

Abstract
How a cancer is initiated and established remains elusive despite all the advances in decades of cancer research. Recently the cancer stem cell (CSC) hypothesis has been revived, challenging the long-standing model of ‘clonal evolution’ for cancer development and implicating the dawning of a potential cure for cancer [1]. The recent identification of pre-cancerous stem cells (pCSCs) in cancer, an early stage of CSC development, however, implicates that the clonal evolution is not contradictory to the CSC hypothesis but is rather an aspect of the process of CSC development [2]. The discovery of pCSC has revealed and will continue to reveal the volatile properties of CSC with respect to their phenotype, differentiation and tumourigenic capacity during initiation and progression. Both pCSC and CSC might also serve as precursors of tumour stromal components such as tumour vasculogenic stem/progenitor cells (TVPCs). Thus, the CSC hypothesis covers the developing process of tumour-initiating cells (TIC) → pCSC → CSC → cancer, a cellular process that should parallel the histological process of hyperplasia/metaplasia (TIC) → pre-cancerous lesions (pCSC) → malignant lesions (CSC → cancer). The embryonic stem (ES) cell and germ line stem (GS) cell genes are subverted in pCSCs. Especially the GS cell protein piwil2 may play an important role during the development of TIC → pCSC → CSC, and this protein may be used as a common biomarker for early detection, prevention, and treatment of cancer. As cancer stem cell research is yet in its infancy, definitive conclusions regarding the role of pCSC cannot be made at this time. However, this review will discuss what we have learned from pCSC and how this has led to innovative ideas that may eventually have major impacts on the understanding and treatment of cancer.
Keywords: tumour-initiating cells, pre-cancerous stem cells, cancer stem cells, tumour development, clonal evolution, genetic instability, Piwil2, tumour vasculogenesis, common cancer biomarker, embryonic stem cell genes, immunoprevention, tumour vaccine
Introduction
A tumour is denoted as a tissue mass resulting from abnormal growth. This growth can be benign or malignant. Benign tumours are usually not invasive and can be removed surgically without recurrence. In contrast, malignant tumours grow uncontrollably, and frequently recur or metastasize after treatment because tumour cells can invade neighbouring or distant tissues. The formation of a malignant tumour includes a lengthy, reversible pre-cancerous stage [3]. Although much has been learned about these processes, the mechanisms underlying initiation, progression, metastasis and recurrence of cancer have not been fully elucidated despite decades of intensive investigations by thousands of dedicated cancer researchers.
Extensive investigations have revealed, however, that cancer is a class of complicated genetic diseases [4]. Carcinogenesis is a complex, multistep process by which a normal cell is stressed and transformed into cancer cells. This often requires concordant expression of a number of genes, including multiple genetic and epigenetic changes in oncogenes, tumour-suppressor genes, cell-cycle regulators, cell adhesion molecules, DNA repair genes and genetic instability as well as telomerase activation [4–6]. However, little is known about how a normal cell is initiated to transform into cancer cells.
Previous research has revealed that a cancer consists of various types of tissue components, including phenotypically heterogeneous cancer cells, stromal cells, and vasculature. While heterogeneous cancer cells are believed to be malignant, the stromal cells and vascular cells of cancer have been considered to be derived from normal progenitors [7, 8], although controversy exists regarding this assumption [9, 10]. Based on the heterogeneity of cancer cells, several models have been proposed to explain cancer development. The stochastic model claims that all cancer cells can reproduce phenotypically heterogeneous cell types in new tumours. However, this model cannot explain why cancer is highly heterogeneous. The CSC hypothesis may resolve the issue: it emphasizes that only a tiny population of cancer cells has the capability to produce phenotypically heterogeneous cells in new tumours; other cells only have limited proliferative capacity. Accordingly these cells have stem-like properties, having the capability of self-renewal and multipotency of differentiation, and thus are called CSCs [11–15]. However, this hypothesis is controversial and has been challenged by recent studies [16, 17], which argue for a long-standing cancer model, well known as clonal evolution [8, 17–19]. The clonal evolution model claims that normal cells mutate and generate abnormal offspring that also mutate, forming a mass of genetically varied cancer cells. At least two hits of oncogenic mutation may be required [4, 20]. Practically, clonal evolution as a mechanism appears to underlie CSC development [2].
Recently we have experimentally identified a new type of cancer cell from murine lymphoma [21], representing an early stage of CSC development but similar to pre-cancer in clinical origin, having the potentials of both benign and malignant differentiation. Therefore we named these cells ‘pCSCs’[2]. The pCSCs have the features of both normal and malignant (cancer) stem cells (Table 1). Regardless of their origin, multiple genetic alterations reflect that the pCSCs have evolutionarily undergone multi-step oncogenic mutations [2, 17]. The development of pCSCs appears to be regulated by a GS cell protein, called Piwil2 [2], which might subvert gene expression including ES cell genes in TICs [2].
1.
Functional comparison between NSCs, pCSCs and CSCs
| Self-Renewal | Multi-potency of Differentiation | Genomic instability | differentiation induced cell death (DICD) | Piwil2 | Tumourigenesis in recipients | ||||
|---|---|---|---|---|---|---|---|---|---|
| Benign | Malignant | ||||||||
| NSCs | + | + | – | – | – | – | – | ||
| pCSCs | + | + | + | + (MIN) | + | +++ | SCID but not BMR mice | ||
| CSCs | + | – | + | +/++? (MIN/CIN?) | –/+? | ++ | SCID & IC mice | ||
MIN: micro-satellite instability; CIN: chromosomal instability.
The terminology of TIC and CSC has been used interchangeably [22, 23]. Since a cancer may develop from the cells with accumulated, mismatching-repaired DNA damages or oncogenic mutations induced by cell stress or carcinogens [24, 25], a TIC should denote a carcinogen-stimulated progenitor that is either abortively or fully developed into CSCs during tumourigenesis. In this review, the term TICs is used practically to denote the cells with oncogenic mutations prior to developing into pCSCs to avoid the conceptual confusion between TICs, pCSCs and CSCs. Theoretically all cells that are hit by carcinogens have the potential to develop into TICs. Practically the cellular process of TIC → pCSC → CSC → cancer should parallel the histological process of hyperplasia/metaplasia (TIC) → pre-cancerous lesions (pCSC) → malignant lesions (CSC → cancer) [2, 3, 26–32].
Although the CSC hypothesis has attracted many cancer researchers during the last few years, the understanding of CSCs is still limited particularly because it has been difficult to isolate CSCs at the single-cell level based on their phenotype and to establish clonal CSC lines. Thus, the cellular and molecular mechanisms underlying CSC development remain elusive. The establishment of a series of stable, clonal pCSC and CSC lines in our laboratory may shed light on CSC research ([2] and L. Chen et al. unpublished data. Herein, I review the progress of CSC research and discuss the potential cellular and molecular mechanisms underlying the initiation and progression of CSCs based on lessons from pCSCs. To strengthen the discussion on the topic, we will introduce some unpublished important observations from my laboratory.
Stem cells and cancer stem cells
There are several models for cancer development. Can these models be unified by the CSC hypothesis? The answer may be ‘yes’.
Properties of stem cells
Stem cells are characterized as the cells that can differentiate into multiple cell types and can maintain the multi-potency of differentiation through self-renewal. They play a crucial role in all aspects of biology, from the development of early embryos to the repair and maintenance of adult tissues. There are at least two types of stem cells: embryonic stem (ES) cells and adult tissue stem (ATS) cells. ES cells are the cells that are derived from the inner cell mass of an early-stage embryo, called a blastocyst. They are pleuripotent and give rise during development to cells of all three primary germ layers: ectoderm, endoderm and mesoderm [33]. A number of transcription factors are required for the maintenance of pleuripotency, such as Oct4, Rex1, Sox2, and TDGF1 [33]. Interestingly these factors have been frequently detected in various types of cancer [34–38].
Unlike ES cells, ATS cells that reside in various tissues in foetal and adult human and animals are multi-potent and can differentiate into tissue-committed cell types, such as haematopoietic stem cells (HSCs) in the bone marrow (BM) and blood, neuronal stem cells in the brain, hepatic stem cells in the liver, and GS cells in the testis and ovary [39–41]. GS cells may be pleuripotent [41]. ATS cells maintain a continuous supply for tissue repair throughout adult life through a process termed self-renewal. Although ATS cells are essentially not pleuripotent, increasing data suggest that they have the potential for transdif-ferentiation [42–44].
Self-renewal is a type of cell division or proliferation specifically used in reference to stem cells. Self-renewed stem cells maintain their property of multi-potency of differentiation, and indefinitely supply pro-genitors required for replenishing relevant tissues. The progenitors may have a multi-potent capacity of differentiation, such as multi-potent progenitors (MPP) in BM [45–47], but they have little or no self-renewal capacity. Thus, self-renewal is a critical feature of stem cells. When stem cells divide, one or both ‘daughter’ cells can retain the properties of parent cells, rather than differentiate into progenitors. Under physiological conditions, stem cells maintain a small but stable and highly efficient pool in tissues (Fig. 1). For example, the HSC pool comprises only about 0.01% of marrow cells, but supplies more than 1 × 109 blood cells of various types per day [48].
1.
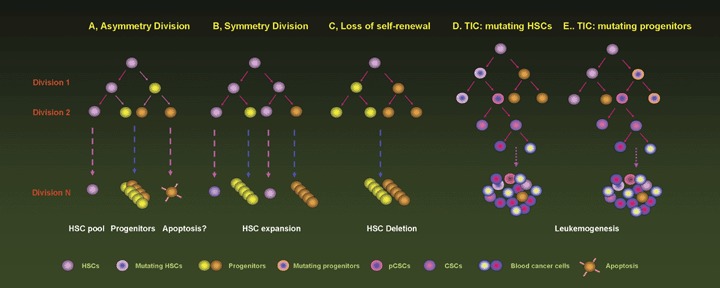
Self-renewal of stem cells and development of CSCs. HSCs maintain their homeostasis via asymmetry (A) and symmetry division (B). In homeostatic environment, HSCs divide asymmetrily to supply progenitors required for replenishment of blood. Once the injured HSCs lose the capacity of self-renewal (C), the healthy HSCs divide symmetrily. Once homeostasis is recovered, HSCs turn off programs for symmetry division. The mutating HSCs (TICs) may develop into pCSCs and CSCs (D), whereas the mutating progenitors (TICs) can acquire the capacity of self-renewal, developing into pCSCs and CSCs (E). Eventually the progenies of CSCs lose control in proliferation, occupying the space for normal HSCs and progenitors. HSC: haematopoietic stem cell; TIC: tumor-initiating cell.
To maintain homeostasis of the stem cell pool, stem cells usually divide asymmetrically. Asymmetric cell division produces two daughter cells that exhibit distinct fates: one self-renewed daughter stem cell remains the same as parent cells, while the other may differentiate into progenitor cells or undergo programmed cell death (apoptosis). The progenies further differentiate into lineage-restricted progenitors, such as common myeloid (CMP) and lymphoid progenitors (CLP) in the haematopoietic system [45–47]. With hierarchical differentiation, HSCs gradually lose their capability of self-renewal. While the stem cell pool is reduced, stem cells also divide symmetrically, or proliferate, to expand or recover the reduced pool. In this case, the two daughter cells retain the same properties as the parent cell. If the stem cells lose the capacity of self-renewal, the stem cell pool will be withered, leading to diseases (Fig. 1).
The fate of stem cells is determined by environmental cues referred to as the stem cell niche, which consists of stromal or accessory cells, cytokines and developmental growth factors [39, 49, 50]. Physiologically the balance between self-renewal and differentiation of stem cells is strictly regulated by the stem cell niche [49, 51]. The property of self-renewal of stem cells is also called ‘stemness’, which is controlled by a number of stemness genes some of which are yet to be determined [52–56]. Interestingly the genes that regulate self-renewal of ATS cells are oncogenic, too, and can be detected in various types of cancer cells. The best studied is Bmi-1, an oncogene encoding a polycomb group transcription factor that is required for self-renewal of normal stem cells (NSCs) and CSCs [14, 57, 58]. However, ATS cells such as HSCs appear not to express ES cell-related genes such as Oct4, Rex1, Sox2 and TDGF1 [2].
Cancer stem cell hypothesis
As introduced above, there are several models describing cancer development, such as the stochastic model and the clonal evolution model [4, 18, 20, 59]. The mainstay of these models is that they essentially involve multiple genetic alterations, thus converging to the model of clonal evolution [18]. These models claim that all cancer cells have the capability to reconstitute a new tumour [17]. However, the CSC hypothesis emphasizes that only a small population of stem-like cancer cells can reconstitute new tumours with all the cell types of the original tumour, while other cancer cells have limited capacity for proliferation and lack multi-potency of differentiation [14]. However, most important is that the CSC hypothesis should explain the entire process of tumour development (Fig. 2). In the clinic, pre-cancer, an important stage of cancer development, is highly heterogeneous and reversible [3, 27, 29, 30]. Accordingly pre-cancer should be mediated by pCSCs rather than CSCs, and pCSCs should have the potential for regression or progression, such as the capacity of benign and malignant differentiation as we have observed in animals [2].
2.
