

Abstract
The identification, purification and characterization of cancer stem cells (CSCs) holds tremendous promise for improving the treatment of cancer. Mounting evidence is demonstrating that only certain tumour cells (i.e. the CSCs) can give rise to tumours when injected and that these purified cell populations generate heterogeneous tumours. While the cell of origin is still not determined definitively, specific molecular markers for populations containing these CSCs have been found for leukaemia, brain cancer and breast cancer, among others. Systems approaches, particularly molecular profiling, have proven to be of great utility for cancer diagnosis and characterization. These approaches also hold significant promise for identifying distinctive properties of the CSCs, and progress is already being made.
Keywords: transcriptomics, proteomics, molecular signature, networks
Introduction
Cancer is the second deadliest disease in the United States, accounting for one out of every four deaths. Despite tremendous efforts dedicated to conquering this disease, the age-adjusted mortality rate has remained almost unchanged for most cancers for the past five decades, with one compelling instance being a devastating brain tumour – glioblastoma – which carries a median survival rate of merely 9–12 months regardless of treatment. Among the conglomerate of cellular, molecular and genetic complications contributing to the persistence of cancer, the cellular origin provides a promising avenue that could lead to targeted elimination of cancer. Cancers often occur in tissues with constant proliferation. A paradigm learned from developmental biology suggests that only the clonogenic stem cells in the hierarchical tissue developmental processes are long-lived, whereas their differentiated progenies lose proliferating potential. Emerging evidence suggests that cancers may actually be a malignance derived from a small cell population dubbed cancer stem cells (CSCs), among the heterogeneous tumour cells, that demonstrate self-renewal and multiple lineage differentiation properties [1]. Discovered initially in leukaemia [2], cancer stem cell populations have been isolated from several tissues, including breast, brain and colorectal cancers [3–5]. The identification of these cancer stem cell populations sets the stage for systems level analysis, and holds potentially tremendous ramifications in terms of our understanding of the molecular pathogenesis of cancer, patient stratification, and therapeutic intervention.
As with any complex biological system, cancer (including CSCs) can be viewed and interrogated at the genome-scale using systems biology approaches. Systems approaches stress three concepts regarding biological information [6]. First, there are two fundamental types of biological information – the digital information of the genome and the environmental information that is outside our DNA. Second, the digital genome information encodes two types of biological networks – protein interactions and gene regulatory networks. Protein networks transmit and use biological information for development, physiology and metabolism. Gene regulatory networks – transcription factors and RNAs that regulate networks of other transcription factors and other RNAs – receive information from, for example, signal-transduction networks, integrate and modulate it, and convey the processed information to networks of genes and proteins that execute developmental and physiological functions. In biological systems, these two types of networks are closely integrated. The organization of these networks can be inferred from various different types of measurements including, for example, global measurements of dynamically changing levels of mRNAs and proteins during developmental and physiological responses, as well as large-scale measurements of protein-protein and protein-DNA interactions. Third, there are many hierarchical levels of organization and information (for example, DNA, RNA and protein networks, cell signalling and metabolic networks and organization and responses of organ systems). To understand biological systems, information must be gathered from as many information levels as possible and integrated into models that generate testable hypotheses about how biological systems function.
In this review article we delineate our view for investigating CSCs using powerful integrated transcriptomic, proteomic and computational approaches. We will focus on gene expression profile signatures which will provide fundamental insights into the networks in the application of cancer diagnosis, patient stratification, and treatment management. We will also discuss emerging new experimental technologies and computational modelling approaches that empower systems strategies for tackling cancer stem cell challenges.
Molecular profiling for cancer classification
Systems approaches have proven of great utility in the study of cancer, with increasing power expected to continue to emerge in the future. Despite notable and significant challenges that remain [7, 8], one area that has shown significant promise is in the mining of global gene expression data sets to identify molecular signatures that can be used for diagnosis and treatment selection [9].
These studies typically involve the collection of samples from two or more classes (e.g. cancer versus normal, or responsive versus non-responsive to treatment) and the use of a set of data on which to train the classifier and another set on which to test. In the absence of a true test set, resampling methods such as cross-validation are generally used to estimate likely performance of the classifier on future data. Challenges often arise in these studies when different measurement platforms are used in training and test sets. The ability to generate an accurate classifier is a function of factors such as (1) the size of the training set relative to the number of features, (2) the computational method used and (3) the inherent distinctness of the selected phenotypes. Typically, the number of samples is far less than the number of transcripts, leading to overfitting being a significant problem. This leads to the need for computational methods that aid in avoiding overfitting when selecting a classifier. A variety of methods have been applied to cancer diagnoses including approaches based on support vector machines [10, 11] and relative expression reversals [12–15], among many others.
Application of these methods has led to the discovery of molecular classifiers of varying degrees of accuracy to identify prognostic signatures for breast cancer [16–32], ovarian cancer [33–35], colon cancer [36, 37], prostate cancer [14, 38–43] and brain cancer [44, 45], among others. Given the success of such global approaches to identify signatures for cancer tumours, there is reason to suppose that such approaches will prove very useful in uncovering new biology, markers, and treatments in the emerging area of CSCs. Indeed, progress in this direction is already being made.
Molecular profiling of cancer stem cells
Following on the successes of molecular profiling in identifying prognostic signatures for many cancers, researchers have begun to perform profiling of CSCs as well. We will discuss such efforts in the context of three cancers: leukaemia, brain and breast. In addition to profiling for signatures of specific CSCs, interesting work has also been done to find general signatures for ‘stemness’ in tumours. For example, an 11-gene signature for ‘stem-ness’ in multiple cancer types has been identified [46] that predicts short interval to disease recurrence, distant metastasis and death from cancer. This signature reflects a BMI-1 oncogene-driven gene expression pathway, where the BMI-1 gene is essential for the self-renewal of haematopoietic and neural stem cells. Using Kaplan–Meier analysis, this signature for ‘stem-ness’ was found to show predictive ability in 11 different cancers, including five epithelial cancers (prostate, breast, lung, ovairna and bladder) and six nonepithelial (lymphoma, mesothelioma, medulloblastoma, glioma and acute myeloid leukaemia). Thus, there is evidence that the property of ‘stemness’ (defined with this signature) is predictive of outcome in a wide variety of tumours. If validated, this observation could have a major impact on patient care [47]. Recent studies have also shown that cancer and normal stem cells share the same self-renewal mechanisms, such as the Bmi1 and Wnt canonical pathways [48, 49], further strengthening the link between stem cells and CSCs.
