2019–24. doi: 10.1073/pnas.94.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 16.Butcher SP, Henshall DC, Teramura Y, et al. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–46. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson TM, Steiner JP, Lyons WE, et al. The immunophilins, FK506 binding protein and cyclophilin, are discretely localized in the brain: relationship to calcineurin. Neuroscience. 1994;62:569–80. doi: 10.1016/0306-4522(94)90389-1. [DOI] [PubMed] [Google Scholar]
- 18.Springer JE, Azbill RD, Nottingham SA, et al. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–51. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang HG, Pathan N, Ethell IM, et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 20.Yang L, Omori K, Suzukawa J, et al. Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci Lett. 2004;357:73–5. doi: 10.1016/j.neulet.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 21.Datta SR, Katsov A, Hu L, et al. 14–3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Molecular cell. 2000;6:41–51. [PubMed] [Google Scholar]
- 22.Datta SR, Ranger AM, Lin MZ, et al. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell. 2002;3:631–43. doi: 10.1016/s1534-5807(02)00326-x. [DOI] [PubMed] [Google Scholar]
- 23.Zha J, Harada H, Yang E, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 24.Tan Y, Demeter MR, Ruan H, et al. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000;275:25865–9. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 25.Tao R, Aldskogius H. Influence of FK506, cyclosporin A, testosterone and nimodipine on motoneuron survival following axotomy. Restor Neurol Neurosci. 1998;12:239–46. [PubMed] [Google Scholar]
- 26.Zhang BW, Zimmer G, Chen J, et al. T cell responses in calcineurin A alpha-deficient mice. J Exp Med. 1996;183:413–20. doi: 10.1084/jem.183.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2(+/-) mouse model of tuberous sclerosis. Nature Med. 2008;14:843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kociok N, Krohne TU, Poulaki V, et al. Geldanamycin treatment reduces neovascularization in a mouse model of retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2007;245:258–66. doi: 10.1007/s00417-006-0355-x. [DOI] [PubMed] [Google Scholar]
- 29.Patel S, Pandey AK, Bajpayee M, et al. Cypermethrin-induced DNA damage in organs and tissues of the mouse: evidence from the comet assay. Mutat Res. 2006;607:176–83. doi: 10.1016/j.mrgentox.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Waza M, Adachi H, Katsuno M, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nature Med. 2005;11:1088–95. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 31.Coggeshall RE, Lekan HA. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol. 1996;364:6–15. doi: 10.1002/(SICI)1096-9861(19960101)364:1<6::AID-CNE2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.De Bilbao F, Dubois-Dauphin M. Time course of axotomy-induced apoptotic cell death in facial motoneurons of neonatal wild type and bcl-2 transgenic mice. Neuroscience. 1996;71:1111–9. doi: 10.1016/0306-4522(95)00505-6. [DOI] [PubMed] [Google Scholar]
- 33.Kou SY, Chiu AY, Patterson PH. Differential regulation of motor neuron survival and choline acetyltransferase expression following axotomy. J Neurobiol. 1995;27:561–72. doi: 10.1002/neu.480270410. [DOI] [PubMed] [Google Scholar]
- 34.De Bilbao F, Guarin E, Nef P, et al. The mouse cpp32 mRNA transcript is early up-regulated in axotomized motoneurons following facial nerve transection. Neurosci Lett. 1999;266:65–8. doi: 10.1016/s0304-3940(99)00264-5. [DOI] [PubMed] [Google Scholar]
- 35.Guarin E, Seuret P, Nef S, et al. cpp32 messenger RNA neosynthesis is induced by fatal axotomy and is not regulated by athanatal Bcl-2 over-expression. Neuroscience. 1999;90:653–64. doi: 10.1016/s0306-4522(98)00445-x. [DOI] [PubMed] [Google Scholar]
- 36.Kanungo AK, Hao Z, Elia AJ, et al. Inhibition of apoptosome activation protects injured motor neurons from cell death. J Biol Chem. 2008;283:22105–12. doi: 10.1074/jbc.M800988200. [DOI] [PubMed] [Google Scholar]
- 37.Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. J Neurosci. 1993;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sargsyan SA, Monk PN, Shaw PJ. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. 2005;51:241–53. doi: 10.1002/glia.20210. [DOI] [PubMed] [Google Scholar]
- 39.Gold BG, Armistead DM, Wang MS. Non-FK506-binding protein-12 neuroimmunophilin ligands increase neurite elongation and accelerate nerve regeneration. Journal of neuroscience research. 2005;80:56–65. doi: 10.1002/jnr.20447. [DOI] [PubMed] [Google Scholar]
- 40.Guo X, Dawson VL, Dawson TM. Neuroimmunophilin ligands exert neuroregeneration and neuroprotection in midbrain dopaminergic neurons. Eur J Neurosci. 2001;13:1683–93. doi: 10.1046/j.0953-816x.2001.01542.x. [DOI] [PubMed] [Google Scholar]
- 41.Gold BG, Zeleny-Pooley M, Wang MS, et al. A nonimmunosuppressant FKBP-12 ligand increases nerve regeneration. Exp Neurol. 1997;147:269–78. doi: 10.1006/exnr.1997.6630. [DOI] [PubMed] [Google Scholar]
