

Abstract
Elevated levels of NF-κB are frequently detected in many inflammatory diseases and cancers. Blocking the IKK–NF-κB pathway has been seen as a promising approach for new therapies. By employing the dominant-negative mutant of IKKβ, our data revealed that loss of IKKβ activity reduces not only the proliferation and invasion of lung adenocarcinoma A549 cells in vitro but also the tumour formation, metastasis and angiogenesis in mouse xenograft model. Treatment of IKKβ inhibitors (CYL-19s and CYL-26z) leads to the arrest of cell cycle progression at G1 and G2/M, followed by apoptosis. IKKβ inhibitors can increase the protein stability, nuclear accumulation and promoter-binding activity of p53, leading to the p21 gene transcription. Furthermore, knockdown of IKKβ by siRNA increased the stability and expression of p53 and p21 promoter activity. In addition, IKKβ inhibitor–induced p53 and p21 expressions were augmented in the presence of IKKβ siRNA. Correlation between p53 acetylation and its protein stabilization was also seen after treatment with IKKβ inhibitors. These results suggest that loss of IKKβ activation is important for the enhancement of p53 stability, leading to p21 expression and cell cycle arrest and apoptosis of tumour cells.
Keywords: IKKβ, p53, p21
Introduction
NF-κB transcriptional factors play important roles in regulation of immune and inflammatory responses and protection of cells from apoptosis [1–3]. The mammalian NF-κB family has five members: NF-κB1 (p105 and p50), NF-κB2 (p100 and p52), RelA (p65), RelB and c-Rel, and the most abundant form is the heterodimer of p65 and p50 [1, 3, 4]. In unstimulated cells, they are sequestered in cytosol as inactive homo- or hetero-dimers by binding to IκB inhibitory proteins [3, 5]. When cells are exposed to various stimuli, IκB is rapidly phosphorylated by a large cytoplasmic IκB kinase (IKK) complex, which consists of the kinase catalytic sub-units IKKα and IKKβ (also known as IKK1 and IKK2) and the essential regulatory sub-unit IKKγ (also known as NEMO) [6, 7]. IKKβ is the kinase required for the phosphorylation of IκB, leading to the polyubiquitination by E3-SCFβ-trCP ubiquitin ligase complex and subsequent degradation by 26S proteasome [8]. In contrast, IKKα is not required for the IκBα degradation but is involved in the processing of p100 to p52 [9]. These effects induce translocation and activation of NF-κB in the nucleus and activate the transcription of genes related to inflammation and immune responses, cell growth, differentiation, apoptosis and transformation [10, 11].
p53 tumour suppressor stands at the crossroads of cellular responses to various stresses [12]. Under normal conditions, p53 is maintained at low level through its interaction with MDM2 [13, 14], which mediates ubiquitination and proteasome-dependent degradation of p53. However, in response to DNA damage, both the quantity and activity of p53 are greatly increased. As a transcription factor, p53 can induce expression of many different downstream genes, including p21, GADD45 and Bax, to elicit various responses, such as cell-cycle arrest, apoptosis and DNA repair [15]. p53 accumulation and activation are thought to be regulated through post-translational modifications such as phosphorylation, acetylation and ubiquitination. Phosphorylation of p53 usually modulates its stability and sequence-specific DNA-binding activity [12]. For instance, phosphorylation of Ser15, Thr18, Ser20 and Ser37 stabilizes p53 by disrupting interaction between p53 and MDM2 [16], whereas phosphorylations at the p53 C-terminus such as Ser315 and Ser392 are reported to regulate the oligomerization state and sequence-specific DNA-binding ability of p53 [17].
Acquisition of resistance to chemotherapeutic agents has emerged as a significant impediment to effectively treat cancers. Some of the cytotoxicity of chemotherapy is attributed to apoptosis through activation of tumour-suppressor protein p53 [18]. The cell death process is positively and negatively regulated by different pathways, suggesting that modulation of the balance between death and survival signals could provide new strategies to improve the efficacy of current chemotherapeutic regimens. Constitutive activation of NF-κB enables malignant cells to escape apoptosis and might be crucial for the development of drug resistance in cancer cells. Inhibition of NF-κB activation can shift the death-survival balance towards apoptosis. It has been demonstrated that compared with wild-type mouse embryonic fibroblasts (MEFs), Ikkα/β–/– MEFs, in which NF-κB activation is inhibited, are more sensitive to anti-cancer agent–induced p53 and cell death [19]. Therefore, blocking the IKK–NF-κB pathway and activating the p53 pathway are promising approaches for developing new anti-cancer therapies.
Here we present a critical role of IKKβ in carcinogenesis. Because IKKβ-dependent NF-κB activation has been proposed to link inflammation and cancers, stable clones with IKKβ mutation of Ser177/Ser181 to AA or Tyr188/Tyr199 to FF, two synthetic α-methylene-γ-butyrolactone derivatives, CYL-19s and CYL-26z with potent inhibitory effect on IKKβ activity [20], and IKKβ siRNA were applied to this study. The cytostatic effect of CYL-19s and CYL-26z on tumour cells was examined. The results showed that loss of IKKβ activity enhances the protein stability of p53, leading to p21 expression, cell cycle arrest and apoptosis.
Materials and methods
Cell culture and chemicals
A549 human lung carcinoma cells were obtained from ATCC. HCT116 and HCT116 p53–/– human colon carcinoma cells were gifts from Dr. M. W. Van Dyke (M.D. Anderson Cancer Center, Houston, TX). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS). IKKβ stable transfectants were selected as previously described [21]. CYL-19s and CYL-26z were synthesized [20] and dissolved as stock solution (50 mM) in dimethyl sulfoxide (DMSO).
MTT assay
The cell growth of the four A549/IKKβ stable cells and the cell viability after CYL-19s and CYL-26z treatment were measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT; Sigma-Aldrich, St. Louis, MO) assay. Cells were plated in triplicate in 24-well plates and treated with increasing concentrations of CYL-19s, CYL-26z or drug diluent (DMSO). After 48 hrs of incubation, 0.5 mg/ml of MTT was added to each well for an additional 4 hrs. The blue MTT formazan precipitate was then dissolved in 100 μl of DMSO. The absorbance at 550 nm was measured on a multi-well plate reader. Cell viability was expressed as a percentage of control.
Animal xenograft assay
Four- to 6-week-old Female Balb/c nude mice were injected with 107 cells of four A549/IKKβ stable transfectants (suspended in 0.1 ml PBS and mixed with 0.1 ml Matrigel [BD Biosciences, San Jose, CA]) in the rear left flank. Mice were observed everyday for 30 days, and tumour growth was measured twice per week with calipers. Tumour volume was calculated using the formula V (mm3) = 0.52*[ab2], where a is the length and b is the width of the tumour. All animal work was performed under protocols approved by the Institutional Animal Care and Use Committee of the College of Medicine, National Taiwan University.