Historical perspective: cancer stem cells in leukaemia
The fundamental concept of CSCs came from early studies of blood cancer (leukaemia) and the blood forming haematopoietic stem cells (HSC). Elegant work by Till and McCulloch in the early 1960s established the existence of bone marrow HSC capable of forming myelo-erythroid colonies in the spleen of lethally irradiated hosts [50]. These cells were later isolated by Weissman and colleagues and shown to be able to self-renewal and exhibit multi-potent differentiation giving rise to all the blood lineages [51, 52]. Studies of human leukaemia using in vitro and in vivo colony-formation assays demonstrated that only a small subset of leukaemia cells possess extensive proliferative capability [53, 54] suggesting that leukaemia may actually be derived from a small leukaemic stem cell (LSC) population. This concept was further proved by the successful isolation of myeloid leukaemia-initiating cells using cell surface phenotype CD34+ CD38− and subsequent in vivo transplantation into severe combined immune-deficient (SCID) mice [2]. One intriguing question that remained unanswered until very recently is whether the LSCs are derived from normal HSCs or from their downstream committed progenitor cells that regain the self-renewal property. Using mouse genetics and clinical studies, Weissman and colleagues demonstrated that both mechanisms exist in leukaemia: while chronic myeloid leukaemia can be derived from LSC residing at the HSC stage [55], the more differentiated progenitor – granulocyte-macrophage progenitor (GMP) – can also gain LSC property during the blast-crisis of myeloid leukaemia [56]. One common characteristic between stem cells and cancer is the unlimited self-renewal capability. The hierarchical organization and the cellular heterogeneity in the haematopoietic systems, and lessons learned from LSC have inspired later isolation of CSCs from solid tumours [3], and will continue to provide theoretical framework for investigating tumouregenesis.
Molecular profiling of brain cancer stem cells
Brain cancer is a highly heterogeneous disease consisting of multiple tumour types affecting both children and adults [57]. The most malignant form of brain cancer, glioblastoma multiforme (GBM), is characterized by a diverse cellular phenotypic and genetic heterogeneity which has been a significant impediment to the development of effective targeted medical therapies. Traditional treatment for GBM consists of surgical resection to reduce the tumour mass followed by radiation and chemotherapy selective for highly-proliferating tumour cells. Despite continuous refinement over the past three decades, these treatments have not significantly impacted time to tumour recurrence or long-term survival in GBM patients. Given the low-proliferate rate of normal brain tissue, it has long been hypothesized that brain cancer may arise from neural stem or progenitor cells which were thought to be more sensitive to oncogenic transformation [58]. Using neural stem cell culture techniques, two different types of malignant brain tumours were shown to possess sub-populations of tumour cells with characteristic stem cell-like properties, such as self-renewal, potential for differentiation and capacity to form neurospheres [59]. The cell (or cells) of origin for these putative brain CSCs has not been determined definitively, but evidence on this subject is starting to accumulate. For example, a number of groups have confirmed that brain tumours contain a small fraction of cells that can be separated on the basis of the cell-surface marker CD133 and that have clonogenic potential as measured by the neurosphere assay. In a key in vivo experiment, a minority population of cells from brain tumours was shown to be tumourigenic by implantation into immuno-compromised mouse brain. In this model, injection of as few as 100 CD133 + cells could generate tumours in vivo, whereas injecting of as many as 10 5 CD133−– cells, which comprise the majority of the GBM tumour cells, did not lead to any viable tumour growth. Interestingly, CD133 had previously been found to be a marker of HSC [60–62] and has now been implicated in the identification of a number of putative tumour stem cells from a variety of human cancers, including leukaemia [63], prostate cancer [64] and colon cancer [4]. At this point, evidence supports that the sub-population of tumourigenic brain cancer cells is a subset of CD133+ cells as not all CD133+ cells are tumourigenic [65]. Several labs are currently refining molecular markers to refine tumour stem cell identification and purification.
The paradigm that brain tumours possess a small fraction of cancer-initiating tumour stem cells which can reconstitute the tumour's cellular and functional hierarchy has important implications for treatment. To further explore the potential differences in sub-populations of GBM cells, Lee et al. used high-throughput genome-wide molecular profiling to characterize GBM-derived tumour stem cells cultured in serum-free conditions optimized to sustain neural stem cells [66]. Genome-wide expression and genotyping studies revealed that these GBM-derived tumour stem cells had extensive similarities to normal neural stem cells and possessed the capacity to initiate brain tumours in a xenograft mouse model. Significantly, the genotype and phenotype of the mouse xenograft brain tumours resulting from implantation of tumour stem cells cultured in serum-free conditions matched that of the parent human GBM tumours from which they were derived. When GBM-derived tumour cell lines were generated using standard serum-based culture conditions not conducive to stem cell growth, the resulting cells lacked the ability to initiate or propagate tumours in the mouse xenograft model. Genome-wide transcription analysis and genotyping studies revealed significant differences between these serum-cultured cell lines and their corresponding parent GBM tumours and matched serum-free cultured tumour stem cell lines. Of note, the serum-cultured cell lines did manifest extensive similarities to the most commonly utilized immortalized cancer cell lines. These studies provided further evidence that GBM has a distinct sub-population of tumour cells with stem cell-like properties, and that it is these cells that are biologically relevant in initiating and sustaining tumour growth. Unfortunately, most preclinical studies have historically been based on targeting therapies to features identified in serum-cultured immortalized cancer cell lines. This landmark large-scale molecular profiling study raises significant concerns about this approach and suggests the use of serum-free cultured tumourstem cells as a more appropriate pre-clinical model [66].