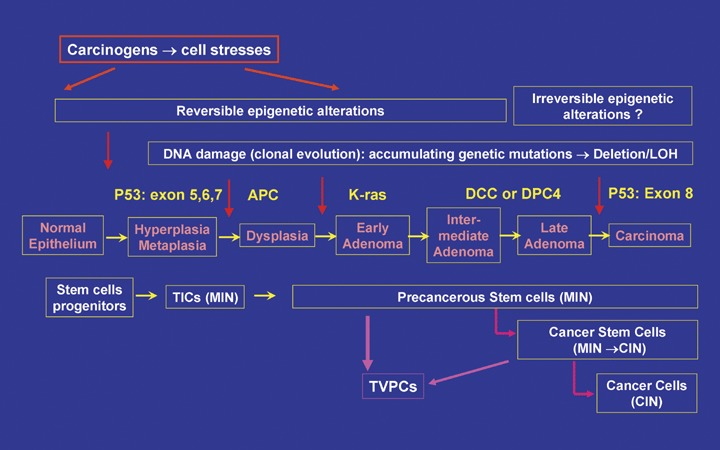
The cancer stem cell hypothesis. Cancer is developed from CSCs that are derived from a TIC insulted by car-cinogens. The TIC can be a stem cell or a progenitor cell and the latter can acquire stemness when progressing to pre-cancerous lesion. With the accumulation of epigenetic and genetic alterations, TICs develop into pCSCs, which have the potential for benign and malignant differentiation depending on environment cues. A qualitative mutation of onco-genes or tumor suppressor genes in pCSCs may render the loss of benign potential of differentiation and commitment to CSCs. The CSC can develop into a tumour with cellular heterogeneity because of its capacity of self-renewal and multi-potency of differentiation, as well as reconstitute a new tumour distant from original tumours. Histologically the proliferation of TICs may result in hyperplasia and metaplasia, the progression of TICs to pCSCs may be responsible for dysplasia or pre-cancer, and the commitment of CSCs ultimately leads to irreversible adenocarcinoma or carcinoma. Moreover, pCSCs and CSCs may also serve as the precursors of tumour stromal components, such as the pre-cursors for tumour vasculogenesis: TVPCs. The cancer stem cell hypothesis explains the whole process of tumour development from a TIC to tumour at the histological, cellular, and molecular levels. Here gastric intestinal cancer is used as a cartoon model [4, 139, 140]. TICs: tumour initiating cells; LOH: Loss of heterozygosity; MIN: micro-satellite instability; CIN: chromosomal instability; TVPCs: tumour vasculogenic stem/progenitor cells.
The concept of the CSC hypothesis is not novel, but remains to be precisely defined. Experiments spanning several decades have shown that only a small subset of cancer cells can form a new tumour when transferred into a new recipient. The clonal nature of these cells implicated in various malignancies led to the postulation of CSCs in the past by several laboratories [11, 31, 60–62]. However, the concept of CSCs was first experimentally documented for human acute myelogenous leukemia (AML) in 1994 [63]. Not until 2003 had CSCs been isolated from solid cancers, including cancers of breast, brain, prostate, colon, ovarian, and pancreas [12, 22, 23, 64–66], although the corresponding stable cell lines have not yet been established in vitro. Recently we have experimentally defined the property of pCSCs [2], rendering possible the integration of the CSC hypothesis with various models of cancer development.
While clonal evolution dissects the molecular basis of cancer development, the CSC hypothesis pinpoints the same cellular origin of the primary, metastatic and recurrent cancer. The gap between these two models can be resolved with identification of pCSCs [2]. The existence of pCSCs in tumours strongly implies that CSC can be derived from the precursors through a mechanism of clonal evolution, or that the target cells of clonal evolution are the pre-cursors of CSCs. Our preliminary studies suggest that the progression of pCSCs to CSCs is associated with hierarchical genetic alterations, a process resembling clonal evolution [2]. Therefore, the concept of clonal evolution may be integrated into the CSC hypothesis: CSCs are a small population of stem cell-like cancer cells, derived from a precursor undergoing clonal evolutionary pre-cancerous mutations. Whether pCSCs progress to CSCs depends on the effect of environmental cues on the clonal evolution [2].
Like ATS cells in tissues, CSCs represent a rare cell population in tumours. They have stem-like properties, reconstituting new tumours with all the cell types presented in the tumour of origin [12, 22, 23, 64, 65]. It should be noted that the stem-like property of pCSCs and CSCs does not mean that they are exactly the same as NSCs with regard to the capacity of self-renewal and multi-potency of differentiation. Their capacity for self-renewal is impaired and multi-potency of differentiation is incomplete, compared to NSCs [2]. While CSCs have been ascribed to metastasis and recurrence of cancer, and considered to be anti-cancer-drug resistant despite of controversies [14, 67, 68], pCSCs exist particularly within the established tumour [2]. The CSC hypothesis may explain why cancer cannot be eradicated by vigorous therapy [69]. Elucidation of molecular mechanisms underlying CSC development would provide novel therapeutic targets for cancer.
Development of pre-cancerous and cancer stem cells
Are CSCs derived from a diseased stem cell or from a progenitor that has acquired stem-like properties during cancer development? Current studies indicate that both are possible. Do pCSCs represent a distinct stage of developing CSCs or are they the same as CSCs? At present pCSCs may be considered as a precursor of CSCs with a clonal evolutionary relationship to the CSCs.
Origin of pre-cancerous and cancer stem cells
While pCSCs are considered as the precursors of CSCs, the ultimate origin of CSCs has not been completely resolved. Based on limited literature, the origin of CSCs can be ATS cells, progenitors or actively replenished proliferating cells such as the precursors of epithelial cells [4]. This may be true. Since multiple genetic mutations are required for cell transformation, sufficient cell cycles are necessary for accumulating the DNA-damage-induced mutations [62, 70]. Both ATS cells and progenitor cells are pro-liferating cells, and therefore go through sufficient cell cycles to accumulate oncogenic mutations during their life. The ATS cells are long-lived cells going through relatively few cell cycles, and may be the primary targets of accumulation of oncogenic mutations [22, 60, 62, 71]. However, the progenitor cells are actively proliferating and differentiating cells, some of them may also have sufficient cell cycles to accumulate oncogenic mutations within the relatively short term compared with ATS cells. This may explain why hyperplasia, metaplasia and dysplasia are frequently observed in the basal cell layer of epithelioid tissues, such as cervix and breast. As a result, the progenitor cells (TICs) may acquire stem-like properties, developing into pCSCs and CSCs. For an example, in blast-crisis chronic myelogenous leukemia (CML), the granulocyte-macrophage progenitors are able to retrodifferentiate into leukaemic stem cells (LSCs) through acquiring the capacity of self-renewal [72]. Therefore, CSCs may be derived from stem cells or proliferating progenitors (Fig. 1).
It is important to further elucidate which types of cells are most susceptible to oncogenic mutations induced by carcinogens. ATS and tissue-uncommitted stem cells might be more susceptible to developing into pCSCs and CSCs than other progenitors [73], and the pCSC might be more plastic than CSCs. Stem cell-derived pCSCs might transdifferentiate into various types of cancer cells [2, 74]. For example, Helicobacter infection-induced gastric CSCs appear to be transdifferentiated from BM HSCs or progenitors [74]. The hepatoid cells derived from haematopoietic pCSCs might retrodifferentiate to pCSCs or CSCs in the presence of the appropriate environmental cue [2] (Fig. 3). Overall, we consider CSCs a subset of cancer cells possessing stem-like properties, and pCSCs are the precursors of CSCs regardless of their origin.
3.
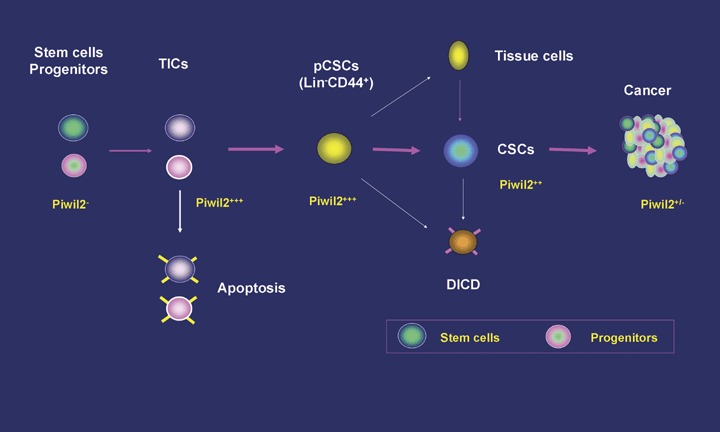
The model of cancer stem cell development. TICs that are adult stem cells or proliferating progenitors insulted by carcinogens may develop into pCSCs or die of programmed cell death during accumulation of epigenetic and genetic alterations. The fates of pCSCs are determined by environmental cues, and they either develop into CSCs or differentiate into benign tissue cells. Both pCSCs and CSCs are susceptible to differentiation-induced cell death (DICD). Piwil2 and piwil2-regulated genes (PRG) such as ES cell genes are subverted in pCSCs, contributing to the stemness of pCSCs, albeit abnormal.
Initiation and progression of pre-cancerous stem cells
Since the CSC hypothesis was revived, CSCs have been experimentally identified in haematopoietic and solid cancers [12, 22, 64, 65, 74, 75]. However, how CSCs are initiated from stem or progenitor cells is largely unknown. Genetic studies have revealed that a malignancy such as human colorectal cancer, a classical genetic model of tumourigenesis [4], undergoes multistep epigenetic alterations and oncogenic genetic mutations [4, 59, 76–78]. Both epigenetic and oncogenetic alterations may occur throughout the colorectal cancer development, from tumour initiation (hyperplastic polyp; HP), pre-cancer (adenomatous polyp; AP) to cancer (adenomatous carcinoma; Ad-ca). For epigenetic alterations, HPs demonstrated DNA hypermethylation in some specified markers [79] but had no significant difference in the global DNA methylation compared to normal mucosal tissues (Shen et al., unpublished data). In the pre-malignant (AP) and malignant (Ad-ca) lesions, global DNA was progressively hypomethylated, while the estrogen receptor (ER)-α gene was hypermethylated in the same manner. Importantly, the global DNA hypomethylation and ER-α gene hypermethylation were synchronically reversed by non-steroidal anti-inflammatory drugs (NSAIDs) such as cyclooxygenase-2 (Cox-2) selective inhibitor celecoxib in AP but not in Ad-ca, suggesting that the epigenetic alterations between pre-cancer and cancer are fundamentally different in responding to anticancer therapy (Fig. 2).
Similarly, genetic alterations may also occur early in some but not all HPs, such as the mutation of oncogene BRAF or KRAS[80]. The loss of heterozy-gosity (LOH) of the APC (adenomatous polyposis coli) gene, which is located on the human 5q21-q22 chromosome, appears to be responsible for epithelial dysplasia (AP) of the colon. The dysplastic lesion progresses to early adenoma with aberrant DNA methylation. The early adenoma advances to intermediate adenoma because of the mutation of the oncogene KRAS, a member of the Ras gene family, which is located on the 12q12.1 chromosome. The mutation of DCC or DPC4/SMAD4 located at the 18q chromosome further drives the pre-malignant lesion to late adenoma. The progression of the late adenoma to carcinoma has been shown to be associated with p53 mutation [4, 59, 76–78]. Among these stages, the dysplasia and early adenoma, but not later adenoma, represent reversible pre-cancerous lesions (Fig. 2). These pre-cancerous lesions, like in other organs such as breast and cervix [27, 81], have the potential to either regress to normal tissue or progress to cancer, depending on the quality of accu-mulated mutations.
It should be noted that the mutation of the so-called oncogene does not necessarily result in cell transformation. Increasing data have revealed that many oncogenes such as Ras seem also to act as watchdogs against carcinogens. The fates (senesces, apoptosis, proliferation or transformation) of Ras-activated cells are dependent on its dose and cellular context [82]. While the multi-potency of differentiation of NSCs is well programmed and accurately regulated, the differentiation programs are supposedly randomly activated in pCSCs and CSCs upon interaction with the tumourigenic niche [2, 83, 84].
According to the CSC hypothesis, multi-step histological and molecular alterations in colorectal cancer implicate the existence of pCSCs. In other words, the formation of pre-cancerous lesions should be mediated by pCSCs (Fig. 2). In PTEN-deficient mice, intestinal stem cells proliferate excessively, resulting in intestinal polyposis, a typical pre-cancerous lesion of colon cancer [28], suggesting that the PTEN-deficient intestinal stem cells act as pCSCs. The identification of ‘cancer-initiating cells’ in patients with colon cancer further supports the hypothesis, although whether these cells are pCSCs and/or CSCs have not been clearly defined [64]. Despite the increased understanding of molecular events of tumour initiation, the early molecular signature common to all types of cancer has not been defined. Thus, how is a TIC changed into a pCSC? The molecular mechanism underlying the switch is unknown. To elucidate this mechanism, it will be critical to directly isolate pCSCs or establish more pCSC lines from either animal or human pre-cancerous lesions.
Clonal evolutionary relationship of pre-cancerous stem cells to cancer stem cells
Isolation of pCSCs from established tumours as well as stem-like cancer cells from human tumour cell lines [2, 66, 85] suggests that pCSCs persist in various stages of tumour development ([2, 21] and L. Chen et al., unpublished data). Interesting questions are what is the relationship between pCSCs and CSCs in an established tumour and are pCSCs distinct from CSCs? In a setting of an established tumour, pCSCs may dynamically provide precursors of CSCs, amplifying the pool of terminal cancer cells. As a result, the tumour then contains many populations with different genetic mutations [2].