In vitro invasion assay
The invasion assay was carried out using Transwell® cell culture chambers (Corning Costar, Cambridge, MA). Briefly, polyvinylpyrrolidone-free polycarbonate filters (8.0 μm pore size, Nuclepore, Pleasanton, CA) were pre-coated with 5 μg of Matrigel on the upper surface. A549/IKKβ stable cells were harvested and then re-suspended in 0.1% FBS/DMEM. Cell suspensions (104 cells) were added to the upper compartment of the chamber. After 24-hr incubation, the top side of the insert membrane was scrubbed free of cells with a cotton swab, and the bottom side was fixed with 3.7% paraformaldehyde, stained with 0.5% crystal violet in 20% methanol. The crystal violet dye retained on the filters was extracted with DMSO and colourimetrically assessed by measuring its absorbance at 590 nm.
In vivo metastasis assay
A549/IKKβ stable cells were resuspended in PBS. Subsequently, 5 × 106 cells in 0.1 ml of PBS were injected into the lateral tail vein of 6-week-old nude mice. Mice were killed after 2 weeks, and all organs were examined for metastasis formation. The lungs were removed and fixed in 10% formalin. The number of lung tumour colonies was counted.
Matrigel angiogenesis assay
Angiogenesis inhibition was quantified using a modification of the Matrigel assay. Mice were injected subcutaneously in the abdominal midline with 0.5 ml of Matrigel alone or with 0.5 ml of condition medium from A549/IKKβ stable cells in Matrigel. Matrigel plugs were harvested on day 14, dissolved in Matrisperse at 4°C and assayed for haemoglobin content using Drabkin’s reagent (Sigma-Aldrich).
Cell cycle analysis
A549 cells were plated in 6-well plates for 24 hrs, and then G0/G1 phase synchronization was achieved by serum-starvation for 72 hrs. Synchronized cells were treated with complete medium containing CYL-19s and CYL-26z (0–10 μM) for 24 hrs. Cell cycle was determined by flow cytometry using a propidium iodide (PI) stain buffer and analyzed on a BD FACSCalibur cytometer with Cellquest software.
Assay for inhibition of [3H]thymidine incorporation
Proliferation of the cells was analyzed by measuring incorporation of [3H]thymidine. A549 cells were plated in 24-well flat-bottom microtiter plates at a density of 5 × 105 cells/well and cultured in medium containing 0.2% FBS for 72 hrs. Synchronized cells were treated with CYL-26z or CYL-19s for 24 hrs after release from the starvation. The cells were labelled with 1 μCi [3H]thymidine/well for 4 hrs at 37°C and then harvested on supporting tubes. Each sample was lysed hypotonically, and the radioactivity was measured in a Beckman model 2200 scintillation counter (Beckman, Fullerton, CA).
RNase protection assay
Total RNA was extracted from A549 cells using TRIZOL™ reagent (Invitrogen, Carlsbad, CA). A RiboQuant Multi-Probe RNase protection assay (RPA) was performed with the hStress-1, hAPO-3d and hCC-2 biotin-label probe sets (BD Pharmingen, San Diego, CA). The probes were hybridized with 3 μg of RNA, and then samples were digested with RNase to remove single-stranded RNA. Remaining probes were resolved on denaturing 5% polyacrylamide gels.
Immunoblotting and immunofluorescence staining
Following treatment with CYL-26z or CYL-19s, total cell lysates were prepared and subjected to SDS-PAGE. Western blot was done with antibodies specific for HA, Lys373/382 acetylated p53, p53, p21, IKKβ, GAPDH or actin (Santa Cruz, Biotechnology, Santa Cruz, CA) as described previously [21]. For immunofluorescence staining, A549 cells, grown on cover slips, were treated with CYL-19s or CYL-26z for 24 hrs in growth medium. The immunofluorescence staining was performed as described previously [21].
Semi-quantitative RT-PCR assay
Total RNA was isolated from A549 cells using TRIZOL™ reagent. Reverse transcription reaction was performed using 2 μg of total RNA and reverse-transcribed into cDNA using oligo dT primer, and then amplified using two oligonucleotide primers derived from published Noxa, Puma, p53 and β-actin sequence, including 5′-AGAGCTGGAAGTCGAGTGT-3′ and 5′-GCACCTTCACATTCCTCTC3′ (Noxa), 5′-GACCTCAACGCACAGTA-3′ and 5′-CTAATTGGGCTCCATCT-3′ (Puma), 5′-AGACCGGCGCACAGAGGAAG-3′ and 5′-CTTTTTGGACTTCAGGTGGC-3′ (p53) or 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ and 5′-CTAGAAGCATTTGCGGGGACGATGGAGGG-3′ (β-actin). PCR is carried out at 94°C for 30 sec, at 55°C for 30 sec and 1 min. at 70°C for 34 cycles. The PCR products are subjected to 1% agarose gel electrophoresis. Quantitative data are obtained using a computing densitometer and ImageQuant Software (Molecular Dynamics, Sunnyvale, CA).
Luciferase assay
A549 cells, grown to 50% confluent in 12-well plates, were transfected with the human luciferase (Luc) or p53-Luc using Tfx-50 (Promega, Madison, WI) according to the manufacturer’s recommendations. Briefly, reporter DNA (0.4 μg) and β-galactosidase DNA (0.2 μg; plasmid pRK containing the β-galactosidase gene driven by the constitutively active SV40 promoter, used to normalize the transfection efficiency) were mixed with 0.6 μl of Tfx-50 in 1 ml of serum-free DMEM. After 10 to 15 min. of incubation at room temperature, the mixture was applied to the cells; then 1 hr later, 1 ml of complete growth medium was added. On the following day, the medium was replaced with fresh medium. Forty-eight hours after transfection, the cells were treated with CYL-26z or CYL-19s for 6 hrs. Cell extracts were then prepared and luciferase and β-galactosidase activities measured, the luciferase activity being normalized to the β-galactosidase activity.
Chromatin immunoprecipitation assay (ChIP)
ChIP analysis was performed as described previously [22]. DNA was amplified across the p53-responsive element 1 (RE1) or RE2 region in the p21 promoter using the primers −2312CAGGCTGTGGCTCTGATGG−2292 and −2131TTCAGAGTAACAGGCTAAGG−2151 (p53RE1), and −1490GGTCTGCTACTGTGTCCTCC−1470 and −1279TATTGTGGGGCTGTTCTGGA−1299 (p53RE2).
siRNA knockdown analysis
IKKβ SMARTpool siRNA (Dharmacon, Lafayette, CO) was transfected with Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s instructions. Briefly, 50% confluent cells in 6-cm dishes were transfected with 200 pmol siRNA in 2 ml of serum-free medium for 6 hrs at 37°C. Then, 2 ml of medium containing 20% FBS was added to the transfection mixture. After 24 hrs, cells were treated with 5 μM CYL-19s and CYL-26z for another 24 hrs. The cells were lysed and the protein expression was analyzed by Western blot.