There is great interest in identifying effective therapies that can target the tumour-initiating and tumour-maintaining sub-population of tumour stem cells identified as CD 133+. Two recent studies used molecular profiling to characterize the differential response of CD133+ cells to therapeutic intervention compared to CD 133– cells. After radiation treatment, GBM tumours exhibited an increase in the fraction of CD133+ cells, more robust activation of DNA repair pathways in CD 133+ than CD 133– cells, and more aggressive growth characteristics on serial transplantation [67]. Pre-treatment with an inhibitor of cell-cycle checkpoints CHK1/CHK2 sensitized CD 133+ cells to radiation, suggesting a role for heightened DNA repair in the relative resistance of CD 133+ tumour stem cells to radiation-induced DNA damage. CD133+ cells also retain the ability to differentiate into more terminally-differentiated and less aggressive cells, a feature lacking in CD133– cells. Bone morphogenic protein (BMP), a potent differentiating molecule, led to significant differentiation of CD133+ cells and decreased tumour growth in an in vivo mouse model [68]. This suggests that the use of differentiating agents may play an important role against tumour stem cells which, which unlike CD 133– cells, retain important differentiation capacity – presumably because different triggering leads to the loss of neoplastic potential. Another area of fertile discovery is the role of epigenetic regulation in the control of stem cell maintenance, self-renewal and differentiation. Transcriptional changes that result from the pharmacological reversal of epigenetic gene silencing, typically with either a histone deacetylase inhibitor or a demethylating agent, allows for the identification of epigenetically-regulated genes on a genome-wide scale using molecular profiling techniques [69, 70]. Recent large-scale genomic studies have confirmed the importance of epigenetic gene silencing in the biological behaviour of GBM, including response to therapy [69, 71, 72]. Using a similar approach, several investigators have identified key genes which are epigenetically regulated during differentiation in neural stem cells and have been implicated in neoplasia [73–75]. Epigenetic modifications make attractive therapeutic targets as they are readily reversed and the underlying DNA sequence is not altered. Taken together, these studies highlight the importance of targeting therapies to the CD 133 + sub-population of cells which can exploit the tumour stem cell properties and vulnerabilities important in the effective therapeutic treatment of GBM.
The fraction of cells in a GBM tumour that express CD133 has been observed to be as little as 1% in low-grade tumours and as high as 30% for high-grade glioblastoma. However, in any particular grade, such as glioblastoma, there is high variability in this fraction [65]. Interestingly, time to progression and overall survival are not greatly affected by extent of resection in GBM patients. Despite the removal of the bulk tumour, consisting mostly of CD133– cells, the tumour recurs within 1 year on average. Thus, it is not yet clear whether the fraction of CD133 + cells in the bulk tumour will prove to be useful as a prognostic marker. It could be that the important neoplastic-associated stem cells constitute only a small fraction of the total CD133 + cells. Assuming that CD133 + cells (or a sub-fraction) are the biologically active tumour stem cells responsible for tumour recurrence, identifying and targeting the location of these cells in vivo, within the tumour or in the surrounding brain after tumour resection, will be of great importance. A recent study indicates that tumour stem cells exist in the ‘perivascular niche’, and that endothelial cells potentiate the tumour stem cells capacity to initiate and maintain tumour growth [76]. Application of inhibitors of endothelial cells significantly depleted tumour blood vessels resulting in a dramatic reduction of tumour stem cells but not CD 133– cells in the bulk tumour. This suggests that targeting the unique vulnerabilities of tumour stem cell populations should include further characterization of the micro-environment that support and sustains tumour stem cell self-renewal and differentiation. Large-scale molecular profiling of purified sub-populations of tumour cells from GBM will play a critical role in further defining these molecular targets [77].
Molecular profiling of breast cancer stem cells
CSCs have also been identified and characterized for breast tumours, a much more common malignancy than brain tumours. Breast cancer tumours contain a sub-population of highly tumourigenic cells characterized by CD44 expression and no or low CD24 expression (CD44+ CD24–/low), as demonstrated by their distinguishing capability to generate tumours in immunodeficient mice [3]. These CD44+ CD24–/low cells have also been demonstrated to have the capacity to give rise to the various cell types characteristic of the bulk tumour. As with CD133 + cells in brain tumours, the CD44 + CD24 –/low cells in breast tumours contain the population of CSCs, but may not uniquely identify them. Indeed, additional cell-surface markers may further distinguish the tumourigenic breast cell population. For example, it has been shown that among CD44+ CD24–/low cells, those that were also ESA+ had enriched tumour generating potential compared with those that were ESA–. Thus, one hypothesis would be that ESA+ CD44+ CD24–/low cells may represent a purer population of a subset of the CSCs [3]. This interesting question for all different tumour stems cells is the extent to which neoplastic stem cell population may be purified by additional cell-surface markers. Also of interest is that CD44+ CD24–/low cells generate additional CD44+ CD24–/low cells as well as phenotypically distinct cells, providing evidence that breast CSCs have the hallmark stem cell characteristics of self-renewal and the capacity to generate heterogeneous populations.
It has been shown that these tumourigenic breast cancer cells can propagate in culture and maintain properties of normal human mammary gland stem/progenitor cells that have the distinct ability to grow in selective culture conditions as non-adherent spherical clusters of cells [78, 79]. Most importantly, after in vitro culturing these cells were found to remain CD44+ CD24–/low and maintain their ability to generate tumours in immunodeficient mice with as few as 1000 injected cells [78, 79]. Thus, there is strong evidence that breast CSCs have indeed been isolated and can even be propagated in culture while maintaining their cancer stem cell characteristics.