According to the CSC hypothesis (Fig. 2), while CSCs are supposed to inevitably reconstitute new tumours in the recipients [1], pCSCs may not necessarily reconstitute the tumours when transferred to recipients. The outcome of the transplant depends on environmental niches [2]. Additional genetic mutations may occur when pCSCs progress to cancer [2]. We have noticed that the karyotype of pCSCs are relatively stable compared to differentiated or terminal cancer cells [2]. While all the pCSC clones isolated from the same host exhibited the same karyotype, each differentiated cancer cell clone from the same mouse exhibited a remarkably different karyotype. The karyotype of the pCSCs were diploid with multiple chromosomal translocations, whereas the differentiated cancer cells had various aneuploid kary-otypes among the clones ([2] and L. Chen et al., unpublished data). This suggests that chromosomal segregation is randomly disturbed during cell cycles when pCSCs progress to cancer cells. Consistent with this notion, few additional variable genetic alterations occurred when pCSCs progress to cancer [2]. These data suggest that pCSC clones stochastically evolved to CSCs with only a few additional genetic alterations. The notion is further supported by the observations in human and animal breast carcinoma. In a genetically engineered mouse (GEM) model of human ductal carcinoma, pre-malignant lesions that have major variable molecular and genetic changes with significant phenotype developed into invasive carcinoma only with few additional genetic changes [30, 86]. In human beings, CD44+ CD24− CSCs from individual breast tumours were clonally related but not always identical to CD44− CD24+ cells [17], suggesting that few additional genetic changes rendered CD44+ CD24− CSCs into CD44− CD24+ cancer cells [12, 17]. Taken together, clonal evolution may occur throughout the process of TICs → pCSCs → CSCs → differentiated cancer cells.
Characteristics of pre-cancerous versus cancer stem cells
The clonal evolutionary relationship between pCSCs and CSCs does make it difficult to phenotypically discriminate pCSCs from CSCs but does not obscure functional distinctiveness between pCSCs and CSCs.
Are there demarcations between pre-cancerous and cancer stem cells?
Clinically all types of cancer seem to have a lengthy pre-cancer stage, which is histologically distinct from cancer [3, 27]. The pre-cancer is reversible and may regress or progress to cancer. Once a pre-cancer develops into an invasive cancer, the latter is irreversible and often fatal. It has been shown, however, that the majority of cancer phenotypes can be demonstrated early in the pre-cancerous lesions in the GEM model [3, 27], consistent with the notion that CSCs are hierarchically developed from pCSCs [2, 14, 75]. Thus, the difference between pCSCs and CSCs may be minimal in phenotype, but distinct especially in biological function [2, 21].
Ideally, a comparison between pCSCs and CSCs with regard to the phenotype, epigenetic and genetic alterations, and the capacity of tumour reconstitution may generate substantial information to distinguish pCSCs from CSCs. However, while CSCs have been identified in various types of human cancer [12, 22, 64–66, 74, 75], only few pCSC lines or CSC lines have been established from these cancers, hampering their characterization. In addition, it would be difficult to distinguish human pCSCs and CSCs using tumour reconstitution assay in animals as we have done for murine pCSCs and CSCs [2]. In the murine system, pCSCs and CSCs can be discriminated based on their capacity for tumourigenesis in the animal models with different environmental cues [2] (Table 1). The current human CSC xenotransplantation models do not in practice discriminate CSCs from pCSCs [12, 22, 64, 65, 74, 75]. Thus, animal models with a competent human immune system are required for the human CSC researcher, although it is a difficult task to establish such a model. At present, investigation of pCSCs and CSCs of animals may shed light on human pCSCs and CSCs [2].
Mainstay of pre-cancerous stem cells: the potential for both benign and malignant differentiation
In our laboratory, we have experimentally defined pCSCs, which have the potential for both benign and malignant differentiation, depending on environmental cues [2]. This can be a functional demarcation between pCSCs and CSCs. The pCSC-derived benign cells may not be typically ‘normal’. They can be subnormal but not malignant or tumourigenic. Both pCSC and pCSC-derived benign cells might remain quiescent or dormant in tissue or undergo apoptosis because of differentiation-induced cell death (DICD) [2]. The quiescent benign cells, like parental pCSCs, might retrodifferentiate into CSCs, resembling a ‘time bomb’ waiting to explode once tumourigenic niches are formed (Fig. 3).
Once pCSCs have progressed to CSCs or cancer cells, they should have lost the capacity of benign differentiation (Table 1; Fig. 3). The functional definition for pCSCs appears to be reliable, because we have successfully established a CSC clone (326T) from a mouse with thymoma, which does not have the potential for benign differentiation (L. Chen et al., unpublished data).
A tumourigenic assay, therefore, is important for distinguishing pCSCs from CSCs (Table 1; Fig. 4). Three murine models, including severe combined immunodeficient disease (SCID), lethally irradiated bone marrow-reconstituted (BMR) and immunocompetent (IC) mice, may be useful and necessary for the assay [2]. SCID mice provide an environment defective in immunosurveillance [87], allowing pCSC expansion in vivo to form tumours; BMR mice provide a recovering immune system and regenerative niches to check pCSCs that are progressing to cancer cells and to drive pCSCs to replenish the regenerating tissues, respectively; and IC mice provide a fully competent immune system to combat the pCSCs that are progressing to CSCs. Thus, SCID mice are used to test the tumourigenic capacity of the tested cells; BMR mice can be used to determine the capacity of benign differentiation and self-renewal of pCSCs, and IC mice can be used to determine whether the cells tested are pCSCs or CSCs (Fig. 4). In fact, while pCSCs are checked by immunosurveillance [2], CSCs are able to evade immunosurveillance and develop into tumours in IC mice (L. Chen et al., unpublished data).
4.
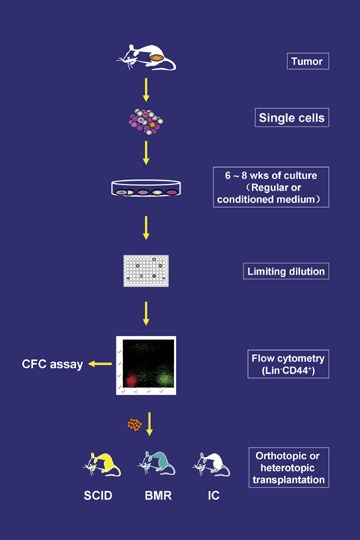
Establishment of clonal pCSC and CSC lines. Single tumour cells are cultured in regular or conditioned medium until tumour cells grow out and demonstrate a stable cell line. Then the cells are cloned by limiting dilution. The cloned cell lines are analysed phenotypically by flow cytometry, and the Lin – CD44+ cells with ambiguous stem cell markers are subjected to in vitro CFC assay and in vivo functional assay. SCID: SCID mice; BMR: lethally irradiated bone marrow-reconstituted mice; IC: immunocompetent mice.
Conserved versus volatile phenotypes for pre-cancerous and cancer stem cells
Traditionally, phenotype is used to define the lineage, population or subset of cells. The phenotype of human CSCs appears to be variable probably because of their tissue of origin and random genomic alterations [12, 22, 64, 65, 74, 75]. Most CSCs have been characterized using the putative markers that identify normal ATS cells [22, 23, 63, 64, 88].
The putative markers for CSCs appear to be variable with the tissue of origin, except for CD44. In human AML, CSCs or LSCs exhibited the phenotype of CD34+ CD38−, indistinguishably from normal HSCs [63, 75]. In breast cancer, CD44+ CD24−/low Lin− cells have CSC activity [12, 63]. In multiple myeloma, about 5% of the cells in the bulk population were syndecan-1 (CD138) negative, and possessed in vitro clonogenic potential [89]. Stem cells from brain, prostate, and colon cancers have been shown to express the cell surface marker CD133 [22, 23, 64, 88]. Laboratory tumour cell lines, the majority of which are transplantable and contain a small sub-population of stem-like cells that is enriched in a side population (SP), expressed a high level of CD44 [85, 90, 91]. Usually the SP has stem cell activity [66, 85, 90] and is associated with the expression of ATP-binding cassette (ABC) transporter Bcrp1/ABCG2 [92]. However, ABCG2− stem-like cancer cells seem to be more primitive than ABCG2+ CSCs [90]. The pCSC clones established in our laboratory did not express ABCG2 [2], in contrast to a CSC clone (326T), which we have recently established (unpublished data). All the human CSCs identified so far might contain pCSCs. Whether ABCG2 expression is a marker that distinguishes pCSCs from CSCs requires further investigation [90].
The haematopoietic murine pCSC lines established in our laboratory are CD45− c-kit− Sca-1− Lin− (KSL)− CD44+ CD24−[2], subtly distinct from the CSC clone (326T): CD45+ KSL− CD44+ CD24+ . The density of CD44 on CSCs was relatively lower than on pCSCs (L. Chen et al., unpublished data). This observation suggests that the adhesion molecule CD44, a transmembrane receptor that is over-expressed in most primary cancers and associated with tumour progression [93–96], is ubiquitously expressed on pCSCs and CSCs. CD117 (c-kit) and Sca-1 (stem cell antigen-1), which are the markers for HSCs [97–99], have been implicated as markers for progression of various types of cancer [100–102]. In contrast to HSCs [97, 98], both the pCSCs and CSCs isolated from murine lymphoma and thymoma, respectively, did not express CD117 and Sca-1, both of which, however, were up-regulated when pCSCs were progressing to cancer [2], consistently with clinical observations of human cancers [100–102]. Therefore, haematopoietic pCSCs and CSCs appear to share a common phenotype: KSL− Lin− CD44+; however, no definitive marker(s) so far can be used to discriminate between pCSCs and CSCs. Broadly the common phenotypic features between haematopoietic pCSCs and CSCs may be Lin− CD44+ with variable levels of stem-like markers. More pCSC and CSC lines are needed to verify the stem-like phenotype of cancer cells: Lin− CD44+ . Once the stem-like phenotype of cancer cells has been determined, in vivo tumourigenesis assays appear to be a gold standard to discriminate pCSCs from CSCs [2].
Genetic instability of pre-cancerous and cancer stem cells
Logically the molecular signature of pCSCs and CSCs should be determined by a spectrum of subverted activation or suppression of oncogenes caused by genetic instability. Two forms of genomic instability have been described for colorectal cancers: chromosomal instability (CIN) and micro-satellite instability (MIN/MSI) [103–105]. CIN in tumour cells invariably leads to aneuploidy, while cells with MIN are usually near diploid [106]. Either CIN or MIN may lead to reverted activation or suppression of oncogenes. Although MIN and CIN were initially reported as mutually exclusive, the association of CIN or MIN with certain tumour types remains controversial [107]. Analysis of karyotype of pCSCs and cancer cells from the same mouse revealed that all the pCSC clones analysed were pseudodiploid with multiple chromosomal translocations, whereas cancer cells were usually aneuploid [2]. Interestingly, a CSC clone (326T) recently established in our laboratory is also diploid, but the frequency of chromosomal translocations was not increased compared to pCSCs (L. Chen et al., unpublished data), suggesting that the malignancy of cancer cells is determined by qualitative (oncogenic) rather than quantitative (non-oncogenic) alterations in genome. Certainly increased genomic instability is believed to be correlated with the increased probability of oncogenic mutations. CIN, usually defined as an accelerated rate of chromosome missegregation during cell division, is thought to result from mitotic defects at checkpoint genes [103]. Taken together, the genetic instability of pCSCs and CSCs may exhibit as MIN, which might transit to CIN when pCSCs and/or CSCs progress to cancer (Fig. 2). Thus, MIN (diploidy or pseudodiploidy) might be a genomic feature of pCSCs and CSCs, whose ability to transit to CIN may account for the malignant potential and poor prognosis of cancer. To verify the hypothesis, more pCSC and CSC lines need to be established and analysed in animals and human beings.
The transition from MIN to CIN may increase the gene dose of mitotic defects in a given pCSC or CSC, promoting its proliferation and differentiation. In fact, the low frequency of pCSCs and CSCs in tumours reflects their limited capacity for proliferation, compared to other cancer cells [2, 12, 23, 63]. Thus, the ploidy of cancer cells might be one of the parameters for distinguishing pCSCs or CSCs from their progenies, although the genomic features are indistinguishable between pCSCs and CSCs.
Subverted expression of embryonic and germ line stem cell genes in pre-cancerous stem cells
Since pCSCs and CSCs have the capacity of self-renewal similarly to NSCs, the molecules that regulate the self-renewal of stem cells may also be utilized by pCSCs and CSCs. Like ATS cells such as BM CD34− Lin− , pCSCs expressed all the stem cell-related and tumourigenesis-related genes tested except for ABCG-2 [2, 90], including Bmi-1[108], Notch-1[109], Fzd2[110], Fzd5[111], β-catenin[112], Smo[113], c-Myc[114], Flt3[115], Bcl-2[116] and stat-3[52]. Importantly pCSCs also expressed ES cell- and GS cell-related genes, including POUF1/Oct-4[117], TDGF1/Cripto[118], Zfp42/REX1[119], SOX2[120] and piwil2[121], which are not expressed in normal ATS cells [2]. However, only the GS cell gene piwil2 is stably expressed in all clones of pCSCs examined. ES cell-related genes Pouf1/Otc4, TDGF1, and Zfp42/REX1, which are required for maintenance of pleuripotency, were ambiguously expressed. These ES cell-related genes might confer ‘pleuripotency’ upon pCSCs, because the haematopoietic pCSCs were able to transdifferentiate into various types of benign tissue cells in BMR mice or even in SCID mice while coexisting with cancer cells [2].
In contrast to pCSCs, CSCs expressed fewer ES cell-related genes and significantly lower levels of piwil2. Among the ATS cell-related and tumourigenesis- related genes mentioned above, Smo and Notch-1 were significantly up-regulated in CSCs (L. Chen et al., unpublished data). Although the significance of the difference in expression of these genes is not yet clear, the difference certainly implicates a distinct molecular signature between pCSCs and CSCs. The establishment of clonal pCSCs and CSCs in our laboratory ensures further precisely defining the molecular signatures of pCSCs versus CSCs.
Contributions of pre-cancerous and cancer stem cells to tumour malignancy
One of the factors that determine tumour malignancy is the growth rate of parenchymal cancer cells, which are supported by stromal components such as vasculature. Our study suggests that pCSCs may serve as precursors of both parenchymal and stromal cells.
Can pre-cancerous and cancer stem cells serve as the precursors of tumour stromal cells and tumour vasculogenic stem/progenitor cells?