Statistical analysis
Data were analyzed using Student’s t-test. P-values <0.05 were considered significant.
Results
Cell proliferation, tumour formation, invasion, metastasis and angiogenesis induced by A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) alveolar epithelial cells
IKKβ plays a major role in the activation of classical NF-κB pathway. It has been well established that IKKβ activation requires phosphorylation of Ser177 and Ser181 in its activation loop [23]. In addition to these sites, phosphorylation of IKKβ at Tyr188 and Tyr199 were also identified to be crucial for NF-κB activation [24, 25]. Which sites play a more important role in the activation of NF-κB and their role in carcinogenesis remain unclear. To address these questions, HA-tagged IKKβ WT and two kinase-deficient IKKβ(AA) and IKKβ(FF) mutants, whose Ser177/Ser181 and Tyr188/Tyr199 were substituted with Ala and Phe, respectively, were used to establish stable transfectants in A549 cells, named as A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF), for cell proliferation, tumour formation, invasion, metastasis and angiogenesis assays. Expression of HA tag in A549 cells were confirmed by Western blot (Fig. 1A). A549/IKKβ(WT) cells showed a greater growth rate than A549/IKKβ(Mock) cells; however, A549/IKKβ(AA) and A549/IKKβ(FF) cells showed slower growth rate than that of A549/IKKβ(WT) cells (Fig. 1B). To further investigate the tumour formation, A549/Mock, A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) cells were introduced into nude mice via subcutaneous administration. As shown in Fig. 1C, tumour formation was seen in mice bearing A549/IKKβ(WT) cells 30 days after injection, but was not seen in mice bearing A549/Mock, A549/IKKβ(AA) and A549/IKKβ(FF) cells. The invasive capacity of A549/IKKβ(WT) cells across Matrigel was greater than that of A549/Mock cells, but that was lower in A549/IKKβ(AA) and A549/IKKβ(FF) cells (Fig. 1D). The role of IKKβ in metastatic colonization was assessed by intravenous injection of stable clone cells into the lateral tail vein of nude mice. Mice injected with A549/IKKβ(WT) cells had numerous large lung metastatic nodules, whereas those injected with A549/IKKβ(AA) and A549/IKKβ(FF) cells had fewer and smaller nodules (Fig. 1E). These results suggest that loss of IKKβ activity was a negative regulator in tumourigenesis. We further asked whether loss of IKKβ activity reduced angiogenesis in vivo. Matrigels in conjunction with medium from stable cells were subcutaneously injected into nude mice. The solid gel plug was removed from the mice for haemoglobin examination. As shown in Fig. 1F, the haemoglobin concentration within Matrigel mixed with condition medium from A549/IKKβ(WT) cells was greater than that from A549/Mock cells, but the haemoglobin concentration was lower within Matrigel with medium from A549/IKKβ(AA) and A549/IKKβ(FF) cells. Taken together, these results indicated that these serine and tyrosine phosphorylations in the activation loop are equally important for IKKβ-mediated tumour proliferation, invasion, metastasis and angiogenesis and that inhibition of IKKβ activity may be a promising anti-cancer strategy.
Fig 1.
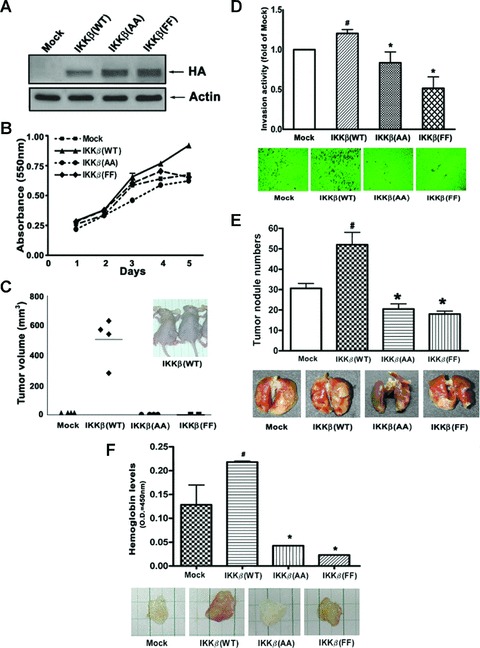
Effects of IKKβ mutation on cell proliferation, tumour formation, invasion, metastasis and angiogenesis. (A) Whole-cell lysates from A549/Mock, A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) cells were prepared and subjected to immunoblotting using antibody specific for HA or actin. (B) The proliferation of A549/Mock, A549/ IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) cells was measured by MTT assay from day 1 to day 5. (C) A549/Mock, A549/IKKβ(WT), A549/ IKKβ(AA) and A549/IKKβ(FF) cells were xenotransplanted into the right hip region of nude mice as described in the Materials and Methods. The tumour volume was calculated by V= 0.52× (the length of width)2× (the length of length). (D) A549/Mock, A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) cells were incubated on transwell culture inserts coated with Matrigel and the invasion activity was measured at the absorbance of 590 nm. Quantitative results of different stable clone cells were present as fold by comparing with the A549/IKKβ(Mock). #P < 0.05 as compared with Mock. *P < 0.05 as compared with A549/IKKβ(WT). (E) A549/Mock, A549/IKKβ(WT), A549/IKKβ(AA) and A549/IKKβ(FF) cells were intravenously introduced into nude mice for 14 days. The mice were sacrificed and lungs were removed and photographed. Quantitative results of tumour nodules on the lung surface were counted and statistically analyzed. #P < 0.05 as compared with Mock. *P < 0.05 as compared with A549/IKKβ(WT). (F) Matrigel angiogenesis assay was conducted by injecting conditional medium (CM) mixed Matrigel into nude mice. After 7 days, the mice were killed and dissected. The haemoglobin levels of the Matrigel plugs were measured with the Drabkin reagent kit 525. #P < 0.05 as compared with Mock. *P < 0.05 as compared with A549/IKKβ(WT).