Machine-learning from global gene expression data has been used to identify a gene signature from tumourigenic breast-cancer cells that is associated with both overall and metastasis-free survival in patients with breast cancer [24]. This signature was found through comparing the gene-expression profiles of the CD44+ CD24–/low breast CSCs, which demonstrate enriched invasiveness [80], with normal breast epithelium. The differentially expressed genes between these two groups were then used to generate a 186-gene signature for ‘invasiveness’ that showed a significant association with overall and metastasis-free survival of patients with breast cancer [24]. This invasiveness signature was then also shown to be associated with prognosis in other cancers, including medulloblastoma, lung cancer and prostate cancer, demonstrating the general features at the gene expression level of invasiveness. It will be interesting to convert these tumourigenic gene signatures into their corresponding biological networks so as to begin to understand the unique and shared features of CSCs.
Emerging technologies for proteome characterization
While transcriptomic approaches have proven useful for identifying informative molecular signatures for cancers and CSCs, proteomic characterizations have lagged behind. The reason for this is clearly the more difficult challenge of measuring proteins compared to transcripts. Emerging proteomics technologies hold the promise of greatly improving our ability to make detailed assessments of protein-based molecular signatures, similar to those that have had success thus far for gene expression. One key advantage of protein signatures relative to gene expression is that proteins can be found in the blood and other accessible bodily fluids more readily due to slower degradation rates than their mRNA counterparts. Thus, protein signatures represent an important class of molecular signature for disease diagnosis. For CSCs, these approaches will help to elucidate the differences between the proteome of CSCs relative to their non-tumourigenic counterparts. Ultimately, the reconstruction of protein and gene regulatory networks of CSCs holds the promise of rationally-designed therapeutics for personalized medicine by allowing the therapeutic chosen to be governed by informative molecular signatures measured from each patient at the time of diagnosis. A few promising proteomics technologies are described below (Fig. 1).
1.
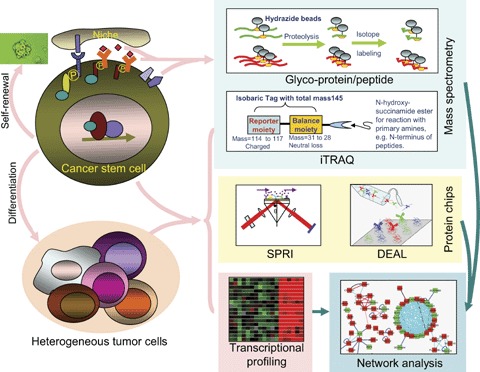
Systems biology strategies for studying cancer stem cells.
Isobaric tagging for relative and absolute quantitation (iTRAQ)
Stable isotope labelling enables the quantitative analysis of protein concentrations through mass spectrometry (MS). One state-of-the-art technique for quantitative MS is iTRAQ [81], which uses stable isotope labelling of proteolytic peptides. This technique modifies primary amino acid groups of peptides by linking a mass balance group (carbonyl group), and a reporter group (based on N-methylpiperazine) by forming an amide bond. The iTRAQ reagents are designed to be isobaric, which enables differentially labelled peptides to appear as single peaks in MS, which is important for reducing peak overlapping in the MS scans. When MS/MS is used for analysis with iTRAQ-tagged peptides, the mass balancing carbonyl moiety is released as a neutral fragment, thereby liberating isotopeencoded reporter ions that provide relative quantitative information on protein abundance. Because four different iTRAQ reagents are currently available, comparative analysis of a set of two to four samples is feasible within a single MS run [82]. The, iTRAQ technology represents the state-of-the-art in quantitative proteomics and represents a promising technology for using proteomics to differentiate key differences in protein networks of CSCs from normal stem cells or other cells in the tumour.
Glyco-peptide capture
MS-based methods will allow for the identification of proteins spanning approximately three orders of magnitude in concentration from a given sample. Therefore, methods that can select specified fractions of the proteome are important for simplifying the sample sufficiently to identify the proteins of interest. One recently developed approach is the shotgun glyco- peptides capture approach [83]. This approach selects for N-linked or O-linked glycosylated peptides, which are enriched for secreted proteins and cell-surface markers. Thus, this approach could be used to identify candidates for unique cell-surface markers for CSCs that differ from the bulk tumour by comparing the glyco-captured proteomes from both sample sets.
Antibody microarrays using surface plasmon resonance imagining (SPRI)
Protein chip methods hold potential for broad quantitative screens of proteins, and a variety of techniques have been developed based on antibody binding [84–86]. Antibody microarrays have been used for biomarker discovery and protein profiling of serum from patients with prostate, lung, pancreas and bladder cancer [87–90]. One emerging approach with tremendous promise is SPRI [82, 91, 92], which enables real-time, label-free measurement of protein expression. SPR-based chips have a detection sensitivity of 10–100 times less than ELISA [82], but have a spatial resolution down to approximately 4 μm [93]. It is thus possible to print up to 800 unique anti-bodies on Lumera Nanocapture Gold™ microarray slides and monitor the abundance of the target proteins in real time [82]. Because the same slide can be regenerated for reuse many times (Z. Hu, C. Lausted, unpublished observations), this means that the approach has the capacity necessary to screen through hundreds of patient samples. Thus, this approach holds tremendous promise in the case of CSCs to be able to screen through large numbers of proteins, including secreted proteins and cell-surface markers and not only measure their presence, but also abundance and the dynamics of their binding. The limitation of this technique is its dependence on the affinity and specificity of the antibodies it employs for detection – cross re-activities in complex protein mixtures (like blood) can pose significant problems.
DNA-encoded antibody libraries (DEAL)
One recently developed technique that offers great potential for analyses of CSCs is DEAL, which enables cell localization and single-cell measurements of protein, RNA, and single-stranded DNA simultaneously on a single chip [94]. DEAL is a highly sensitive measurement technique, with a reported detection limit of 10 fM for the protein IL-2-150 times more sensitive than ELISA. This sensitivity can be applied to the isolation of rare cells based on combinations of cell-surface markers, enabling the isolation and addressing of individual CSCs. DEAL can also be used to make single cell measurements of secreted proteins from each of these isolated single cells. Thus, DEAL offers superb sensitivity and the ability to perform spatially multiplexed detection for characterization of CSCs.