A tumour is de facto a neoplastic organ that loses control of growth. Its malignancy is essentially determined by its capacity for growth, angiogenesis, invasiveness, and metastasis. As an entity of neo-organ, a tumour consists of various components, including stromal components such as fibroblastic stromal cells and vasculature and parenchymal cancer cells. However, the origin of the stromal components is essentially unknown. The prevailing concept is that stromal cells and vascular endothelial cells are derived from normal precursors [8], which are stimulated by growth factors such as vascular endothelial growth factor (VEGF) produced by tumour cells including CSCs [122]. However, the concept of tumour angiogenesis has been challenged by clinical investigation, which demonstrated that the tumour vasculature can be derived from tumour cells, called vasculogenic mimicry [123–125], although this is controversial [126]. Tumour angiogenesis denotes a process of the formation of tumour vasculature from pre-existing blood vessels or circulating endothelial progenitor cells (EPCs), which is mainly mediated by the VEGF family produced by host or tumour cells [127, 128]. In contrast, tumour vasculogenesis is a process of the formation of new blood vessels from tumour-derived TVPCs, which can be mediated by the same factors for angiogenesis. The discovery of pCSCs implicates that tumour stromal cells and vasculature may be derived from the same precursors of cancer cells. pCSCs have been shown to develop into hepatoid cells and endothelial-like cells in regenerative tissues [2]. This finding prompted us to explore whether pCSCs contribute to tumour vasculature. In support of the hypothesis, we have observed that pCSCs not only produce VEGF and angiogenic factors but also serve as TVPCs (Fig. 2). In the pCSC-derived tumours, tumour neovascularization seems mainly mediated by the mechanism of tumour vasculogenesis rather than by angiogenesis, suggesting that tumour vasculogenesis is crucial for tumour growth [193]. Encouraged by this finding, we have successfully established a cell line from a murine lymphoma, composed of mesenchymal stem cells, lymphoid blast cells and endothelial cells (Q. Yan et al., unpublished data). Experiments are in progress to determine whether these cell components are derived from the same precursor. Thus, like normal organs, which are replenished by tissue stem cells, the tumour stromal components may be replenished by pCSCs and/or CSCs, although a normal precursor could not be excluded.
Does the frequency of pre-cancerous and cancer stem cells in tumours determine the growth rate of cancer?
The frequency of pCSCs and CSCs in tumours may be highly variable; for example, the frequency of CSCs in human colon cancer varies 10 times or more between individuals [23]. It is not clear whether the variation is associated with the cancer prognosis. However, in breast cancer, CD44+ CSCs seem to correlate with poor clinical outcome [17], suggesting that the frequency of CSCs might be a predictor of cancer prognosis.
In contrast to the putative CSCs in solid tumours [12, 23], the frequency of LSCs is extremely low in the human AML, approximately 0.1–1 per million AML blasts [63, 75]. The cell capable of initiating human AML in non-obese diabetic mice with severe combined immunodeficiency disease (NOD/Scid) mice possesses the differentiative and proliferative capacities and the potential for self-renewal expected of an LSC [75]. By tracking individual human LSCs in NOD/Scid mice serially transplanted with AML cells, LSCs were found not to be functionally homogeneous but, like the normal HSC compartment, comprise distinct hierarchically arranged LSC classes [129]. In this experimental model, two important features for LSCs/CSCs were revealed: some LSCs are quiescent or divided rarely and underwent self-renewal rather than commitment after cell division; and normal developmental processes are not completely abolished during leukaemogenesis [129]. These features implicate that some so-called LSCs might be at the pre-cancerous stage of development.
Consistently, pCSCs isolated from mouse lymphoma had very low engraftment in the BMR and blastocyst-complemented chimera (BCC) mice [2]. The low engraftment of pCSCs may be related to their unique property, in that pCSCs are distinct from both NSCs and CSCs. Essentially the programs of self-renewal and multi-potency of differentiation in pCSCs are impaired. The defects might be lethal for some renewed individual cells in a non-tumourigenic niche. Thus, pCSCs might not be as efficient as NSCs in engraftment in the presence of non-tumourigenic cues and not as ready as CSCs to evade immune surveillance [2]. In contrast, they are potently engrafted and dramatically expanded in immuno-compromised recipients or tumourigenic niches. The growth rate of pCSC-derived tumours in SCID mice is much higher than that of the tumours derived from differentiated cancer cell lines of the same origin [2], strongly suggesting that both frequency of pCSCs and environmental niches determine the growth rate of tumour.
Are pre-cancerous and cancer stem cells associated with metastatic cancer?
It is well received that metastasis or recurrence usually occurs in the patients with invasive or advanced cancers, but rarely in those with pre-cancerous lesions. However, metastatic tumour cells usually are multi-potent with characteristics of embryonic pro-genitors and are reprogrammable in embryonic environments [130], resembling pCSCs [2]. Based on the CSC hypothesis, the pre-cancerous lesion is believed to be mediated by pCSCs. However, the pre-cancerous lesion lacking metastasis does exclude the possibility of pCSCs migrating or metas-tasizing to distant organs once a pre-cancer pro-gresses to invasive cancer. Actually in some cases, metastasis may occur at the same time or before the primary cancer is discovered. This might be accounted for by pCSC and CSC metastasis. After arriving in a secondary site the metastasized pCSCs might proliferate and differentiate into CSCs and cancer cells, undergo benign differentiation or apoptosis, be eliminated by the mechanism of immune surveillance, or remain as solitary dormant cells, depending on environmental niches [2, 29, 81]. Activation of dormant pCSCs and CSCs at secondary sites may account for the recurrence of cancer [131]. It is possible that the dormant pCSCs and CSCs could be activated by intensive cancer therapy such as chemotherapy and radiotherapy [69, 132]. Elucidation of these potential mechanisms will greatly benefit cancer therapy. In fact, we have observed that pCSCs migrated away from the site of injection in SCID mice could differentiate into benign or malignant cells at secondary sites, such as in the liver [2].
Understanding of the mechanisms underlying pCSC and CSC migration and colonization is important for developing strategies to prevent cancer metastasis. Recently it has been demonstrated that some tissue stem cells such as neural stem cells can selectively migrate to the site of cancer – the phenomenon of moving towards the diseased area is called pathotropism [133]. This implicates an important biological mechanism underlying cancer metastasis. Do pCSCs and CSCs have the ability of pathotropism? It is worthwhile to clarify the issue. For example, is the site of inflammation a destination of pCSC and CSC migration? If so, what is the driving force for the migration? As discussed above, pCSCs, CSCs, and various types of differentiated cancer cells may coexist in established tumours. It is likely that pCSCs and CSCs are the origin of metastatic cancer, which not only provide the precursors of differentiated cancer cells, but also supply the need for the buildup of cancer stromal components such as tumour neovasculature. Better characterization of the capability and destination of pCSC and CSC migration as well as the niches required for their expansion and differentiation will facilitate the understanding of the mechanisms underlying cancer metastasis.
Clonal evolution underlies the development of TICs → pCSCs → CSCs
A critical question is how the oncogenic program is initiated or activated in TICs? Epigenetic alterations induced by carcinogens in TICs may be an initial step of carcinogenesis. Is clonal evolution contradictory to the CSC hypothesis? The answer may be ‘no’. In contrast, increasing evidence indicates that clonal evolution is a feature of TICs → pCSCs → CSCs, and can be integrated into the context of the CSC hypothesis.
Tumour initiation (TIC → pCSC): epigenetic alterations precede genetic mutations
As discussed above, the CSC hypothesis holds that CSCs can give rise to a tumour in tumourigenic niches (Fig. 2). The CSCs are supposed to be developed from a TIC (which can be a stem or progenitor cell) with the capacity of self-renewal although this ability is impaired. However, little is known about how a TIC develops into CSCs, although extensive investigations have revealed that human tumourigenesis is a complex, multi-step process often requiring concordant expression of a number of genes [4–6, 134].
Regardless of the origin of CSCs, the initial step of carcinogenesis may start from epigenetic changes of the affected cells or TICs [135], which are induced by cellular stresses or carcinogens. In vitro studies revealed that exposure of mammary stem/progenitor cells to estrogen led to aberrant methylation of tumour suppressor genes and clonal expansion of progeny epithelial cells (Tim H. Wang, personal communication). These epigenetic changes prevailed in primary breast tumours, and were even detected in ‘normal breast tissue’ adjacent to tumour [136]. The phenomenon, which is called ‘field or geographical effect’, was also observed in colon cancer [137, 138]. These observations implicate that epigenetic alteration precedes genomic alteration. The ‘field effect’ might be associated with the aberrant methylation of proliferating epithelial progenitors in the area, or the existence of pCSCs, which, like normal ATS cells, could replenish normal tissue surrounding the tumour. It would be interesting further to investigate whether genetic mutations occurred in the breast stem/progenitors exposed to estrogen in vitro.
The immediate outcome of epigenetic changes is probably genetic mutations of oncogenes. These mutations, however, may not necessarily lead to malignant changes of the TICs. In some experimental models, it is clear that two hits of mutation produce only a benign precursor lesion and additional genetic events are necessary for a malignancy [20]. Analysis of human pre-cancerous and cancerous gastric lesions revealed that quality rather than quantity of an oncogene mutation determined the outcome of the mutation. While p53 mutation was found in gastric metaplasia, dysplasia and carcinoma at comparable frequency, the malignancy of the mutation was determined by the exons affected. Mutation of exons 5, 6 and 7 of p53 is usually associated with pre-cancerous lesions, whereas exon 8 is closely linked with gastric cancer. LOH or deletion of p53 and APC (anaphase-promoting complex) is always associated with malignant phenotypes: poor differentiation, vascular invasion, lymph node metastasis, and short survival time [139, 140]. These observations suggest that non-deletional mutation versus LOH of tumour suppressor genes may determine whether pCSCs could progress to CSCs in gastrointestinal cancer.
Taken together, the benign differentiation potential of pCSCs may explain the development of pre-cancerous lesions, whereas the loss of the potential may lead to the progression of pCSCs to CSCs, the latter being responsible for the malignant lesion. It would be interesting to investigate whether the LOH of tumour suppressor genes is a turning point for pCSCs progressing to CSCs. A CSC may have gone through cellular stress (carcinogens) → epigenetic alteration → non-deletional multiple genetic mutations → LOH of tumour suppressor genes (Fig. 2). As discussed earlier, CIN/aneuploidy is likely found in the progenies of CSCs. This model might lead us to elucidate the mechanisms underlying CSC initiation.
Tumour progression (pCSC → CSC): additional dynamic genetic alterations
In tumourigenic environments, pCSCs undergo phenotypic, morphologic and genomic changes while forming tumours [2]. The phenotype of pCSCs derived from lymphoma changed from CD45− KSL− to CD45+ KSL+[2]. In this case, the lineage markers including CD3, B220, CD11b, Gr-1, Ter-119 and NK1.1 were all up-regulated concomitantly with up-regulation of HSC markers c-kit and Sca-1 [98]. c-kit, a transmembrane tyrosine kinase receptor encoded by proto-oncogene c-kit, and Sca-1, a glycosylphos-phatidylinositol- linked cell surface protein, have been identified as markers of cancer progression in various types of non-haematopoietic cancers [100–102]. Thus, c-kit and Sca-1 are unlikely the predictors for CSCs. Consistently, the 326T clone of CSC expressed neither c-kit nor Sca-1 (L. Chen et al., unpublished data).
Morphological changes were also observed while pCSCs were progressing to cancer in vivo and in vitro. Various types of cancers derived from pCSCs were found in SCID mice. Upon differentiating cytokine stimulation, some pCSCs exhibited differentiated morphology, although the differentiation is eventually aborted [2].
Although it is not clear whether the phenotypic and morphologic changes of pCSC-derived cells are associated with genomic alterations, it is conceivable that genomic alteration is dynamic when pCSCs progress to CSCs or cancer. Increased deletional mutation may be associated with increasing malignant potency of pCSCs in the tumourigenic niches depending on the genes involved, such as those involved in cell cycling, cell senescence (telomerase), cell adhesion and migration as well as tumour neovascularization [4–6, 134]. Although the multiple genomic alterations are random, the quantitative alterations may eventually lead to qualitative changes. For example, the pCSC (clone 2C4)-derived tumour cell line exhibited an additional deletion of chromosome 10 and an add(14) in 90.6% (29/32) of the cells, while the other three cells had the dup(14) and a third copy of the del(15), thus showing karyotypic evolution in both clones of this line compared with the parental pCSC line [2]. These changes occurred dynamically in vivo within only a few weeks of transfer, whereas no significant change was observed for all clones of pCSCs during >3 years of in vitro maintenance (our unpublished observations). These observations indicate that additional genetic alterations were required when pCSCs progress to cancer, although the precise genes affected by the evolution need to be explored further.
The dynamic genomic alterations of pCSCs progressing to cancer reflect a clonal evolutionary mechanism underlying CSC development. Clonal evolution has been considered as the mechanism underlying tumour development including metastasis before the CSC hypothesis was revived [17, 19], and is continuing to challenge the CSC hypothesis. The validity of the CSC hypothesis has been called into question by Shipitsin et al., based on the genetic difference between CD24+ and CD44+ cells in breast cancers [17]. However, the same authors recognized that the clonal evolution of genome involved intratumoural heterogeneity [17]. This suggests that CD24+ cancer cells could be progenies of CD44+ cancer cells, similarly to the pCSCs, which exhibited additional genetic changes along with phenotypic changes when they were progressing to cancer [2]. Thus, clonal evolution is not contradictory to the CSC hypothesis, and can be included in the context of CSC hypothesis underlying CSC development. Since the pCSCs that we tested are clonal lines derived from the single-cell level [2], the conclusion is more reliable compared with that from bulk cell populations.