IKKβ inhibitors induced cell cycle arrest and apoptosis
Blocking cell cycle progression is a strategy for cancer treatment. More and more cytotoxic drugs have been shown to cause cell death, at least partially by inducing cell cycle arrest or apoptosis [26, 27]. To study whether the anti-tumour activity by loss of IKKβ was achieved through the influence of cell cycle progression, pharmacologic IKK inhibitors were further used in cell cycle analysis. Two potent and specific IKKβ inhibitors, the synthetic α-methylene-γ-butyrolactone derivatives-CYL-19s and CYL-26z, were developed and demonstrated to possess potent inhibitory effect on cancer cell invasion in our previous study [20]. These two compounds were used for the further study of the molecular events elicited by loss of IKKβ activity. A549 cells synchronized at G1 phase by serum starvation for 3 days were treated with completed medium containing 0.1% DMSO or 1, 5, 10 μM of these two compounds for 24 hrs. As shown in Fig. 2A, about 21% of the cells entered S and G2/M phase, respectively, after serum replenishment, and these effects were inhibited by CYL-19s and CYL-26z in a dose-dependent manner, indicating that CYL compounds block G1/S transition. The result from [3H]thymidine incorporation also showed the inhibition of serum-induced DNA synthesis by CYL-19s and CYL-26z dose dependently (Fig. 2B), supporting their effects on cell cycle arrest. When asynchronous cells were treated with 5 μM of CYL compounds for 24 and 48 hrs, these two compounds dramatically induced the fraction of sub-G1 cells with slight increase in G2/M phase (Fig. 2C), indicating that IKKβ inhibition induced not only cell cycle arrest but also apoptosis. These anti-tumour properties of CYL-19s and CYL-26z were further confirmed by MTT assay. CYL-19s and CYL-26z markedly inhibited the cell growth in a dose-dependent manner. The respective growth inhibition was 20, 24, 64, 83 and 84% induced by 1, 3, 10, 30 and 50 μM of CYL-19s. An equivalent effect was seen by CYL-26z (Fig. 2D). Altogether, these findings revealed that inhibition of IKKβ by CYL-19s and CYL-26z exhibit anti-tumour activity via induction of cell cycle arrest at both G1 and G2/M phase, inhibition of DNA synthesis and initiation of apoptosis.
Fig 2.
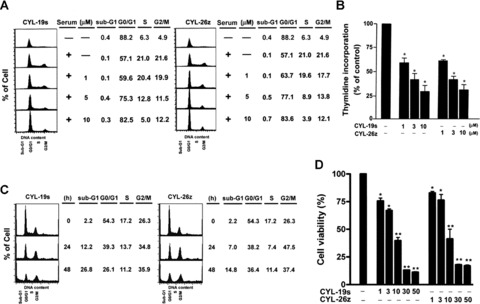
Effects of IKKβ inhibitors on cell cycle progression and DNA synthesis in A549 epithelial cells. (A) Cells were synchronized at G1 phase by serum starvation for 3 days and then treated with 1, 5, 10 μM CYL-19s or CYL-26z for 24 hrs. Then, cells were harvested and analyzed by flow cytometry. (B) For measuring the effect of CYL-19s and CYL-26z on DNA synthesis, cells were labelled with [3H]-thymidine for 8 hrs. Then, the cells were harvested and radioactivity was measured as described in Materials and Methods. *P < 005 as compared with basal. (C) Cells were treated with 5 μM CYL-19s or CYL-26z for 24 and 48 hrs. Then, cells were harvested and analyzed by flow cytometry. (D) A549 cells were treated with various concentrations of CYL-19s and CYL-26z for 48 hrs. The cell viability was measured by MTT assay. *P < 0.05, **P < 0.01 as compared with basal.
IKKβ inhibitors increased the level of p21
To characterize which gene related to cell cycle and apoptosis was regulated by CYL-19s and CYL-26z, RPA was performed. As shown in Fig. 3A, CYL-19s and CYL-26z induced p21, Bax and DR5 mRNA expression but had no effect on p53 mRNA expression. We also analyzed the mRNA levels of pro-apoptotic Bcl-2 family genes, Noxa and Puma, which are reported to be p53 target genes [28]. Their expression was not induced by the treatment with 5 μM CYL-19s and CYL-26z (Fig. 3B). The increase in p21 protein expression was confirmed by Western blot. The expression of p21 was enhanced by CYL-19s or CYL-26z in a dose- and time-dependent manner (Fig. 3C–E).
Fig 3.
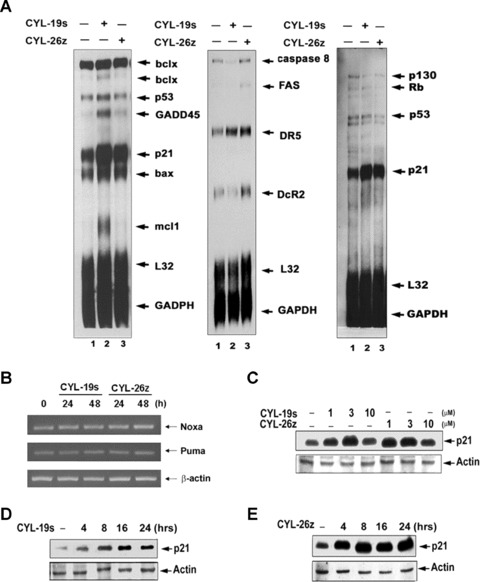
IKKβ inhibitors stimulate p21 expression in A549 epithelial cells. (A) RNase-protected probes hybridized with total RNA from A549 cells treated with 5 μM CYL-19s and CYL-26z for 24 hrs were subjected to RNase protection assay (RPA) as described in Materials and Methods, GADPH and L32 were shown as internal controls. (B) Cells were treated with 5 μM CYL-19s and CYL-26z for 24 and 48 hrs, and then mRNA were prepared and subjected to RT-PCR. For p21 protein expression, cells were treated with indicated concentration of CYL-19s and CYL-26z for 24 hrs (C), or with 10 μM CYL-19s and CYL-26z for indicated time (D and E). Whole-cell lysates were subjected to immunoblotting using antibody specific for p21 or actin.
p21 is a well-known target of p53. To determine whether p53 is responsible for the CYL-19s- and CYL-26z-induced p21 expression, p21 constructs (illustrated as Fig. 4A) were transiently transfected into A549 cells. Both CYL-19s- and CYL-26z induced the p21 promoter activity by about 2.5-fold, and these effects were abolished once p53-responsive element 1 (p53RE1) on p21 promoter was deleted (Fig. 4B). Knockdown of IKKβ by siRNA also increased p21 promoter activity (Fig. 4C), demonstrating that loss of IKKβ induced p21 expression. To confirm the role of p53 in CYL compound-induced p21 expression, the p21 protein level was further examined in CYL compound-treated HCT116 wild-type (wt) and p53–/– cells. CYL-19s and CYL-26z can induce p21 expression in HCT116 wt cells, but the effect was not observed in HCT116 p53–/– cells (Fig. 4D), confirming the induction of p21 expression through a p53-dependent pathway. p53 transactivation is correlated with its DNA-binding activity, therefore, effect of CYL-19s and CYL-26z on the recruitment of p53 to p21 promoter was examined by chromatin immunoprecipitation (ChIP) assay. As shown in Fig. 4E, the binding of p53 to two responsive elements on p21 promoter (p53RE1 and p53RE2) was enhanced by CYL-19s and CYL-26z in a time-dependent manner. Therefore, our results revealed that these IKKβ inhibitors induced p21 expression in a p53-dependent manner. Interestingly, the increased level of p53 protein was also observed in the CYL-19s and CYL-26z-treated HCT116 wt cells. Since the result from RPA revealed treatment of these two compounds did not affect p53 mRNA level (Fig. 3A), it is suggested that inhibition of IKKβ may increase p53 expression at post-translational level.