- 42.Gold BG, Densmore V, Shou W, et al. Immunophilin FK506-binding protein 52 (not FK506-binding protein 12) mediates the neurotrophic action of FK506. J Pharmacol Exp Ther. 1999;289:1202–10. [PubMed] [Google Scholar]
- 43.Gold BG, Zhong YP. FK506 requires stimulation of the extracellular signal-regulated kinase 1/2 and the steroid receptor chaperone protein p23 for neurite elongation. Neurosignals. 2004;13:122–9. doi: 10.1159/000076565. [DOI] [PubMed] [Google Scholar]
- 44.Enan E, Matsumura F. Specific inhibition of calcineurin by type II synthetic pyrethroid insecticides. Biochemical pharmacology. 1992;43:1777–84. doi: 10.1016/0006-2952(92)90710-z. [DOI] [PubMed] [Google Scholar]
- 45.Simpson EM, Linder CC, Sargent EE, et al. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 46.Komura H, Iwaki M. Species differences in in vitro and in vivo small intestinal metabolism of CYP3A substrates. J Pharm Sci. 2008;97:1775–800. doi: 10.1002/jps.21121. [DOI] [PubMed] [Google Scholar]
- 47.Moos T, Mollgard K. Cerebrovascular permeability to azo dyes and plasma proteins in rodents of different ages. Neuropathol Appl Neurobiol. 1993;19:120–7. doi: 10.1111/j.1365-2990.1993.tb00416.x. [DOI] [PubMed] [Google Scholar]
- 48.Borel JF, Feurer C, Magnee C, et al. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology. 1977;32:1017–25. [PMC free article] [PubMed] [Google Scholar]
- 49.Ratajczak T, Carrello A. Cyclophilin 40 (CyP-40), mapping of its hsp90 binding domain and evidence that FKBP52 competes with CyP-40 for hsp90 binding. J Biol Chem. 1996;271:2961–5. doi: 10.1074/jbc.271.6.2961. [DOI] [PubMed] [Google Scholar]
- 50.Carrello A, Ingley E, Minchin RF, et al. The common tetratricopeptide repeat acceptor site for steroid receptor-associated immunophilins and hop is located in the dimerization domain of Hsp90. J Biol Chem. 1999;274:2682–9. doi: 10.1074/jbc.274.5.2682. [DOI] [PubMed] [Google Scholar]
- 51.Owens-Grillo JK, Hoffmann K, Hutchison KA, et al. The cyclosporin A-binding immunophilin CyP-40 and the FK506-binding immunophilin hsp56 bind to a common site on hsp90 and exist in independent cytosolic heterocomplexes with the untransformed glucocorticoid receptor. J Biol Chem. 1995;270:20479–84. doi: 10.1074/jbc.270.35.20479. [DOI] [PubMed] [Google Scholar]
- 52.Powers JF, Brachold JM, Schelling K, et al. Potentiation of mitogenesis in adult rat chromaffin cell cultures by immunosuppressive agent FK506. Neurosci Lett. 2004;356:5–8. doi: 10.1016/j.neulet.2003.10.083. [DOI] [PubMed] [Google Scholar]
- 53.Kuo CJ, Chung J, Fiorentino DF, et al. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–3. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 54.Eastwood SL, Salih T, Harrison PJ. Differential expression of calcineurin A subunit mRNA isoforms during rat hippocampal and cerebellar development. Eur J Neurosci. 2005;22:3017–24. doi: 10.1111/j.1460-9568.2005.04518.x. [DOI] [PubMed] [Google Scholar]
- 55.Kuno T, Mukai H, Ito A, et al. Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J Neurochem. 1992;58:1643–51. doi: 10.1111/j.1471-4159.1992.tb10036.x. [DOI] [PubMed] [Google Scholar]
- 56.Herdegen T, Skene P, Bahr M. The c-Jun transcription factor–bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–31. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- 57.Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, et al. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993;24:23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- 58.Kenney AM, Kocsis JD. Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J Neurosci. 1998;18:1318–28. doi: 10.1523/JNEUROSCI.18-04-01318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–85. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tymianski M, Wallace MC, Spigelman I, et al. Cell-permeant Ca2+ chelators reduce early excitotoxic and ischemic neuronal injury in vitro and in vivo. Neuron. 1993;11:221–35. doi: 10.1016/0896-6273(93)90180-y. [DOI] [PubMed] [Google Scholar]
- 61.A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTF Treatment Study Group. Neurology. 1996;46:1244–9. doi: 10.1212/wnl.46.5.1244. [DOI] [PubMed] [Google Scholar]
- 62.Henderson JT, Seniuk NA, Richardson PM, et al. Systemic administration of ciliary neurotrophic factor induces cachexia in rodents. J Clin Invest. 1994;93:2632–8. doi: 10.1172/JCI117276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koike K, Hashimoto K, Fukami G, et al. The immunophilin ligand FK506 protects against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum. Neuropharmacology. 2005;48:391–7. doi: 10.1016/j.neuropharm.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 64.Furuichi Y, Maeda M, Moriguchi A, et al. Tacrolimus, a potential neuroprotective agent, ameliorates ischemic brain damage and neurologic deficits after focal cerebral ischemia in nonhuman primates. J Cereb Blood Flow Metab. 2003;23:1183–94. doi: 10.1097/01.WCB.0000088761.02615.EB. [DOI] [PubMed] [Google Scholar]