Biomolecular networks in cancer
Systems approaches to cancer [95–99] require not only the identification of the key components of a system through global analyses, but also information about how these components interact in biological networks. Network models of multiple types have been applied to cancer systems. The most commonly applied to cancer are interaction networks, including protein–protein interaction networks, protein-DNA interaction networks and so forth. Gene expression data can be used to identify differentially expressed genes in which can then be visualized on interaction networks, as has been done for lung cancer [100]. Various properties of these networks have been studied [99], with reported findings including, for example, the enrichment of cancer-related genes among the ‘hubs’ of the networks. While these interaction networks are very useful tools for visualizing large data sets, they are not computable, predictive network models, which are those that hold the most promise for predictive medicine and drug development. Predictive models stemming from mathematical descriptions of biochemical reaction networks and statistical influence models should prove highly useful [101].
Another area of network modelling that should prove very beneficial in research of cancer and CSCs is that of metabolic networks. Key metabolic differences have been shown to exist in cancers which can be exploited using Positron Emission Tomography (PET) to do in vivo imaging of tumours [102] and even to predict treatment response [103, 104]. If key metabolic differences can be found between CSCs and the rest of the tumour, such approaches could potentially even be used to identify the location of cancer stem cell populations in vivo. One enabling resource for large-scale quantitative modelling of metabolic networks in cancer is the recent stoichiometric reconstruction of known human metabolism at the genome-scale [105]. With this global reconstruction, gene expression and other data can be used to create initial models of the genome-scale metabolic networks of a variety of human cell types, including for CSCs. These biochemical reaction networks can be used to make numerous quantitative simulations that have been shown previously to match well with experimental data in model organisms [106]. These successes with model organisms have also been extended to models of simple human systems such as the erythrocyte [107, 108] and mitochondria [109], with the global metabolic reconstruction poised to allow for larger human metabolic networks to now be modelled. These studies may well provide insights into the unique metabolic features of cancer cells – allowing one to identify both metabolic features that are shared among cancer cells and features that are unique to individual types of cancer.
More detailed dynamic models of specific biochemical networks in cancer have been made for important signalling networks in cancer, leading to insightful biological observations for, among many others, the NF-κB signalling network [110–112]. As isolated cancer stem cell populations become better characterized, it will be possible to model these systems to identify differences in their regulation in CSCs and further identify possible therapeutic targets. Dynamic simulations of large-scale signalling networks in cancer cells has also been performed [113].
Large amounts of high-throughput data (i.e. transcriptomes) can also be used to infer networks that can explain statistical dependencies seen in the data, indicate candidate novel interacting partners, and quantitatively predict the gene expression resulting from knockouts or environmental perturbation. For model systems, such approaches are now being successfully applied at the genome-scale for gene-regulatory networks [114, 115]. Such approaches are now also being applied to mammalian systems as was done for normal and cancerous B cells with the development of an algorithm called Reconstruction of Accurate Cellular Networks (ARACNe) [116]. As cancer stem cell populations are profiled extensively, these same approaches will be useful to identify predictive networks for CSCs. Comparing these networks to those in normal stem cells and other tumour cells should prove highly informative for identifying drug targets unique to the cancer stem cell population of interest. By generating networks of CSCs in particular and comparing them with networks of normal stem/progenitor cells we should be able to greatly enhance our understanding of what could lead to these cells becoming cancerous.
Computational modelling and systems approaches will be key to catalyzing the future of drug discovery [117, 118], and drug discovery focused specifically on CSCs offers tremendous promise for advancing cancer therapies. Thus, computational modelling of cancer stem cell networks to identify potential therapeutic targets and to predict the effect of drug-induced perturbations is critical to this field moving forward.
Perspective and concluding remarks
The identification and prospective isolation of CSCs from leukaemia and a number of solid tumours has spawned a new paradigm in cancer research. From the perspective of systems biology – with the goal of predictive, preventive, personalized, and participatory (P4) medicine – we envision increasingly global assessment of CSCs and their microenvironments (niche) at the level of complete transcriptome, proteome and epigenome, using empowering new high-throughput technologies. The resulting gene expression profile signatures of cancer stem cell would serve as more accurate indicatives for cancer diagnosis and prognosis. Emerging proteomic technologies employing MS and protein chip platforms would allow for identification of better cell-surface markers and their interaction with the resident stem cell niche and potential diagnostic markers from both body fluids and tumour tissues. Incorporating these data into biological networks will provide fundament insights into the biology of CSCs and their abilities for renewal and differentiation. These combined efforts will ultimately lead to new therapeutic strategy specifically targeting CSCs for unprecedented personalized cancer therapy.