Fig 4.
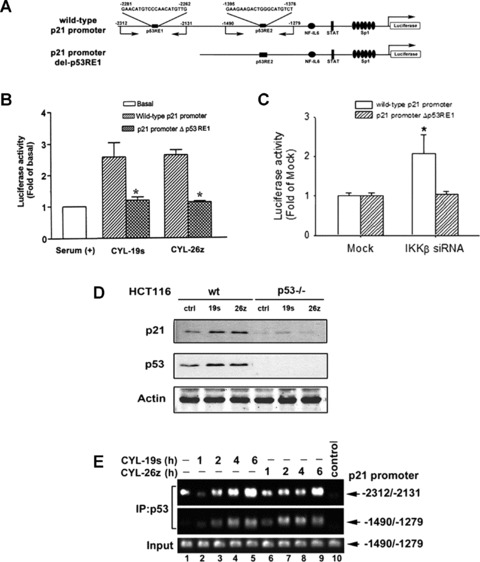
IKKβ inhibitors-induced p21 expression required the p53. (A) Schematic illustration of various transcription factor binding sites on p21-Luc promoter. The p53-responsive element 1 (p53RE1)-deleted mutant was illustrated and referred as p21 promoter del-p53RE1. The targeting sites of primers used in ChIP assay were also indicated. (B) A549 cells were transfected with p21 promoter-Luc reporter (wild-type [wt] or its p53-deleted mutant), and then treated with 10 μM CYL-19s and CYL-26z for 24 hrs. Luciferase activity was measured, normalized with β-galactosidase activity and expressed as the mean S.E.M. of three independent experiments performed in triplicate. *P < 0.05 as compared with wt promoter. (C) A549 cells were transfected with IKKβ siRNA and p21 promoter-Luc reporter (wt or its p53-deleted mutant). Then, luciferase activity was measured, normalized with β-galactosidase activity and expressed as the mean S.E.M. of three independent experiments performed in triplicate. *P < 0.05 as compared with Mock. (D) HCT116 wt or HCT116 p53–/– cells were treated with 5 μM CYL-19s and CYL-26z. Whole-cell lysates were examined by anti-p21 or anti-p53 antibody. (E) A549 cells were treated with 10 μM CYL-19s and CYL-26z for the indicated times, and then ChIP assays were performed. Chromatin was immunoprecipitated with anti-p53 antibody or without antibody (control). One percent of the precipitated chromatin was assayed to verify equal loading (Input). Five-fold dilution was subjected to PCR using primer to the p21 promoter (–2312/–2131 and –1490/–1279).
IKKβ inhibitors increased the stability and activity of p53
To further confirm the level of p53 transcript was not affected in the presence of CYL-19s and CYL-26z, a semi-quantitative RT-PCR assay was performed to examine the p53 mRNA in A549 cells. In consistence with the result from RPA, the level of p53 mRNA was not significantly affected in both CYL-19s- and CYL-26z-treated A549 cells (Fig. 5A). However, an increase in protein expression of p53 indeed was seen in response to CYL-19s and CYL-26z stimulation after a 4-hr treatment and sustained for 24 hrs (Fig. 5B). Furthermore, the promoter activity using a p53-responsive promoter-luciferase plasmid (p53-Luc) was also activated by CYL-19s and CYL-26z (Fig. 5C). To further confirm inhibition of IKKβ-induced p53 and p21 expression, siRNA was used to knockdown IKKβ. As shown in Fig. 5D, the protein expression of p53 and p21 was enhanced by IKKβ siRNA, and CYL-19s and CYL-26z-induced p53 and p21 expression were augmented by IKKβ siRNA as well.
Fig 5.
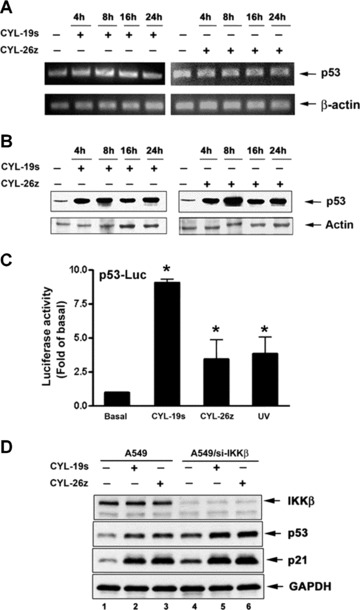
IKKβ inhibitors increase the level and activity of p53. (A) Cells were treated with 5 μM CYL-19s or CYL-26z for the indicated time, and then mRNA were prepared and subjected to RT-PCR. (B) A549 cells were treated with 10 μM CYL-19s or CYL-26z for the indicated time. Whole-cell lysates were subjected to immunoblotting using antibody specific for p53 or actin. (C) A549 cells were transfected with p53 responsive-Luc reporter (p53-Luc) then treated with 5 μM CYL-19s and CYL-26z for 24 hrs or exposed to 50 J/m2 UV (as positive control). Luciferase activity was measured, normalized with β-galactosidase activity and expressed as the mean S.E.M. of three independent experiments performed in triplicate. *P < 0.05 as compared with basal. (D) A549 cells were transfected with IKKβ siRNA then treated with 5 μM CYL-19s or CYL-26z for 24 hrs. Whole-cell lysates were subjected to immunoblotting using antibody specific for IKKβ, p53, p21 or GAPDH.