Clonal evolution and niche effect
Whether a TIC or pCSC evolves to CSCs depends on environmental cues [2, 17]. In other words, is the progression of pCSCs to cancer not necessarily intrinsic? Tumourigenic niches have important impacts on pCSCs developing into CSCs or cancer [2, 81]. The factors that affect DNA remodelling, rearrangements and/or chromosomal segregation, during cell cycles are critical for the evolution. These factors can induce further aberrant DNA remodelling, rearrangement and/chromosomal segregation in pCSCs, eventually leading to uncontrollable cell growth. Two recent studies have suggested that bad niches may drive good stem cells into bad ones [141–143]. Similarly, the pCSCs, a bad seed in a ‘good’ bed such as in BMR mice, may develop into benign tissue cells, whereas they develop into malignant cells in a ‘bad’ bed such as in SCID mice [2]. In BMR mice, tissue injury might provide an environment for pCSCs to transdifferentiate into corresponding tissue cells, while the recovering immune system can effectively eliminate malignant progenies of pCSCs, if any develops. In contrast, SCID mice provided tumourigenic niches in the absence of an adaptive immune system [2]. Certainly the precise niches affecting pCSC development need to be further characterized at the cellular and molecular levels.
Molecular pathways for the development of pre-cancerous and cancer stem cells
Are there distinct molecular pathways between pCSCs, CSCs and NSCs? Current studies suggest that ES and GS cell-related genes are subverted in pCSCs and CSCs. Among them, piwil2 may be important for the development of TICs → pCSCs → CSCs in various types of cancer.
Expression of embryonic, germ line and adult tissue stem cell-related genes in pre-cancerous and cancer stem cells
As discussed earlier, the self-renewal, pleuripotency and multi-potency of ES, GS and ATS cells are maintained and regulated by distinct genes. The pCSCs not only expressed ATS cell-related genes such as Bmi-1, Notch-1and Smo, but also ES cell-related genes Oct-4, TDGF-1, and REX1 and GS cell-related gene piwil2 [2]. While the levels of Bmi-1 and Notch-1 are comparable to CD34− Lin− HSCs, the level of Smo appears to be lower than in ATS cells [2], but up-regulated in CSCs (L. Chen et al., unpublished data). The expression of ES cell-related genes in pCSCs is unstable, probably because of the high sensitivity of these genes to culture conditions or environmental cues. Piwil2 is stably expressed in pCSCs at a high level [2], suggesting that piwil2 might be a reliable marker for pCSCs. Consistently, piwil2 was ubiquitously detected in human pre-cancerous lesions of various organs [193]. ES and GS cell-related genes are usually undetectable in normal ATS cells, despite few reports implicating that ATS cells may also express ES cell-related genes [36]. Whether the expression is due to TICs and/or pCSCs in the individuals tested is an interesting issue.
CSCs, like pCSCs, expressed ES, GS and ATS cell-related genes as well. However, the expression pattern of these genes seems to be less stable compared to that observed in pCSCs. As compared to pCSCs, CSCs expressed fewer ES cell genes within an individual clone, up-regulated Smo transcripts, and down-regulated piwil2 transcripts ([2] and L. Chen et al., unpublished data). The ectopic expression of ES and GS cell genes in pCSCs and CSCs can be considered as ‘atavism’ of ATS/progenitor cells, which confer upon ATS cells the properties of ES cells, such as indefinite growth or immortalization.
Molecular pathways for self-renewal of pre-cancerous and cancer stem cells
Since pCSCs and CSCs have the capacity of self-renewal similarly to stem cells, the molecules that regulate the self-renewal of stem cells are also involved in this process in pCSCs and CSCs. Extra-cellular signals such as Notch, Wnt and Hedgehog have been found to link with self-renewal and maintenance of HSCs [112, 144–148]. In fact, pCSCs expressed comparable levels of transcripts of the Notch-1 and β-catenin, though relatively low levels of Smo compared to CD34− Lin− HSCs [2] . However, Notch-1 expression was enhanced in CSCs (L. Chen et al., unpublished data). Notch signalling controls cell fate decisions during embryonic development and ATS cell self-renewal and differentiation. In cancer, Notch-1 is frequently deregulated, serving as either an oncogene [149, 150] or a tumour suppressor [150, 151]. Thus, its role in the development of pCSCs and CSCs needs to be determined.
Bmi-1, a member of the polycomb protein group (PcG) gene, appears to play a critical role for the self-renewal of NSCs and CSCs [55, 56, 108, 152, 153]. The PcG genes, which are transcriptional repressors, have an essential role in embryogenesis, regulation of the cell cycle and lymphopoiesis. Increased expression of Bmi-1 promotes HSC self-renewal, probably through enhancing symmetrical cell division of HSCs and mediating a higher probability of inheritance of stemness through cell division. Loss-of-function analyses revealed that among the PcG genes, absence of Bmi-1 is preferentially linked with a profound defect in HSC self-renewal [55]. Introducing genes known to produce AML into Bmi-1–/– haematopoietic stem cells from foetal livers induced AML with normal kinetics. However, Bmi-1–/– LSCs from primary recipients were unable to produce AML in secondary recipients. The result indicates that Bmi-1 is also required for self renewal of LSCs in the murine AML model [153].
Since the molecules governing the self-renewal of ATS cells were also identified in pCSCs and CSCs [2, 60, 154], it is likely that pCSCs and CSCs use the same molecular pathways as ATS cells to maintain their capability of self-renewal [60, 154]. However, ectopic expression of ES and GS cell genes is unique for pCSCs and CSCs. Although the piwi protein is required for self-renewal of stem cells in various organisms [155], piwil2 was not required for the self-renewal of adult HSCs, because the number and repopulation activity of HSCs from the BM of piwil2-disrupted mice was not affected [121]. Thus, the role of piwil2 in the self-renewal of pCSCs and CSCs remains elusive.
Role of piwil2 in the development of pre-cancerous and cancer stem cells
Increasing data suggest that all tumours may arise from a cell undergoing cellular stress → epigenetic alterations → genetic mutations → inactivation of tumour suppressors and/or activation of oncogenes. However, it has been difficult to define a genetic marker for CSC development, because the onco-genic genes expressed in CSCs are usually over-lapped with those expressed in normal ATS cells, such as Bmi-1 and c-Myc[2, 108]. Although ES cell-related genes, such as Oct-4, TDGF-1, REX1, and SOX2 may be subverted in pCSCs and CSCs, they are ambiguously and unstably expressed in these cells [2] or cancer cells [35–37]. In contrast, the GS cell gene or protein of piwil2 has been stably detected in pCSCs [2] and pre-cancerous lesions [193], suggesting that piwil2 may be unique for pCSC and CSC development.
piwil2 (alias mili in mouse or hili in humans), which is exclusively expressed in GS cells of testis [121, 156], is a member of the PIWI/Argonaute gene family [157]. The genes of the piwi family are defined by highly conserved PAZ (Piwi/Argonaute/Zwille) and Piwi (P-element induced wimpy testis) domains and play important roles in stem cell self-renewal [155], gametogenesis [121], small RNA-mediated gene silencing [158], and translational regulation in various organisms [158].
The PAZ domain is found only in Piwi/Argonaute proteins and Dicer. Argonaute proteins are the catalytic components of the RNA-induced silencing complex (RISC) [159, 160], the protein complex responsible for the gene silencing via a mechanism of RNA interference (RNAi) [161]. Dicer contains two RNase III domains and one PAZ domain, and is a ribonuclease in the RNase III family that cleaves double- stranded RNA (dsRNA) and pre-micro-RNA (miRNA) into short double-stranded siRNA [162, 163]. Dicer catalyzes the first step of RNA interference to generate siRNA and initiates the formation of the RISC. Argonaute proteins in the RISC serve as endonucleases to degrade messenger RNA (mRNA) via binding siRNA fragments, which are subsequently complemented to mRNAs [164].
The piwi is a protein domain homologous to piwi proteins in Drosophila and, like the PAZ domain, is present in PIWI/Argonaute gene family proteins [157], which bind and cleave RNA [162, 164]. The proteins containing a PIWI PAZ domain (PPD) are called PPD proteins. In addition to RNAi, PPD proteins might play a role as cell-cycle regulators independent of RNAi and a role in chromosomal remodelling [164]. Piwi proteins, a subfamily of PIWI/Argonaute gene family, contain two important members, piwil1 (alias miwi in mouse or hiwi in human beings) and piwil2 [157].
There is no evidence so far that piwi protein is a component of the RISC [165, 166]. It has been reported that piwi proteins, unlike Argonaute proteins, seem not to be associated with micro-RNA (miRNA) or siRNA [166]. However, piwi proteins do have target RNA cleavage activity, independent of Dicer [165]. Both piwil1 and piwil2 were found to bind a novel class of RNA called piwi-interacting RNA (piRNA) or repeat-associated small interfering RNAs (rasiRNAs) in mammal testis [165–169]. They may silence the selfish genetic elements, such as retro-transposons, in the germ line cells of testis [167–171].
Dysregulated piwi protein expression appears to be associated with tumourigenesis [2, 172, 173]. Although piwil1 was up-regulated in seminomas [172], it was not detected in pCSCs [2]. In contrast, piwil2 was stably expressed in pCSCs and pre-cancerous lesions [2, 193]. The piwil2 was also detected in various human and animal tumour cell lines with variable levels (L. Chen et al., unpublished data and [173]), probably related to the number of pCSCs and CSCs in each line. piwil2, which is mapped to chro-mosome 8 in human beings and 14 in mouse and encodes a 3069-nucleotide mRNA, and a 971- amino-acid protein (109.5 kD), is exclusively expressed in GS cells of testis [121, 156, 157]. It is likely that ectopic expression of piwil2 may contribute to the development of pCSCs and CSCs, because knocking down of piwil2 mRNA led to contained pCSC proliferation in vitro, and consistent over-expression of piwil2 in BM cells led to enhanced proliferation [2]. Over-expression of piwil2 in NIH3T3 cells led to inhibition of apoptosis and promotion of proliferation via the Stat3/Bcl-X(L) signalling pathway [173].
However, the role of piwil2 in pCSC and CSC development may be more complex than we expected. For example, while ectopic expression of piwil2 in Lin− BM cells promoted their proliferation, the event was followed by the formation of an embryonic body-like colony, and eventually these cells became apoptotic ([2] and Y. Yin et al.,unpublished data). The morphologic changes of piwil2-expressing cells were fre-(c) quently observed before cell death, suggesting a disturbed process of cell differentiation. In an autoreactive T cell-induced pre-cancer model, piwil2 was ectopically activated in pre-cancerous lesions of various organs, suggesting that piwil2 rapidly responded to cellular stress caused by carcinogens or inflammatory cytokines, and might be expressed before a TIC progresses to pCSCs. Moreover, several iso-forms of piwil2 have been identified and cloned in our laboratory, which might play differential roles in pCSC and CSC development. Importantly piwil2 regulated the expression of a number of genes, including genes of ES cells, ATS cells and oncogenes (Y. Yin et al., unpublished data). These observations suggest the diverse roles of piwil2 during pCSC and CSC development, which are probably related to its interaction with piRNA [167, 168].
The mechanisms underlying piwil2 regulation of pCSC development are largely unknown. Since other PIWI/Argonaute family members are responsible for RNAi, DNA remodelling and cell cycling under physiological condition, ectopic expression of piwil2 may implicate that these pathways are disturbed by piwil2 in the TICs or pCSCs, leading to tumourigenesis. This may be an important model to explore common molecular pathways for cancer development of all origins.
Do embryonic stem cell-related genes confer upon pre-cancerous stem cells the capacity of transdifferentiation?
We have reported that pCSCs can transdifferentiate into various types of tissue cells in BMR mice, suggesting that these cells are uncommitted stem-like cells. One the other hand, these pCSCs were of haematopoietic origin [2, 21], suggesting that they may have already been committed to the haematopoietic lineage. How can a pre-determined pCSC transdifferentiate into other tissue cells? This is an interesting issue, and the apparent contradiction may be reconciled by the ‘atavic’ expression of ES cell genes.
Transdifferentiation has been observed in various types of ATS cells [42, 43, 174–178] and probably in CSCs [35, 74], although there have been great controversies [44, 179, 180]. Our observations suggest that the transdifferentiation may be related to sub-verted expression of ES cell genes in pCSCs, which confers upon pCSCs the ‘pleuripotency’ of differentiation, albeit incomplete [2]. Certainly, the transdifferentiation is pathologic, and may not be reproducible with normal counterpart ATS cells [179]. ES cell genes have been detected in normal and bulk cancer cells [36, 181]. While there is no doubt that the cancer cells may contain pCSCs and CSCs, whether these normal cells contain TICs or pCSCs needs to be elucidated.
The experimental models for cancer stem cell research
CSC research is in its infancy. Since identification of CSCs in leukemia in 1994 [63] and in solid tumours in 2003 [12], we experimentally defined pCSCs in 2007 [2]. Practically distinguishing pCSCs from CSCs may be critical for differentiating diagnosis between pre-cancer and cancer as well as for early detection, prevention and treatment of cancer. However, experimental models for pCSC and CSC research are limited [8]. We would like to share our experiences with the reader (Fig. 4).
The roadblock for discrimination of human pre-cancerous and cancer stem cells
The criteria for defining pCSCs and CSCs should include their unique phenotype, molecular signature and potential for reconstituting tumour. The pheno-type and molecular signature of pCSCs may be indistinguishable from CSCs, because of their hierar-chical development [129]. It is conceivable that the CSCs isolated from human haematopoietic and solid cancers might be a mixture of pCSCs and CSCs [12, 23, 63]. Owing to the low frequency and phenotypic heterogeneity of pCSCs and CSCs in tumours, it would be difficult to phenotypically discriminate pCSCs from CSCs, or CSCs from cancer cells [17, 182]. Thus, it is necessary to establish a series of clonal cell lines for better characterization of pCSCs and CSCs at the cellular and molecular levels.