Acknowledgments
NDP was supported by the Sam E. and Kathleen Henry postdoctoral fellowship from the American Cancer Society and a developmental research grant from the Pacific Ovarian Cancer Research Consortium (Urban, P50 CA083636). LH and QT are supported by NIH grants PO1DK53074 and U54 CA119347. The authors also acknowledge financial support from the James S. McDonnell Foundation.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 5.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 6.Hood L, Heath JR, Phelps ME, Lin B. Systems biology and new technologies enable predictive and pre-ventative medicine. Science. 2004;306:640–3. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 7.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guide-lines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99:147–57. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 8.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J Clin Oncol. 2005;23:7332–41. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 9.Quackenbush J. Microarray analysis and tumor classification. N Engl J Med. 2006;354:2463–72. doi: 10.1056/NEJMra042342. [DOI] [PubMed] [Google Scholar]
- 10.Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–14. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 11.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, Poggio T, Gerald W, Loda M, Lander ES, Golub TR. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA. 2001;98:15149–54. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geman D, D'Avignon C, Naiman DQ, Winslow RL. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol. 2004;3:19. doi: 10.2202/1544-6115.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan AC, Naiman DQ, Xu L, Winslow RL, Geman D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics. 2005;21:3896–904. doi: 10.1093/bioinformatics/bti631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu L, Tan AC, Naiman DQ, Geman D, Winslow RL. Robust prostate cancer marker genes emerge from direct integration of inter-study microarray data. Bioinformatics. 2005;21:3905–11. doi: 10.1093/bioinformatics/bti647. [DOI] [PubMed] [Google Scholar]
- 15.Price ND, Trent J, El-Naggar AK, Cogdell D, Taylor E, Hunt KK, Pollock RE, Hood L, Shmulevich I, Zhang W. Highly accurate two-gene classifier for differentiating gastrointestinal stromal tumors and leiomyosarcomas. Proc Natl Acad Sci USA. 2007;104:3414–9. doi: 10.1073/pnas.0611373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park ES, Lee JS, Woo HG, Zhan F, Shih JH, Shaughnessy JD, Frederic Mushinski J. Heterologous tissue culture expression signature predicts human breast cancer prognosis. PLoS ONE. 2007;2:e145. doi: 10.1371/journal.pone.0000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler AS, Chang HY. From description to causality: mechanisms of gene expression signatures in cancer. Cell Cycle. 2006;5:1148–51. doi: 10.4161/cc.5.11.2798. [DOI] [PubMed] [Google Scholar]
- 18.Buyse M, Loi S, Van't Veer L, Viale G, Delorenzi M, Glas AM, D'Assignies MS, Bergh J, Lidereau R, Ellis P, Harris A, Bogaerts J, Therasse P, Floore A, Amakrane M, Piette F, Rutgers E, Sotiriou C, Cardoso F, Piccart MJ. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–92. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- 19.Dai H, Van't Veer L, Lamb J, He YD, Mao M, Fine BM, Bernards R, Van De Vijver M, Deutsch P, Sachs A, Stoughton R, Friend S. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65:4059–66. doi: 10.1158/0008-5472.CAN-04-3953. [DOI] [PubMed] [Google Scholar]
- 20.Foekens JA, Atkins D, Zhang Y, Sweep FC, Harbeck N, Paradiso A, Cufer T, Sieuwerts AM, Talantov D, Span PN, Tjan-Heijnen VC, Zito AF, Specht K, Hoefler H, Golouh R, Schittulli F, Schmitt M, Beex LV, Klijn JG, Wang Y. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J Clin Oncol. 2006;24:1665–71. doi: 10.1200/JCO.2005.03.9115. [DOI] [PubMed] [Google Scholar]
- 21.Glinsky GV, Higashiyama T, Glinskii AB. Classification of human breast cancer using gene expression profiling as a component of the survival predictor algorithm. Clin Cancer Res. 2004;10:2272–83. doi: 10.1158/1078-0432.ccr-03-0522. [DOI] [PubMed] [Google Scholar]
- 22.Goncalves A, Esterni B, Bertucci F, Sauvan R, Chabannon C, Cubizolles M, Bardou VJ, Houvenaegel G, Jacquemier J, Granjeaud S, Meng XY, Fung ET, Birnbaum D, Maraninchi D, Viens P, Borg JP. Postoperative serum proteomic profiles may predict metastatic relapse in high-risk primary breast cancer patients receiving adjuvant chemotherapy. Oncogene. 2006;25:981–9. doi: 10.1038/sj.onc.1209131. [DOI] [PubMed] [Google Scholar]
- 23.Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- 24.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–26. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 25.Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–16. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Nuyten DS, Kreike B, Hart AA, Chi JT, Sneddon JB, Wessels LF, Peterse HJ, Bartelink H, Brown PO, Chang HY, Van De Vijver MJ. Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res. 2006;8:R62. doi: 10.1186/bcr1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedren S, Bergh J. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–64. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomassen M, Tan Q, Eiriksdottir F, Bak M, Cold S, Kruse TA. Prediction of metastasis from low-malignant breast cancer by gene expression profiling. Int J Cancer. 2007;120:1070–5. doi: 10.1002/ijc.22449. [DOI] [PubMed] [Google Scholar]
- 29.Van De Vijver MJ, He YD, Van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, Van Der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 30.Van't Veer LJ, Dai H, Van De Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, Van Der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 32.Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, Glas AM, Perou CM, Van't Veer LJ. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005;65:9155–8. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- 33.De Cecco L, Marchionni L, Gariboldi M, Reid JF, Lagonigro MS, Caramuta S, Ferrario C, Bussani E, Mezzanzanica D, Turatti F, Delia D, Daidone MG, Oggionni M, Bertuletti N, Ditto A, Raspagliesi F, Pilotti S, Pierotti MA, Canevari S, Schneider C. Gene expression profiling of advanced ovarian cancer: characterization of a molecular signature involving fibroblast growth factor 2. Oncogene. 2004;23:8171–83. doi: 10.1038/sj.onc.1207979. [DOI] [PubMed] [Google Scholar]