To explore whether CYL-19s and CYL-26z can modulate p53 turnover, its decay rate was examined. After treatment with CYL-19s and CYL-26z for 4 hrs, cells were treated with cycloheximide (CHX) for the indicated time. As shown in Fig. 6A, the degradation of p53 was greatly reduced in the presence of CYL-19s and CYL-26z. Knockdown of IKKβ by siRNA also increased p53 stability (Fig. 6B). Thus, CYL-19s and CYL-26z-mediated p53 activation resulted from the decrease in its degradation. The accumulation of p53 in the nucleus was also seen using an immunofluorescence assay (Fig. 6C). Post-translational modification of p53 such as phosphorylation and acetylation at the C-terminal is closely related to its stabilization and transcriptional activation [29]. Our data also showed that the acetylation of p53 on Lys373/382 in either the nucleus or total cell lysates (TCL) was seen upon CYL-19s and CYL-26z stimulation (Fig. 6D), suggesting that inhibition of IKKβ may increase p53 protein stability through these post-translational modification.
Fig 6.
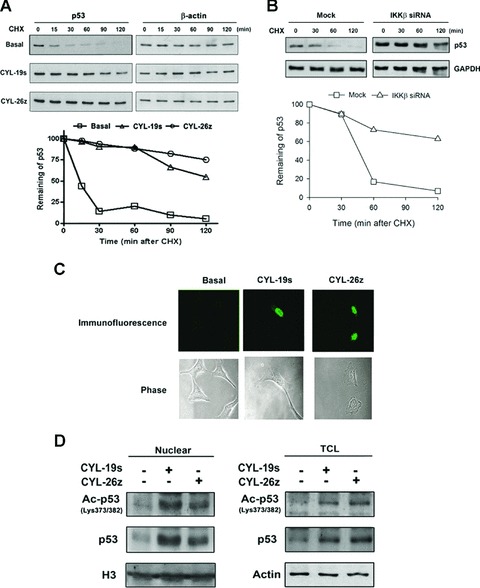
IKKβ inhibitors increase the stability of p53. (A) Cells were treated with CYL-19s or CYL-26z for 4 hrs, and then cells were treated with 500 nM CHX and harvested at the indicated time periods. Whole-cell lysates were subjected to immunoblotting using antibody against p53 or actin. p53 expression normalized with actin was quantified using ImageQuant and the p53 remaining is indicated graphically. (B) A549 cells were transfected with IKKβ siRNA then treated with 500 nM CHX. Cells were harvested at the indicated time periods. Whole-cell lysates were subjected to immunoblotting using antibody against p53 or GAPDH. p53 expression normalized with GAPDH was quantified using ImageQuant and the p53 remaining is indicated graphically. (C) Cells for p53 immunofluorescence staining were fixed and stained as described in Materials and Methods. (D) Cells were treated with 10 μM CYL-19s or CYL-26z for 24 hrs, and then nuclear extracts or total cell lysates were prepared and subjected to immunoblotting using antibody against acetyl-p53 (Lys373/382), p53, H3 or actin.
IKKβ inhibitors exhibit p53-dependent anti-tumour activity
To further confirm the role of p53 and p21 in the anti-tumour effect of CYL-19s and CYL-26z, HCT116 WT, p53–/– and p21–/– cells were treated with different concentrations (1, 5 and 10 μM) of CYL-19s and CYL-26z for 3 days, and the cell viability was determined by MTT assay. The cell viability in HCT116 wt cells was inhibited by these two compounds dose dependently (Fig. 7A). Cells with p53 deficiency were more resistant to CYL-19s- and CYL-26z-induced decrease in cell viability, which was correlated with the decrease in subG1 fraction in p53–/– cells (Fig. 7A and B). However, the decreased cell viability in p21–/– cells was not recovered (Fig. 7A), because CYL compounds accumulated more cells at subG1 (CYL-19s-treated cells; 16.9–29.6%) or G2/M phase (CYL-26z-treated cells; 34.2–56.4%) (Fig. 7B). These findings revealed that the anti-tumour activity of CYL-19s and CYL-26z acts via p53-dependent but p21-independent apoptosis.
Fig 7.
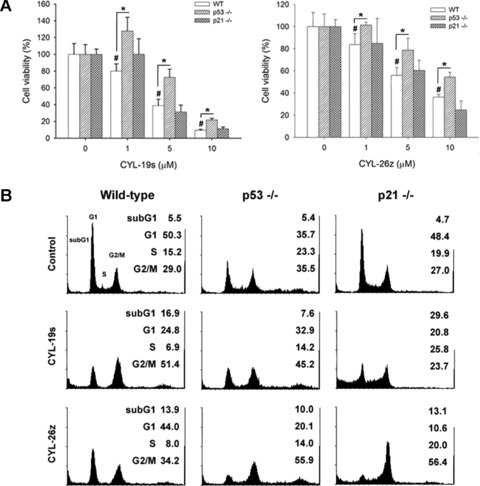
IKKβ inhibitors induce p53-dependent cytotoxicity. (A) HCT116 wild-type (WT), p53–/– and p21–/– cells plated at a density of 5 × 103 cells/well in a 96-well plate were treated with 1, 5 or 10 μM for 3 days. MTT assay was performed and absorbance was measured at 550 nm. #P < 0.05, compared with basal; *P < 0.05, compared with wild-type cells. (B) HCT116 WT, p53–/– and p21–/– cells were cultured in 6-cm dish and treated with 5 μM CYL-19s and CYL-26z for 24 hrs. Cells were harvested and examined by flow cytometry as described in Materials and Methods.
Discussion
NF-κB is a major link between inflammation and cancers [30]. The association of NF-κB activation with tumour promotion, progression and metastasis is well documented in several mouse models [31, 32]. Selectively targeting IKKβ is an effective approach to inhibit NF-κB activity because IKKβ plays a major role in the canonical pathway. Thus, unwanted side effects related to inhibition of the non-canonical pathway, such as adaptive immunity, can be avoided [33]. In the present study, we used A549 cells over-expressing IKKβ(WT), IKKβ(AA) and IKKβ(FF) to investigate the role of IKKβ in tumourigenesis, including proliferation, tumour formation, invasion, metastasis and angiogenesis. We found that A549/IKKβ(AA) and A549/IKKβ(FF) cells, specifically lacking IKKβ activity, are able to abrogate tumour promotion. Our previous studies propose that both phosphorylation of Ser177/Ser181 and Tyr188/Tyr199 are required for full activation and biological functions of IKKβ[24, 25]. Here, we demonstrated that mutation of Ser177/Ser181 to AA and Tyr188/Tyr199 to FF showed similar effect to abrogate tumour promotion (Fig. 1). These results suggest that IKKβ plays an important role in tumourigenesis, and both Ser177/Ser181 and Tyr188/Tyr199 phosphorylation sites are required and equally contribute to the oncogenic activity of IKKβ. To clarify the molecular mechanism(s), specific IKKβ inhibitors, CYL-19s and CYL-26z, and IKKβ siRNA were used for further investigation. We have previously found that CYL-19s and CYL-26z suppress inflammation and cancer cell invasion through inhibition of IKKβ activity [20]. Now we have found that inhibition of IKKβ by these inhibitors or siRNA enhances p53 acetylation, stabilization and nuclear accumulation, leads to p21 expression, cell cycle arrest, apoptosis and decreased cell viability.