In the murine system, the potential of pCSCs and CSCs for reconstituting tumour can be tested in SCID, BMR and IC mice [2]. However, it would be difficult to test the potency of human pCSCs versus CSCs, because human tumour xenografts are not histocompatible in BMR and IC mice. Thus, the SCID mice with MHC-matched human immune system are required. At present this is a hurdle that is impossible to run over. However, the difficulty may be overcome by elucidation of niche requirement of pCSCs versus CSCs. Currently human CSCs are verified relying on their capacity of tumour reconstitution in the original (orthotopic) tissue, such as breast CSCs in the mammary gland [12] and brain CSCs in the brain [22]. However, this model does not distinguish pCSCs from CSCs. Pre-cancer appears to be distinct from cancer in engraftment after transplantation [3, 27]. In an animal model of human breast cancer [27, 29], while malignant tissues may grow out both orthotopically and heterotopically (ectopically), pre-cancer tissues grow out orthotopically rather than heterotopi-cally after transplantation [29]. This suggests that pre-malignant and malignant tissues require distinct growth niches. Thus, human pCSCs might be distinguishable form CSCs by their suitability for orthotopic or heterotopic xenotransplantation.
Approaches to establishing clonal pre-cancerous and cancer stem cell lines
To elucidate the cellular and molecular mechanisms underlying CSC development, clonal pCSC and CSC lines are required. By using the cell lines in combining with SCID, BMR, BCC and/or IC mouse models [2], one can investigate the environmental cues that have an effect on the tumourigenesis of pCSCs and/or CSCs, the mechanisms of tumour regression or progression underlying immunoediting [87, 183], and the molecular signatures of pre-cancer and cancer. It might be difficult to investigate the molecular mechanisms underlying the development of pCSCs and CSCs in the light of the known dynamical genetic instability (DGI) of pCSCs and CSCs in vivo[2]. However, the DGI provides otherwise an opportunity to screen genes involving in the progression of pCSCs → CSC → cancer, because MIN in clonal pCSCs and CSCs are relatively genetically stable than CIN in other cancer cells ([2] and L. Chen et al., unpublished data). With the identification of the in vivo biological phenotypes, one will be able to define relevant molecular signatures using various optimal genetic and biochemical approaches.
It is well known that not all tumours can give rise to immortalized cell lines. This may be related to the phenomenon that almost all tumour cells die during cultivation. Based on our experience, tumour cells may proliferate during the first few days of culture followed by massive apoptosis. Only a small number of tumour cells are resistant to spontaneous apoptosis in cultures. These cells may grow out in 6–8 weeks of cultivation [21], likely containing pCSCs and/or CSCs ([2] and L. Chen et al., unpublished data).
We have developed two cultivation systems to establish clonal pCSC and CSC lines ([2, 21] and L. Chen et al., unpublished data). First, we establish immortalized tumour cell lines by long-term cultivation of bulk tumour cells [21]. During 6–8 weeks of cultivation, fibroblast-like cells grow out followed by non-adherent cells. It should be noted that one should not throw away the cultures within 6 weeks of cultivation if few cells are observed. In some cases, regular medium may not support tumour cell growth. To overcome this difficulty, we have developed a condition medium supporting HSC growth [2]. We have successfully established a CSC clone (326T) from a thymoma using the conditioned medium (L. Chen et al., unpublished data). For other types of tumours, relevant growth factors may be required but usually are not necessary. The immortalized cell lines should contain pCSCs and/or CSCs [2, 21, 85, 184]. Alternatively, the purified pCSCs or CSCs based on phenotype may be cultivated directly. Based on our experience, however, we suggest that the success rate might be very low, because of the lack of supporting cells required for stem-like cells to grow out in cultures.
Following establishment of immortalized cell lines, pCSCs and CSCs should be cloned and characterized through a three-step procedure (Fig. 4). First, tumour cell lines are cloned by limiting dilution and then, the resultant clones screened for stem-like cells by lineage exclusion using flow cytometry. pCSCs and CSCs do not express Lin-specific markers but are CD44+[2, 12]. Finally Lin− CD44+ clones with ambiguous expression of stem cell markers are subject to functional assay.
Phenotypic characterization and functional verification of pre-cancerous and cancer stem cells
Lin− CD44+ phenotype seems reliable for pCSCs or CSCs [2, 12]; other markers such as CD133 likely vary with cancer types [12, 22, 23]. It is unlikely that pCSCs and CSCs express a complete set of NSC markers. In contrast, some stem cell markers such as Sca-1 and c-kit [97, 98] may be detected in later-stage cancer cells [2, 100, 102]. Thus, stem cell markers are unreliable for pCSCs and CSCs, and may be used as complementary markers. The loss of some ATS cell markers may be an important feature of pCSCs and CSCs. The SP population of tumour cell lines may contain pCSCs and CSCs, but not all of them, because more primitive CSCs seem not to express ABCG2 [90]. Whether the ABCG2-negative population represents pCSCs needs further investigation [2].
Based on phenotype, one cannot discriminate pCSCs from CSCs. Self-renewal and incomplete multi-potency of differentiation is the stem-like properties of pCSCs and CSCs, whereas benign or malignant differentiation potential is a functional demarcation between pCSCs and CSCs [2] (Table 1; Fig. 3). While serial transplantation is a gold standard for self-renewal activity [2, 21], colony formation in soft agar or colony-forming cell (CFC) assays may also be used for screening of the clones for self-renewal activity [2, 184]. The ability of single cells to achieve both benign and malignant differentiation in vivo is a gold standard for pCSCs. However, while it is difficult to test the freshly isolated pCSCs at a single-cell level [2], clonal cell lines derived from a single cell should be sufficient to verify the existence of pCSCs at the single-cell level [2].
Clinical implications: early detection, prevention and treatment via targeting of pre-cancerous and cancer stem cells
Since the CSC hypothesis has been revived in the last few years, the preventive and therapeutic targets for cancer have been shifting from terminal cancer cells to CSCs. Biomarkers unique for cancer but common to all types of cancer may exist in pCSCs and CSCs, which could be developed into novel tumour vaccines. Traditional chemotherapy has limitations in prevention and treatment of cancer. With the progress in CSC research, efficient immunoprevention and immunotherapy would be developed, despite the current setback in cancer immunotherapy [185, 186].
A novel strategy integrating early detection and prevention for the cure of cancer
The prevention and cure of cancer remains a major public health issue. Currently there is no safe and cost-effective therapeutic mode for human cancer, because of the lack of approaches to early detection and prevention. To prevent or cure cancer, an ideal therapeutic strategy should integrate approaches of early detection and prevention. TICs, pCSCs and CSCs are ideal targets for therapy. The ectopic expression of piwil2 and ES cell-related genes in pCSCs lends credence to early detection and prevention of cancer, because these genes were found to express at the very early stage of cancer development [2, 173, 193].
Traditionally, chemotherapy is widely used for cancer prevention and therapy. However, this therapeutic mode seems to be neither safe nor cost-effective. The fatal shortcomings of chemotherapy include that (a) the molecules targeted are required as well for normal development and/or survival of tissue cells; (b) each anti-cancer drug only targets a limited population of patients with the same cancer because the targeted molecules are not common for all cases of the cancer; and (c) most of the anti-cancer drugs inevitably breach immune systems, leading to immune tolerance or immunodeficiency. Therefore, even if an anti-cancer drug that specifically targets a common biomarker for TICs, pCSCs, and/or CSCs might be ideal for prevention and treatment of cancer, immunoprevention and immunotherapy may be more safe and cost-effective than chemoprevention and chemotherapy by avoiding generation of drug-resistant CSCs [187].
Potential of piwil2 for early detection of cancer
Although a large number of biomarkers for cancer have been identified, none of them are common for all types of cancer especially at early stages of cancer development [95]. For example, HER2, estrogen receptor (ER), progesterone receptor (PR) or Ki67 have been used as markers for breast cancer diagnosis. However, the lack of sensitivity and specificity preclude the use of these markers for early detection of breast cancer [182]. Moreover, these markers are not exclusively expressed in malignant cells and thus they are not appropriate to be used as therapeutic targets of breast cancer. In contrast, piwil2, which was found stably expressed in pCSCs [2], was detected in various stages of breast cancer, including pre-cancerous lesions (Liu et al., unpublished data). Thus, piwil2 has much higher sensitivity and specificity than HER-2, ER, PR and Ki67 for early detection of breast cancer. In addition to breast cancer, piwil2 can be detected in various types of cancer [2, 173, 193], suggesting that piwil2 has the potential to be used as a common biomarker for early detection of cancer.
Potential of pre-cancerous and cancer stem cells as a target for immunoprevention
As discussed above, pCSCs and CSCs may exist in established tumours to replenish growing tumours [2, 21]. Blocking this replenishment might lead to a cure for cancer. Based on the CSC hypothesis, elimination of pCSCs and/or CSCs can prevent the development of cancer. Since the development of pCSCs was suppressed in IC mice [2], we suggested that pCSCs can be used as a tumour vaccine. Pre-immunization of syngeneic mice with pCSCs prevented tumour reconstitution by a syngeneic tumour cell line unrelated to the pCSCs even 8 months later. Importantly, piwil2-specific T cells were detected in the mice which rejected tumours. This finding suggests that immune response to piwil2 may prevent cancer development, and thus piwil2 has the potential to be used as a vaccine for immunoprevention of cancer. The novel concept of immunoprevention through targeting of pCSCs or pre-cancerous lesions was further supported by another cancer model, which demonstrated that a biological response modifier (BRM) called Kaovaccine TM could prevent lung cancer development in the SP-C/p53-273H mice transgenic with mutated human p53 (p53-273H) under the transcriptional control of the lung-specific human surfactant protein C (SP-C) promoter [188], which spon-taneously developed lung adenocarcinoma after 4 months of age. Treatment of the transgenic mice with Kaovaccine TM at 12 months of age effectively prevented the development of lung cancer (J.-X. Gao et al., unpublished data).
Potential of pre-cancerous and cancer stem cells for the development of tumour vaccines
Large numbers of T-cell specific tumour antigens associated with testicular or embryonic antigens have been identified [118, 189–191]. However, immunotherapy based on these tumour antigens has encountered unexpected setback in translational studies. This may be because none of the antigens stably and ubiquitously express in CSCs, and most of the tumour models used for immunotherapy are not physiological with regard to the process of tumour development. The vaccine activity of pCSCs as discussed above implicates that cellular vaccines derived from pCSCs and CSCs might be more efficient in prevention and treatment of human cancer. Thus, it is highly prospective to develop mono- or multivalent tumour vaccines via targeting of the molecules uniquely expressed in pCSCs and CSCs. In addition to piwil2, pCSCs ambiguously expressed ES cell genes, such as Oct-4, TDGF-1 and REX1 [2]. ES cell gene products have been detected in various types of cancer [36, 118, 181, 192], and some of them such as SOX2 have demonstrated strong immunogenicity in patients with monoclonal gammopathy [192]. These interesting findings strongly implicate that tumour vaccines against pCSCs and CSCs might be safe and cost-effective in the prevention and treatment of cancer.
Concluding remarks
Tumours have been considered as neo-organs in the body. Tumour malignancy is determined essentially by the degree of uncontrollable growth, angiogenesis and the capacity for metastasis. As an entity of neo-organ, tumour consists of various components, including parenchymal cells, stromal cells and vasculature. Like normal organs, the components of which are replenished by tissue stem cells, tumour components may be replenished by stem-like cancer cells, that is, pCSCs and CSCs. Therefore, pCSCs and CSCs may be essential for the maintenance of tumour malignancy, which depends on the outcomes of the interaction between stem-like cancer cells with tumourigenic niches.
As an organ, tumour is more likely to mimic bone marrow, in which rare numbers of HSCs are the source for the maintenance of blood components. HSCs can give rise to a large number of blood cells every day, but they never exceed the threshold required for homeostasis. In an established tumour, there is no homeostatic balance between pCSCs/CSCs and cancer cells. Small numbers of pCSCs and CSCs may constantly give rise to various tumour components as long as the tumourigenic niche exists. Thus, pCSCs and CSCs are the primary targets for cancer therapy.
The discovery of pCSCs allows us to further explore the factors that are responsible for benign or malignant differentiation of a developing CSC. A series of pCSC and CSC lines are definitely required for elucidation of the biochemical and genetic events involving clonal evolution during tumour development. The focused research may unveil the common cellular and molecular pathways for TIC → pCSC → CSC → cancer. Especially identification of the molecular signature unique for TICs, pCSCs and CSCs, respectively, would lead to early detection, prevention and treatment of cancer.
Acknowledgments
I sincerely thank Drs. Rulong Shen and Sanford H. Barsky for their collaborative support in the cancer stem cell research, and Drs. Jim Van Brocklyn and Rulong Shen for their critical review of the manuscript. This work received supported from Strategy Initiative (SIG), 2006/2007, Immunology Program Award 2007 (OSUCCC) and American Cancer society IRG-112367.
Note added in proof
After this article was accepted for publication, the following article was published, supporting the view-point proposed in this review that the progression of precancerous stem cells to cancer is associated with the mechanism of clonal evolution: Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukaemia. Science 2008; 319: 336–9.