- 34.Smith DI. Transcriptional profiling develops molecular signatures for ovarian tumors. Cytometry. 2002;47:60–2. doi: 10.1002/cyto.10042. [DOI] [PubMed] [Google Scholar]
- 35.Spentzos D, Levine DA, Ramoni MF, Joseph M, Gu X, Boyd J, Libermann TA, Cannistra SA. Gene expression signature with independent prognostic significance in epithelial ovarian cancer. J Clin Oncol. 2004;22:4700–10. doi: 10.1200/JCO.2004.04.070. [DOI] [PubMed] [Google Scholar]
- 36.Barrier A, Boelle PY, Roser F, Gregg J, Tse C, Brault D, Lacaine F, Houry S, Huguier M, Franc B, Flahault A, Lemoine A, Dudoit S. Stage II colon cancer prognosis prediction by tumor gene expression profiling. J Clin Oncol. 2006;24:4685–91. doi: 10.1200/JCO.2005.05.0229. [DOI] [PubMed] [Google Scholar]
- 37.Giacomini CP, Leung SY, Chen X, Yuen ST, Kim YH, Bair E, Pollack JR. A gene expression signature of genetic instability in colon cancer. Cancer Res. 2005;65:9200–5. doi: 10.1158/0008-5472.CAN-04-4163. [DOI] [PubMed] [Google Scholar]
- 38.Halvorsen OJ, Oyan AM, Bo TH, Olsen S, Rostad K, Haukaas SA, Bakke AM, Marzolf B, Dimitrov K, Stordrange L, Lin B, Jonassen I, Hood L, Akslen LA, Kalland KH. Gene expression profiles in prostate cancer: association with patient subgroups and tumour differentiation. Int J Oncol. 2005;26:329–36. [PubMed] [Google Scholar]
- 39.Lin B, White JT, Lu W, Xie T, Utleg AG, Yan X, Yi EC, Shannon P, Khrebtukova I, Lange PH, Goodlett DR, Zhou D, Vasicek TJ, Hood L. Evidence for the presence of disease-perturbed networks in prostate cancer cells by genomic and proteomic analyses: a systems approach to disease. Cancer Res. 2005;65:3081–91. doi: 10.1158/0008-5472.CAN-04-3218. [DOI] [PubMed] [Google Scholar]
- 40.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D'Amico AV, Richie JP, Lander ES, Loda M, Kantoff PW, Golub TR, Sellers WR. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 41.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 42.Luo J, Dunn T, Ewing C, Sauvageot J, Chen Y, Trent J, Isaacs W. Gene expression signature of benign prostatic hyperplasia revealed by cDNA microarray analysis. Prostate. 2002;51:189–200. doi: 10.1002/pros.10087. [DOI] [PubMed] [Google Scholar]
- 43.Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WL. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest. 2004;113:913–23. doi: 10.1172/JCI20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuller GN, Mircean C, Tabus I, Taylor E, Sawaya R, Bruner JM, Shmulevich I, Zhang W. Molecular voting for glioma classification reflecting heterogeneity in the continuum of cancer progression. Oncol Rep. 2005;14:651–6. [PubMed] [Google Scholar]
- 45.Kim S, Dougherty ER, Shmulevich I, Hess KR, Hamilton SR, Trent JM, Fuller GN, Zhang W. Identification of combination gene sets for glioma classification. Mol Cancer Ther. 2002;1:1229–36. [PubMed] [Google Scholar]
- 46.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–21. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lahad JP, Mills GB, Coombes KR. Stem cell-ness: a “magic marker” for cancer. J Clin Invest. 2005;115:1463–7. doi: 10.1172/JCI25455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 49.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 50.Till JE, Mc CE. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res. 1961;14:213–22. [PubMed] [Google Scholar]
- 51.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 52.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci USA. 1992;89:2804–8. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46:411–22. [PubMed] [Google Scholar]
- 54.Bruce WR, Van Der Gaag H. A Quantitative Assay for the Number of Murine Lymphoma Cells Capable of Proliferation in Vivo. Nature. 1963;199:79–80. doi: 10.1038/199079a0. [DOI] [PubMed] [Google Scholar]
- 55.Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–43. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 56.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 57.Kleihues P, Schauble B, Zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 58.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–7. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 59.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblas-toma. Cancer Res. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 60.Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820–4. doi: 10.1016/j.bbrc.2006.10.128. [DOI] [PubMed] [Google Scholar]
- 61.Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–12. [PubMed] [Google Scholar]
- 62.Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–21. [PubMed] [Google Scholar]
- 63.Wuchter C, Ratei R, Spahn G, Schoch C, Harbott J, Schnittger S, Haferlach T, Creutzig U, Sperling C, Karawajew L, Ludwig WD. Impact of CD133 (AC133) and CD90 expression analysis for acute leukemia immunophenotyping. Haematologica. 2001;86:154–61. [PubMed] [Google Scholar]
- 64.Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S, Rhim JS. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007;67:3153–61. doi: 10.1158/0008-5472.CAN-06-4429. [DOI] [PubMed] [Google Scholar]
- 65.Dirks PB. Brain tumour stem cells: the undercurrents of human brain cancer and their relationship to neural stem cells. Philos Trans R Soc Lond B Biol Sci. 2008;363:139–52. doi: 10.1098/rstb.2006.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and geno-type of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 67.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 68.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 69.Foltz G, Ryu GY, Yoon JG, Nelson T, Fahey J, Frakes A, Lee H, Field L, Zander K, Sibenaller Z, Ryken TC, Vibhakar R, Hood L, Madan A. Genome-wide analysis of epigenetic silencing identifies BEX1 and BEX2 as candidate tumor suppressor genes in malignant glioma. Cancer Res. 2006;66:6665–74. doi: 10.1158/0008-5472.CAN-05-4453. [DOI] [PubMed] [Google Scholar]
- 70.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 72.Mueller W, Nutt CL, Ehrich M, Riemenschneider MJ, Von Deimling A, Van Den Boom D, Louis DN. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene. 2007;26:583–93. doi: 10.1038/sj.onc.1209805. [DOI] [PubMed] [Google Scholar]