Human cancers arise from an imbalance of cell growth and cell death. NF-κB and p53 are the key proteins to govern this balance. Inactivation of p53 and hyperactivation of NF-κB is a common occurrence in human cancers. Thus, bi-targeted anti-cancer drugs that simultaneously activate p53 and inhibit NF-κB are evolving attractive strategy. Recent results suggest that a surprising selection of small molecules have this desirable dual activity [34]. For example, quinacrine, an anti-malarial drug, and other derivatives of 9-aminoacridine are found to simultaneously repress NF-κB and activate p53 in renal cell carcinoma [35]. In the present study, IKKβ inhibitors (CYL-19s and CYL-26z) or knockdown of IKKβ by siRNA increased the p53 expression and stability. How CYL-19s and CYL-26z function to target both pathways is unclear. Our recent results demonstrated that IKKα tips the balance between NF-κB and p53 through phosphorylation of CBP [36], because p65 and p53 negatively regulate each other’s activity by competing for the limiting pool of coactivator p300 or CBP, which are required for transactivation [36, 37]. Since CYL-19s and CYL-26z blunted the nuclear translocation of NF-κB and increased p53, this might favour the interaction of CBP with p53. We found the increase in p53 acetylation after treatment with CYL-19s and CYL-26z, suggesting that enhanced interaction between CBP and p53 might occur. Therefore, CYL-19s and CYL-26z could block the NF-κB-induced survival signals and shift the balance in the favour of p53-mediated death signals. Indeed, CYL-19s and CYL-26z down-regulated the expression of NF-κB-regulated genes involved in invasion (e.g. ICAM-1, COX-2) [20], and up-regulated the expression of p53-regulated genes involved in cell cycle arrest (e.g. p21) and apoptosis (e.g. Bax and DR5) (Fig. 3A). They also induced cytotoxic effect in HT-29 colon cancer cells through the increase of Bax and decrease of Bcl-2 expression (data not shown).
IKKβ has been reported to increase the levels of MDM2, thereby negatively regulating p53 stability [19]. Bcl-3, a member of IκB family, is shown to interact with NF-κB p50 and p52 homodimers to promote the transcriptional activity of NF-κB [38, 39]. Similar to IKKβ, Bcl-3 is also shown to induce MDM2 gene expression and the subsequent suppression of p53 activity [40]. It is possible that loss of IKKβ negatively regulated MDM2 and then stabilized p53 expression [19]. However, a number of reports showed the increase of p53 transcription and stability by activated NF-κB, which can induce the expression of p53 viaκB sites in its promoter [41–44]. Therefore, it needs to be investigated why loss or activation of NF-κB can increase p53 stability. It has been proposed that basal NF-κB activity is important for cell survival under normal conditions. However, in the presence of DNA damage, NF-κB stabilizes p53 and therefore serves an acute pro-apoptotic function [45, 46]. Indeed, NF-κB has been reported to exert both pro-apoptotic and anti-apoptotic activities [47].
Diverse stresses activate p53 by post-translational modifications and nuclear accumulation, contributing to the apoptotic activity of p53 in several cancer cells. Among post-translational modifications, ubiquitination, phosphorylation and acetylation mostly affect its overall appearance and activity [12]. Recent findings suggest that these modifications have a profound effect on p53 stability and function. Phosphorylation of Ser392 stabilizes the formation of p53 tetramer, which is critical for enhancement of DNA binding and activating gene transcription [48, 49]. Phosphorylation of p53 on serines 15 and 20 at the N-terminus and acetylation on lysines 373 and 382 at the C-terminus attenuate its interaction with MDM2, which possesses E3 ligase activity to induce p53 ubiquitination and degradation [29]. Moreover, p53 acetylation stimulates its DNA-binding activity in vivo and promotes apoptosis [50]. Our results showed that CYL-19s- and CYL-26z-enhanced p53 expression and nuclear accumulation were consistent with the nuclear p53 Lys373/382 acetylation. Thus, post-translational modification and subsequent stabilization of p53 by IKKβ inhibitors may explain the ability of CYL-19s and CYL-26z to activate p21 transcription. Our data indicated that CYL-19s- and CYL-26z-induced p21 expression was not seen in HCT116 p53–/– cells (Fig. 4C), and p21 promoter activity induced by CYL compounds was abolished when p53-responsive element 1 (p53RE1) was deleted (Fig. 4B). These findings demonstrated that p53 was the most important regulator of CYL-19s- and CYL-26z-induced p21 expression, although other factors such as Sp1/Sp3 and Smads have also been reported to be critical for the p21 transcription [51]. Interestingly, it was observed that CYL compounds were able to enhance p53 expression in the absence of IKKβ (Fig. 5D), raising the possibility that several mechanisms other than targeting IKKβ might be involved. Our previous study showed that CYL compounds can also attenuate the TNF-α-induced complex formation of IKKα/CBP/p65 but increase the interaction between CBP and p53 [36], implying that the CYL compound may also inhibit IKKα and thereby enhance the interaction between CBP and p53. The results that CYL compounds can induce p53 acetylation further supports this notion (Fig. 6D). However, it still does not rule out the possibility that they might also directly activate the p53 pathway. This can explain why either CYL-19s or CYL-26z alone is more potent than IKKβ siRNA to induce p21. Therefore, CYL-19s and CYL-26z could simultaneously target NF-κB and p53 pathways.
The α-methylene-γ-butyrolactone compounds are electrophilic and apt to react with biological nucleophiles such as the sulfhydryl group of GSH, proteins and DNA [52]. In the present study, we studied the cytostatic effects of these two novel compounds CYL-26z and CYL-19s. They decreased the viability of lung adenocarcinoma via inducing cell cycle arrest and apoptosis. Several bioactive natural products and synthetic chemicals possessing this moiety have been reported to exhibit a diverse range of biological activities, including inhibition of fatty acid metabolism, inflammation and cell proliferation [53, 54]. The α-methylene-γ-butyrolactone derivatives such as C75, parthenolide and some sesquiterpenes have remarkable in vitro and in vivo anti-cancer properties against a wide range of tumour cells [55, 56]. A complete understanding of the molecular mechanism(s) involved in the cell cycle arrest and apoptosis driven by α-methylene-γ-butyrolactone derivatives may be important for devising better strategies for cancer therapy.