References
- 1.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Shen R, Ye Y, Pu XA, Liu X, Duan W, Wen J, Zimmerer J, Wang Y, Liu Y, Lasky LC, Heerema NA, Perrotti D, Ozato K, Kuramochi-Miyagawa S, Nakano T, Yates AJ, Carson Iii WE, Lin H, Barsky SH, Gao JX. Precancerous stem cells have the potential for both benign and malignant differentiation. PLoS ONE. 2007;2:e293. doi: 10.1371/journal.pone.0000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman JJ, Albores-Saavedra J, Bostwick D, Delellis R, Eble J, Hamilton SR, Hruban RH, Mutter GL, Page D, Rohan T, Travis W, Henson DE. Precancer: a conceptual working definition–results of a consensus conference. Cancer Detect Prev. 2006;30:387–94. doi: 10.1016/j.cdp.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Fearon ER, Vogelstein B. A genetic model for col-orectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 7.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 8.Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 2006;66:1891–5. doi: 10.1158/0008-5472.CAN-05-3450. [DOI] [PubMed] [Google Scholar]
- 9.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 10.Larrivee B, Niessen K, Pollet I, Corbel SY, Long M, Rossi FM, Olive PL, Karsan A. Minimal contribution of marrow-derived endothelial precursors to tumor vasculature. J Immunol. 2005;175:2890–9. doi: 10.4049/jimmunol.175.5.2890. [DOI] [PubMed] [Google Scholar]
- 11.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–82. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–5. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 16.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 17.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 19.Kuukasjarvi T, Karhu R, Tanner M, Kahkonen M, Schaffer A, Nupponen N, Pennanen S, Kallioniemi A, Kallioniemi OP, Isola J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–604. [PubMed] [Google Scholar]
- 20.Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol. 1996;122:135–40. doi: 10.1007/BF01366952. [DOI] [PubMed] [Google Scholar]
- 21.Gao JX, Liu X, Wen J, Zhang H, Durbin J, Liu Y, Zheng P. Differentiation of monocytic cell clones into CD8alpha(+) dendritic cells (DC) suggests that monocytes can be direct precursors for both CD8alpha(+) and CD8alpha(–) DC in the mouse. J Immunol. 2003;170:5927–35. doi: 10.4049/jimmunol.170.12.5927. [DOI] [PubMed] [Google Scholar]
- 22.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 23.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 24.Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007;6:1831–6. doi: 10.4161/cc.6.15.4516. [DOI] [PubMed] [Google Scholar]
- 25.Shimada M, Nakanishi M. DNA damage check-points and cancer. J Mol Histol. 2006;37:253–60. doi: 10.1007/s10735-006-9039-4. [DOI] [PubMed] [Google Scholar]
- 26.Bond JH. Colon polyps and cancer. Endoscopy. 2003;35:27–35. doi: 10.1055/s-2003-36410. [DOI] [PubMed] [Google Scholar]
- 27.Cardiff RD, Anver MR, Boivin GP, Bosenberg MW, Maronpot RR, Molinolo AA, Nikitin AY, Rehg JE, Thomas GV, Russell RG, Ward JM. Precancer in mice: animal models used to understand, prevent, and treat human precancers. Toxicol Pathol. 2006;34:699–707. doi: 10.1080/01926230600930129. [DOI] [PubMed] [Google Scholar]
- 28.He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, Wiedemann LM, Barrett TA, Hood L, Wu H, Li L. PTEN-deficient intestinal stem cells initi-ate intestinal polyposis. Nat Genet. 2007;39:189–98. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–305. [PubMed] [Google Scholar]
- 30.Namba R, Maglione JE, Davis RR, Baron CA, Liu S, Carmack CE, Young LJ, Borowsky AD, Cardiff RD, Gregg JP. Heterogeneity of mammary lesions represent molecular differences. BMC Cancer. 2006;6:275. doi: 10.1186/1471-2407-6-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sternlicht M, Mirell C, Safarians S, Barsky SH. A novel strategy for the investigation of clonality in pre-cancerous disease states and early stages of tumor progression. Biochem Biophys Res Commun. 1994;199:511–8. doi: 10.1006/bbrc.1994.1258. [DOI] [PubMed] [Google Scholar]
- 32.Wistuba II. Genetics of preneoplasia: lessons from lung cancer. Curr Mol Med. 2007;7:3–14. doi: 10.2174/156652407779940468. [DOI] [PubMed] [Google Scholar]
- 33.Koestenbauer S, Zech NH, Juch H, Vanderzwalmen P, Schoonjans L, Dohr G. Embryonic stem cells: similarities and differences between human and murine embryonic stem cells. Am J Reprod Immunol. 2006;55:169–80. doi: 10.1111/j.1600-0897.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 34.Xing PX, Hu XF, Pietersz GA, Hosick HL, McKenzie IF. Cripto: a novel target for antibody-based cancer immunotherapy. Cancer Res. 2004;64:4018–23. doi: 10.1158/0008-5472.CAN-03-3888. [DOI] [PubMed] [Google Scholar]
- 35.Monk M, Holding C. Human embryonic genes re-expressed in cancer cells. Oncogene. 2001;20:8085–91. doi: 10.1038/sj.onc.1205088. [DOI] [PubMed] [Google Scholar]
- 36.Tai MH, Chang CC, Kiupel M, Webster JD, Olson LK, Trosko JE. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26:495–502. doi: 10.1093/carcin/bgh321. [DOI] [PubMed] [Google Scholar]
- 37.Raman JD, Mongan NP, Liu L, Tickoo SK, Nanus DM, Scherr DS, Gudas LJ. Decreased expression of the human stem cell marker, Rex-1 (zfp-42), in renal cell carcinoma. Carcinogenesis. 2006;27:499–507. doi: 10.1093/carcin/bgi299. [DOI] [PubMed] [Google Scholar]
- 38.Dong C, Wilhelm D, Koopman P. Sox genes and cancer. Cytogenet Genome Res. 2004;105:442–7. doi: 10.1159/000078217. [DOI] [PubMed] [Google Scholar]
- 39.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 40.Johnson J, Canning J, Kaneko T, Pru JK, Tilly JL. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature. 2004;428:145–50. doi: 10.1038/nature02316. [DOI] [PubMed] [Google Scholar]
- 41.Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, Baba S, Kato T, Kazuki Y, Toyokuni S, Toyoshima M, Niwa O, Oshimura M, Heike T, Nakahata T, Ishino F, Ogura A, Shinohara T. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119:1001–12. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 42.Orkin SH. Stem cell alchemy. Nat Med. 2000;6:1212–3. doi: 10.1038/81303. [DOI] [PubMed] [Google Scholar]
- 43.Shen CN, Slack JM, Tosh D. Molecular basis of transdifferentiation of pancreas to liver. Nat Cell Biol. 2000;2:879–87. doi: 10.1038/35046522. [DOI] [PubMed] [Google Scholar]
- 44.Orlic D. BM stem cells and cardiac repair: where do we stand in 2004? Cytotherapy. 2005;7:3–15. doi: 10.1080/14653240510018028. [DOI] [PubMed] [Google Scholar]
- 45.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 46.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 47.Adolfsson J, Mansson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, Anderson K, Sitnicka E, Sasaki Y, Sigvardsson M, Jacobsen SE. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol. 1995;11:35–71. doi: 10.1146/annurev.cb.11.110195.000343. [DOI] [PubMed] [Google Scholar]
- 49.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–5. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 51.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Constantinescu S. Stemness, fusion and renewal of hematopoietic and embryonic stem cells. J Cell Mol Med. 2003;7:103–12. doi: 10.1111/j.1582-4934.2003.tb00209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 54.Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science. 2002;298:601–4. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- 55.Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, Van Lohuizen M, Nakauchi H. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–51. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 56.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 57.Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, Samans B, Berwanger B, Christiansen H, Lutz W. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–54. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175–9. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knudson AG, Jr, Strong LC, Anderson DE. Heredity and cancer in man. Prog Med Genet. 1973;9:113–58. [PubMed] [Google Scholar]
- 60.Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23:7164–77. doi: 10.1038/sj.onc.1207933. [DOI] [PubMed] [Google Scholar]
- 61.Barsky SH, Grossman DA, Ho J, Holmes EC. The multifocality of bronchioloalveolar lung carcinoma: evidence and implications of a multiclonal origin. Mod Pathol. 1994;7:633–40. [PubMed] [Google Scholar]
- 62.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 63.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 64.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 65.Zhang YQ, Kritzik M, Sarvetnick N. Identification and expansion of pancreatic stem/progenitor cells. J Cell Mol Med. 2005;9:331–44. doi: 10.1111/j.1582-4934.2005.tb00359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci USA. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michor F. Chronic myeloid leukemia blast crisis arises from progenitors. Stem Cells. 2007;25:1114–8. doi: 10.1634/stemcells.2006-0638. [DOI] [PubMed] [Google Scholar]
- 68.Michor F. Reply: the long-term response to imatinib treatment of CML. Br J Cancer. 2007;96:679–80. [Google Scholar]
- 69.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 70.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 71.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 72.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 73.Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp Hematol. 2002;30:896–904. doi: 10.1016/s0301-472x(02)00869-x. [DOI] [PubMed] [Google Scholar]
- 74.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring, Wang TC. Gastric cancer originating from bone marrow- derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 75.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 76.Knudson AG., Jr Hereditary cancers: from discovery to intervention. J Natl Cancer Inst Monogr. 1995;17:5–7. [PubMed] [Google Scholar]
- 77.Parada LF, Land H, Chen A, Morganstern J, Weinberg RA. Cooperation between cellular oncogenes in the transformation of primary rat embryo fibroblasts. Prog Med Virol. 1985;32:115–28. [PubMed] [Google Scholar]
- 78.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 79.Minoo P, Baker K, Goswami R, Chong G, Foulkes WD, Ruszkiewicz AR, Barker M, Buchanan D, Young J, Jass Extensive DNA methylation in normal colorectal mucosa in hyperplastic polyposis. Gut. 2006;55:1467–74. doi: 10.1136/gut.2005.082859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chan TL, Zhao W, Leung SY, Yuen ST. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003;63:4878–81. [PubMed] [Google Scholar]
- 81.Maglione JE, McGoldrick ET, Young LJ, Namba R, Gregg JP, Liu L, Moghanaki D, Ellies LG, Borowsky AD, Cardiff RD, MacLeod CL. Polyomavirus middle T-induced mammary intraepithelial neoplasia outgrowths: single origin, divergent evolution, and multiple outcomes. Mol Cancer Ther. 2004;3:941–53. [PubMed] [Google Scholar]
- 82.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent onco-gene- induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 83.Dingli D, Michor F, Antal T, Pacheco JM. The emergence of tumor metastases. Cancer Biol Ther. 2007;6:383–90. doi: 10.4161/cbt.6.3.3720. [DOI] [PubMed] [Google Scholar]
- 84.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66:4553–7. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 85.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA. 2004;101:781–6. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Namba R, Maglione JE, Young LJ, Borowsky AD, Cardiff RD, MacLeod CL, Gregg JP. Molecular characterization of the transition to malignancy in a genetically engineered mouse-based model of ductal carcinoma in situ. Mol Cancer Res. 2004;2:453–63. [PubMed] [Google Scholar]
- 87.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 88.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 89.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–6. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 91.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 93.Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthases: potential therapeutic targets in cancer. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:3–14. doi: 10.2174/1568006053005056. [DOI] [PubMed] [Google Scholar]
- 94.Nair KS, Naidoo R, Chetty R. Expression of cell adhesion molecules in oesophageal carcinoma and its prognostic value. J Clin Pathol. 2005;58:343–51. doi: 10.1136/jcp.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Agnantis NJ, Goussia AC, Batistatou A, Stefanou D. Tumor markers in cancer patients. An update of their prognostic significance. Part II. in vivo. 2004;18:481–8. [PubMed] [Google Scholar]
- 96.Nagano O, Saya H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004;95:930–5. doi: 10.1111/j.1349-7006.2004.tb03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang L, Bryder D, Adolfsson J, Nygren J, Mansson R, Sigvardsson M, Jacobsen SE. Identification of Lin(–)Sca1(+)kit(+)CD34(+)Flt3– short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105:2717–23. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- 98.Osawa M, Nakamura K, Nishi N, Takahasi N, Tokuomoto Y, Inoue H, Nakauchi H. in vivo self-renewal of c-Kit+ Sca-1+ Lin(low/–) hemopoietic stem cells. J Immunol. 1996;156:3207–14. [PubMed] [Google Scholar]
- 99.Wong SH, Lowes KN, Bertoncello I, Cook MJ, Simmons PJ, Kornberg AJ, Kapsa RM. Evaluation of Sca-1 and c-Kit as selective markers for muscle remodelling by non-hemopoietic bone marrow cells. Stem Cells. 2007;25:1364–74. doi: 10.1634/stemcells.2006-0194. [DOI] [PubMed] [Google Scholar]
- 100.Potti A, Ganti AK, Tuchman SA, Sholes K, Langness E, Koka V, Koch M. HER-2/neu and CD117 (c-kit) overexpression in patients with pesti-cide exposure and extensive stage small cell lung carcinoma (ESSCLC) J Carcinog. 2005;4:8. doi: 10.1186/1477-3163-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Demetri GD. Targeting c-kit mutations in solid tumors: scientific rationale and novel therapeutic options. Semin Oncol. 2001;28:19–26. [PubMed] [Google Scholar]
- 102.Xin L, Lawson DA, Witte ON. The Sca-1 cell surface marker enriches for a prostate-regenerating cell sub-population that can initiate prostate tumorigenesis. Proc Natl Acad Sci USA. 2005;102:6942–7. doi: 10.1073/pnas.0502320102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 104.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 105.Cardoso J, Molenaar L, De Menezes RX, Van Leerdam M, Rosenberg C, Moslein G, Sampson J, Morreau H, Boer JM, Fodde R. Chromosomal instability in MYH- and APC-mutant adenomatous polyps. Cancer Res. 2006;66:2514–9. doi: 10.1158/0008-5472.CAN-05-2407. [DOI] [PubMed] [Google Scholar]