In summary, our results support that loss of IKKβ activity can be viewed as a critical target for cancer therapy. To our knowledge, this is the first report demonstrating that inhibition of NF-κB activity enhances p53 acetylation, stabilization and nuclear accumulation, leading to p21 expression, and cell cycle arrest and apoptosis of tumour cells. Importantly, the ability to simultaneously inhibit NF-κB and activate p53 makes CYL-19s and CYL-26z potentially useful for treating cancers.
Acknowledgments
This work was supported by a research grant from the National Science Council of Taiwan NSC97-2320-B-002-033-MY3 and Joint Project of National Taiwan University, College of Medicine and Chinese Medical University 97F008-105.
References
- 1.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin ASJr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Finco TS, Nantermet PV, et al. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–10. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Israel A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10:129–33. doi: 10.1016/s0962-8924(00)01729-3. [DOI] [PubMed] [Google Scholar]
- 7.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol. 2002;3:20–6. doi: 10.1038/ni0102-20. [DOI] [PubMed] [Google Scholar]
- 9.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 10.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Li ZW, Omori SA, Labuda T, et al. IKK beta is required for peripheral B cell survival and proliferation. J Immunol. 2003;170:4630–7. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- 12.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 13.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 14.Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 16.Shieh SY, Ikeda M, Taya Y, et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature. 1995;376:88–91. doi: 10.1038/376088a0. [DOI] [PubMed] [Google Scholar]
- 18.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–7. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 19.Tergaonkar V, Pando M, Vafa O, et al. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 20.Huang WC, Chan ST, Yang TL, et al. Inhibition of ICAM-1 gene expression, monocyte adhesion and cancer cell invasion by targeting IKK complex: molecular and functional study of novel alpha-methylene-gamma-butyrolactone derivatives. Carcinogenesis. 2004;25:1925–34. doi: 10.1093/carcin/bgh211. [DOI] [PubMed] [Google Scholar]
- 21.Lin YC, Shun CT, Wu MS, et al. A novel anticancer effect of thalidomide: inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clin Cancer Res. 2006;12:7165–73. doi: 10.1158/1078-0432.CCR-06-1393. [DOI] [PubMed] [Google Scholar]
- 22.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;25:6592–602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woronicz JD, Gao X, Cao Z, et al. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–9. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 24.Huang WC, Chen JJ, Chen CC. c-Src-dependent tyrosine phosphorylation of IKKbeta is involved in tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 expression. J Biol Chem. 2003;278:9944–52. doi: 10.1074/jbc.m208521200. [DOI] [PubMed] [Google Scholar]
- 25.Huang WC, Chen JJ, Inoue H, et al. Tyrosine phosphorylation of I-kappa B kinase alpha/beta by protein kinase C-dependent c-Src activation is involved in TNF-alpha-induced cyclooxygenase-2 expression. J Immunol. 2003;170:4767–75. doi: 10.4049/jimmunol.170.9.4767. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad N, Adhami VM, Afaq F, et al. Resveratrol causes WAF-1/p21-mediated G(1)-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin Cancer Res. 2001;7:1466–73. [PubMed] [Google Scholar]
- 27.Chinni SR, Li Y, Upadhyay S, et al. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–36. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 28.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 29.Li M, Luo J, Brooks CL, et al. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002;277:50607–11. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 30.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo JL, Maeda S, Hsu LC, et al. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Moschos SJ, Chaudhary PM, Kirkwood JM. Resolving ‘kinks’ of chemotherapy in melanoma. J Natl Cancer Inst. 2008;100:833–5. doi: 10.1093/jnci/djn189. [DOI] [PubMed] [Google Scholar]
- 34.Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nat Rev Drug Discov. 2008;7:1031–40. doi: 10.1038/nrd2759. [DOI] [PubMed] [Google Scholar]
- 35.Gurova KV, Hill JE, Guo C, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci USA. 2005;102:17448–53. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang WC, Ju TK, Hung MC, et al. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–95. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westerheide SD, Mayo MW, Anest V, et al. The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol. 2001;21:8428–36. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viatour P, Bentires-Alj M, Chariot A, et al. NF-kappa B2/p100 induces Bcl-2 expression. Leukemia. 2003;17:1349–56. doi: 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- 40.Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006;20:225–35. doi: 10.1101/gad.1352206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Lozano G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. 1994;269:20067–74. [PubMed] [Google Scholar]
- 42.Benoit V, Hellin AC, Huygen S, et al. Additive effect between NF-kappaB subunits and p53 protein for transcriptional activation of human p53 promoter. Oncogene. 2000;19:4787–94. doi: 10.1038/sj.onc.1203831. [DOI] [PubMed] [Google Scholar]
- 43.Kirch HC, Flaswinkel S, Rumpf H, et al. Expression of human p53 requires synergistic activation of transcription from the p53 promoter by AP-1, NF-kappaB and Myc/Max. Oncogene. 1999;18:2728–38. doi: 10.1038/sj.onc.1202626. [DOI] [PubMed] [Google Scholar]
- 44.Qin ZH, Chen RW, Wang Y, et al. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci. 1999;19:4023–33. doi: 10.1523/JNEUROSCI.19-10-04023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujioka S, Schmidt C, Sclabas GM, et al. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-kappaB. J Biol Chem. 2004;279:27549–59. doi: 10.1074/jbc.M313435200. [DOI] [PubMed] [Google Scholar]
- 46.Aleyasin H, Cregan SP, Iyirhiaro G, et al. Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage. J Neurosci. 2004;24:2963–73. doi: 10.1523/JNEUROSCI.0155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutta J, Fan Y, Gupta N, et al. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–16. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 48.Stenger JE, Tegtmeyer P, Mayr GA, et al. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J. 1994;13:6011–20. doi: 10.1002/j.1460-2075.1994.tb06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakaguchi K, Sakamoto H, Lewis MS, et al. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry (Mosc) 1997;36:10117–24. doi: 10.1021/bi970759w. [DOI] [PubMed] [Google Scholar]
- 50.Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 51.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–5. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 52.Picman AK, Rodriguez E, Towers GH. Formation of adducts of parthenin and related sesquiterpene lactones with cysteine and glutathione. Chem Biol Interact. 1979;28:83–9. doi: 10.1016/0009-2797(79)90116-9. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Pineres AJ, Castro V, Mora G, et al. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem. 2001;276:39713–20. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- 54.Lyss G, Knorre A, Schmidt TJ, et al. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J Biol Chem. 1998;273:33508–16. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
- 55.Kuhajda FP, Pizer ES, Li JN, et al. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA. 2000;97:3450–4. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wen J, You KR, Lee SY, et al. Oxidative stress-mediated apoptosis. The anticancer effect of the sesquiterpene lactone parthenolide. J Biol Chem. 2002;277:38954–64. doi: 10.1074/jbc.M203842200. [DOI] [PubMed] [Google Scholar]