- 106.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 107.Georgiades IB, Curtis LJ, Morris RM, Bird CC, Wyllie AH. Heterogeneity studies identify a subset of sporadic colorectal cancers without evidence for chromosomal or microsatellite instability. Oncogene. 1999;18:7933–40. doi: 10.1038/sj.onc.1203368. [DOI] [PubMed] [Google Scholar]
- 108.Raaphorst FM. Self-renewal of hematopoietic and leukemic stem cells: a central role for the Polycomb-group gene Bmi-1. Trends Immunol. 2003;24:522–4. doi: 10.1016/s1471-4906(03)00241-2. [DOI] [PubMed] [Google Scholar]
- 109.Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes Dev. 2005;19:2331–42. doi: 10.1101/gad.1337005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang HY. WNT-frizzled signaling via cyclic GMP. Front Biosci. 2004;9:1043–7. doi: 10.2741/1310. [DOI] [PubMed] [Google Scholar]
- 111.Ishikawa T, Tamai Y, Zorn AM, Yoshida H, Seldin MF, Nishikawa S, Taketo MM. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 2001;128:25–33. doi: 10.1242/dev.128.1.25. [DOI] [PubMed] [Google Scholar]
- 112.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 113.Sakurada K, Ohshima-Sakurada M, Palmer TD, Gage FH. Nurr1, an orphan nuclear receptor, is a transcriptional activator of endogenous tyrosine hydroxylase in neural progenitor cells derived from the adult brain. Development. 1999;126:4017–26. doi: 10.1242/dev.126.18.4017. [DOI] [PubMed] [Google Scholar]
- 114.Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–5. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- 115.Drexler HG, Quentmeier H. FLT3: receptor and ligand. Growth Factors. 2004;22:71–3. doi: 10.1080/08977190410001700989. [DOI] [PubMed] [Google Scholar]
- 116.Cotter FE. Unraveling biologic therapy for Bcl-2- expressing malignancies. Semin Oncol. 2004;31:18–21. doi: 10.1053/j.seminoncol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 117.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–55. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 118.Baldassarre G, Romano A, Armenante F, Rambaldi M, Paoletti I, Sandomenico C, Pepe S, Staibano S, Salvatore G, De Rosa G, Persico MG, Viglietto G. Expression of teratocarcinoma-derived growth factor-1 (TDGF-1) in testis germ cell tumors and its effects on growth and differentiation of embryonal carcinoma cell line NTERA2/D1. Oncogene. 1997;15:927–36. doi: 10.1038/sj.onc.1201260. [DOI] [PubMed] [Google Scholar]
- 119.Palmqvist L, Glover CH, Hsu L, Lu M, Bossen B, Piret JM, Humphries RK, Helgason CD. Correlation of murine embryonic stem cell gene expression profiles with functional measures of pluripotency. Stem Cells. 2005;23:663–80. doi: 10.1634/stemcells.2004-0157. [DOI] [PubMed] [Google Scholar]
- 120.Orkin SH. Chipping away at the embryonic stem cell network. Cell. 2005;122:828–30. doi: 10.1016/j.cell.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 121.Kuramochi-Miyagawa S, Kimura T, Ijiri TW, Isobe T, Asada N, Fujita Y, Ikawa M, Iwai N, Okabe M, Deng W, Lin H, Matsuda Y, Nakano T. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development. 2004;131:839–49. doi: 10.1242/dev.00973. [DOI] [PubMed] [Google Scholar]
- 122.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–8. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 123.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS, Hendrix MJ. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–52. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pezzolo A, Parodi F, Corrias MV, Cinti R, Gambini C, Pistoia V. Tumor origin of endothelial cells in human neuroblastoma. J Clin Oncol. 2007;25:376–83. doi: 10.1200/JCO.2006.09.0696. [DOI] [PubMed] [Google Scholar]
- 125.Nolan DJ, Ciarrocchi A, Mellick AS, Jaggi JS, Bambino K, Gupta S, Heikamp E, McDevitt MR, Scheinberg DA, Benezra R, Mittal V. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–58. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fausto N. Vasculogenic mimicry in tumors. Fact or artifact? Am J Pathol. 2000;156:359. doi: 10.1016/S0002-9440(10)64738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21:505–15. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- 128.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 129.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 130.Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7:246–55. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 131.Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 132.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 133.Fodde R. Stem cells and metastatic cancer: fatal attraction? PLoS Med. 2006;3:e482. doi: 10.1371/journal.pmed.0030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tahara E. Genetic pathways of two types of gastric cancer. IARC Sci Publ. 2004:327–49. [PubMed] [Google Scholar]
- 135.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 136.Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM, Centeno B, Weber F, Leu YW, Shapiro CL, Eng C, Yeatman TJ, Huang TH. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res. 2006;12:6626–36. doi: 10.1158/1078-0432.CCR-06-0467. [DOI] [PubMed] [Google Scholar]
- 137.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–8. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 138.Giovannucci E, Ogino S. DNA methylation, field effects, and colorectal cancer. J Natl Cancer Inst. 2005;97:1317–9. doi: 10.1093/jnci/dji305. [DOI] [PubMed] [Google Scholar]
- 139.Hao Y, Zhang J, Yi C, Qian W. Abnormal change of p53 gene in gastric and precancerous lesions and APC gene deletion in gastric carcinoma and near tissues. J Tongji Med Univ. 1997;17:75–8. doi: 10.1007/BF02888238. [DOI] [PubMed] [Google Scholar]
- 140.Lu Y, Li Z, Sun M. Multiple gene alterations involved in the processor of human gastric carcinogenesis. Zhonghua Yi Xue Za Zhi. 1995;75:679–82, 710–1. [PubMed] [Google Scholar]
- 141.Perry JM, Li L. Disrupting the stem cell niche: good seeds in bad soil. Cell. 2007;129:1045–7. doi: 10.1016/j.cell.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 142.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment- induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–95. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood. 2002;99:2369–78. doi: 10.1182/blood.v99.7.2369. [DOI] [PubMed] [Google Scholar]
- 145.Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–16. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 146.Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- 147.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 148.Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–22. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 149.Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O'Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR, Kaye FJ. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling path-way. Nat Genet. 2003;33:208–13. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 150.Maillard I, Pear WS. Notch and cancer: best to avoid the ups and downs. Cancer Cell. 2003;3:203–5. doi: 10.1016/s1535-6108(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 151.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, Van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 152.Dick JE. Stem cells: self-renewal writ in blood. Nature. 2003;423:231–3. doi: 10.1038/423231a. [DOI] [PubMed] [Google Scholar]
- 153.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 154.Passegue E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci USA. 2003;100:11842–9. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cox DN, Chao A, Baker J, Chang L, Qiao D, Lin H. A novel class of evolutionarily conserved genes defined by piwi are essential for stem cell self-renewal. Genes Dev. 1998;12:3715–27. doi: 10.1101/gad.12.23.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kuramochi-Miyagawa S, Kimura T, Yomogida K, Kuroiwa A, Tadokoro Y, Fujita Y, Sato M, Matsuda Y, Nakano T. Two mouse piwi-related genes: miwi and mili. Mech Dev. 2001;108:121–33. doi: 10.1016/s0925-4773(01)00499-3. [DOI] [PubMed] [Google Scholar]
- 157.Sasaki T, Shiohama A, Minoshima S, Shimizu N. Identification of eight members of the Argonaute family in the human genome small star, filled. Genomics. 2003;82:323–30. doi: 10.1016/s0888-7543(03)00129-0. [DOI] [PubMed] [Google Scholar]
- 158.Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev. 2002;16:2733–42. doi: 10.1101/gad.1026102. [DOI] [PubMed] [Google Scholar]
- 159.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–41. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 160.Song JJ, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Biol. 2003;10:1026–32. doi: 10.1038/nsb1016. [DOI] [PubMed] [Google Scholar]
- 161.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 162.Kolb FA, Zhang H, Jaronczyk K, Tahbaz N, Hobman TC, Filipowicz W. Human dicer: purification, properties, and interaction with PAZ PIWI domain proteins. Methods Enzymol. 2005;392:316–36. doi: 10.1016/S0076-6879(04)92019-8. [DOI] [PubMed] [Google Scholar]
- 163.Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep. 2004;5:189–94. doi: 10.1038/sj.embor.7400070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jaronczyk K, Carmichael JB, Hobman TC. Exploring the functions of RNA interference pathway proteins: some functions are more RISCy than others? Biochem J. 2005;387:561–71. doi: 10.1042/BJ20041822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Grivna ST, Pyhtila B, Lin H. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc Natl Acad Sci USA. 2006;103:13415–20. doi: 10.1073/pnas.0605506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC. Specific association of piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev. 2006;20:2214–22. doi: 10.1101/gad.1454806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M, Tuschl T. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–7. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- 168.Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–7. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- 169.Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–14. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316:744–7. doi: 10.1126/science.1142612. [DOI] [PubMed] [Google Scholar]
- 171.Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 172.Qiao D, Zeeman AM, Deng W, Looijenga LH, Lin H. Molecular characterization of hiwi, a human member of the piwi gene family whose overexpression is correlated to seminomas. Oncogene. 2002;21:3988–99. doi: 10.1038/sj.onc.1205505. [DOI] [PubMed] [Google Scholar]
- 173.Lee JH, Schutte D, Wulf G, Fuzesi L, Radzun HJ, Schweyer S, Engel W, Nayernia K. Stem-cell protein Piwil2 is widely expressed in tumors and inhibits apoptosis through activation of Stat3/Bcl-XL pathway. Hum Mol Genet. 2006;15:201–11. doi: 10.1093/hmg/ddi430. [DOI] [PubMed] [Google Scholar]
- 174.Iking-Konert C, Ostendorf B, Sander O, Jost M, Wagner C, Joosten L, Schneider M, Hansch GM. Transdifferentiation of polymorphonuclear neutrophils to dendritic-like cells at the site of inflammation in rheumatoid arthritis: evidence for activation by T cells. Ann Rheum Dis. 2005;64:1436–42. doi: 10.1136/ard.2004.034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lagasse E, Connors H, Al-Dhalimy M, Reitsma M, Dohse M, Osborne L, Wang X, Finegold M, Weissman IL, Grompe M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–34. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 176.Alison MR, Poulsom R, Jeffery R, Dhillon AP, Quaglia A, Jacob J, Novelli M, Prentice G, Williamson J, Wright NA. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406:257. doi: 10.1038/35018642. [DOI] [PubMed] [Google Scholar]
- 177.Poulsom R, Forbes SJ, Hodivala-Dilke K, Ryan E, Wyles S, Navaratnarasah S, Jeffery R, Hunt T, Alison M, Cook T, Pusey C, Wright NA. Bone marrow contributes to renal parenchymal turnover and regeneration. J Pathol. 2001;195:229–35. doi: 10.1002/path.976. [DOI] [PubMed] [Google Scholar]
- 178.Camargo FD, Green R, Capetanaki Y, Jackson KA, Goodell MA. Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat Med. 2003;9:1520–7. doi: 10.1038/nm963. [DOI] [PubMed] [Google Scholar]
- 179.Weissman IL. Normal and neoplastic stem cells. Novartis Found Symp. 2005;265:35–50. [PubMed] [Google Scholar]
- 180.Nygren JM, Jovinge S, Breitbach M, Sawen P, Roll W, Hescheler J, Taneera J, Fleischmann BK, Jacobsen SE. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 181.Mongan NP, Martin KM, Gudas LJ. The putative human stem cell marker, Rex-1 (Zfp42): structural classification and expression in normal human epithelial and carcinoma cell cultures. Mol Carcinog. 2006;45:887–900. doi: 10.1002/mc.20186. [DOI] [PubMed] [Google Scholar]
- 182.Duffy MJ. Role of tumor markers in patients with solid cancers: a critical review. Eur J Intern Med. 2007;18:175–84. doi: 10.1016/j.ejim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 183.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 184.Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26:1357–60. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 185.Bhardwaj N. Harnessing the immune system to treat cancer. J Clin Invest. 2007;117:1130–6. doi: 10.1172/JCI32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schreiber RD. Cancer vaccines 2004 opening address: the molecular and cellular basis of cancer immunosur-veillance and immunoediting. Cancer Immun. 2005:5. [PubMed] [Google Scholar]
- 187.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–7. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 188.Duan W, Ding H, Subler MA, Zhu WG, Zhang H, Stoner GD, Windle JJ, Otterson GA, Villalona-Calero MA. Lung-specific expression of human mutant p53-273H is associated with a high frequency of lung adenocarcinoma in transgenic mice. Oncogene. 2002;21:7831–8. doi: 10.1038/sj.onc.1205909. [DOI] [PubMed] [Google Scholar]
- 189.Old LJ. Cancer/testis (CT) antigens–a new link between gametogenesis and cancer. Cancer Immun. 2001;1:1. [PubMed] [Google Scholar]
- 190.Dean M. Cancer stem cells: redefining the paradigm of cancer treatment strategies. Mol Interv. 2006;6:140–8. doi: 10.1124/mi.6.3.5. [DOI] [PubMed] [Google Scholar]
- 191.Sell S. Cancer stem cells and differentiation therapy. Tumour Biol. 2006;27:59–70. doi: 10.1159/000092323. [DOI] [PubMed] [Google Scholar]
- 192.Spisek R, Kukreja A, Chen LC, Matthews P, Mazumder A, Vesole D, Jagannath S, Zebroski HA, Simpson AJ, Ritter G, Durie B, Crowley J, Shaughnessy JD, Jr, Scanlan MJ, Gure AO, Barlogie B, Dhodapkar MV. Frequent and specific immunity to the embryonal stem cell-associated antigen SOX2 in patients with monoclonal gammopathy. J Exp Med. 2007;204:831–40. doi: 10.1084/jem.20062387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shen R, Ye Y, Chen L, Yan Q, Barsky SH, Gao J-X. Precancerous stem cells can serve as tumor vasculogenic progenitors. PLoS One. doi: 10.1371/journal.pone.0001652. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]