- 73.Lotem J, Sachs L. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene. 2006;25:7663–72. doi: 10.1038/sj.onc.1209816. [DOI] [PubMed] [Google Scholar]
- 74.Roloff TC, Nuber UA. Chromatin, epigenetics and stem cells. Eur J Cell Biol. 2005;84:123–35. doi: 10.1016/j.ejcb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 75.Kondo T. Epigenetic alchemy for cell fate conversion. Curr Opin Genet Dev. 2006;16:502–7. doi: 10.1016/j.gde.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 76.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 77.Mischel PS, Cloughesy TF, Nelson SF. DNA-microarray analysis of brain cancer: molecular classification for therapy. Nat Rev Neurosci. 2004;5:782–92. doi: 10.1038/nrn1518. [DOI] [PubMed] [Google Scholar]
- 78.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 79.Ponti D, Zaffaroni N, Capelli C, Daidone MG. Breast cancer stem cells: an overview. Eur J Cancer. 2006;42:1219–24. doi: 10.1016/j.ejca.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 80.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R, Jr, Badve S, Nakshatri H. CD44+/CD24– breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8:R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 82.Hu Z, Hood L, Tian Q. Quantitative proteomic approaches for biomarker discovery. Proteomics. 2007 doi: 10.1002/prca.200700109. In review. [DOI] [PubMed] [Google Scholar]
- 83.Sun B, Ranish JA, Utleg AG, White JT, Yan X, Lin B, Hood L. Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Mol Cell Proteomics. 2007;6:141–9. doi: 10.1074/mcp.T600046-MCP200. [DOI] [PubMed] [Google Scholar]
- 84.Song S, Li B, Wang L, Wu H, Hu J, Li M, Fan C. A cancer protein microarray platform using antibody fragments and its clinical applications. Mol Biosyst. 2007;3:151–8. doi: 10.1039/b608973a. [DOI] [PubMed] [Google Scholar]
- 85.Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:RESEARCH0004. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olle EW, Sreekumar A, Warner RL, McClintock SD, Chinnaiyan AM, Bleavins MR, Anderson TD, Johnson KJ. Development of an internally controlled antibody microarray. Mol Cell Proteomics. 2005;4:1664–72. doi: 10.1074/mcp.M500052-MCP200. [DOI] [PubMed] [Google Scholar]
- 87.Gao WM, Kuick R, Orchekowski RP, Misek DE, Qiu J, Greenberg AK, Rom WN, Brenner DE, Omenn GS, Haab BB, Hanash SM. Distinctive serum protein profiles involving abundant proteins in lung cancer patients based upon antibody microarray analysis. BMC Cancer. 2005;5:110. doi: 10.1186/1471-2407-5-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miller JC, Zhou H, Kwekel J, Cavallo R, Burke J, Butler EB, Teh BS, Haab BB. Antibody microarray profiling of human prostate cancer sera: antibody screening and identification of potential biomarkers. Proteomics. 2003;3:56–63. doi: 10.1002/pmic.200390009. [DOI] [PubMed] [Google Scholar]
- 89.Orchekowski R, Hamelinck D, Li L, Gliwa E, Van Brocklin M, Marrero JA, Vande Woude GF, Feng Z, Brand R, Haab BB. Antibody microarray profiling reveals individual and combined serum proteins associated with pancreatic cancer. Cancer Res. 2005;65:11193–202. doi: 10.1158/0008-5472.CAN-05-1436. [DOI] [PubMed] [Google Scholar]
- 90.Sanchez-Carbayo M, Socci ND, Lozano JJ, Haab BB, Cordon-Cardo C. Profiling bladder cancer using targeted antibody arrays. Am J Pathol. 2006;168:93–103. doi: 10.2353/ajpath.2006.050601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koga H, Kyo M, Usui-Aoki K, Inamori K. A chip-based miniaturized format for protein-expression profiling: the exploitation of comprehensively produced antibodies. Electrophoresis. 2006;27:3676–83. doi: 10.1002/elps.200500821. [DOI] [PubMed] [Google Scholar]
- 92.Huber W, Mueller F. Biomolecular interaction analysis in drug discovery using surface plasmon resonance technology. Curr Pharm Des. 2006;12:3999–4021. doi: 10.2174/138161206778743600. [DOI] [PubMed] [Google Scholar]
- 93.Lyon LA, Musick MD, Natan MJ. Colloidal Au-enhanced surface plasmon resonance immunosensing. Anal Chem. 1998;70:5177–83. doi: 10.1021/ac9809940. [DOI] [PubMed] [Google Scholar]
- 94.Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR. DNA-encoded antibody libraries: a unified platform for multiplexed cell sorting and detection of genes and proteins. J Am Chem Soc. 2007;129:1959–67. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alberghina L, Chiaradonna F, Vanoni M. Systems biology and the molecular circuits of cancer. Chembiochem. 2004;5:1322–33. doi: 10.1002/cbic.200400170. [DOI] [PubMed] [Google Scholar]
- 96.Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J. Cancer: a Systems Biology disease. Biosystems. 2006;83:81–90. doi: 10.1016/j.biosystems.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 97.Khalil IG, Hill C. Systems biology for cancer. Curr Opin Oncol. 2005;17:44–8. doi: 10.1097/01.cco.0000150951.38222.16. [DOI] [PubMed] [Google Scholar]
- 98.Stilwell JL, Guan Y, Neve RM, Gray JW. Systems biology in cancer research: genomics to cellomics. Methods Mol Biol. 2007;356:353–65. doi: 10.1385/1-59745-217-3:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang E, Lenferink A, O'Connor-McCourt M. Cancer systems biology: exploring cancer-associated genes on cellular networks. Cell Mol Life Sci. 2007;64:1752–62. doi: 10.1007/s00018-007-7054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21:4205–8. doi: 10.1093/bioinformatics/bti688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Price ND, Shmulevich I. Biochemical and statistical network models for systems biology. Curr Opin Biotechnol. 2007;18:365–70. doi: 10.1016/j.copbio.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Czernin J, Phelps ME. Positron emission tomography scanning: current and future applications. Annu Rev Med. 2002;53:89–112. doi: 10.1146/annurev.med.53.082901.104028. [DOI] [PubMed] [Google Scholar]
- 103.Hsueh WA, Kesner AL, Gangloff A, Pegram MD, Beryt M, Czernin J, Phelps ME, Silverman DH. Predicting chemotherapy response to paclitaxel with 18F-Fluoropaclitaxel and PET. J Nucl Med. 2006;47:1995–9. [PubMed] [Google Scholar]
- 104.Su H, Bodenstein C, Dumont RA, Seimbille Y, Dubinett S, Phelps ME, Herschman H, Czernin J, Weber W. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006;12:5659–67. doi: 10.1158/1078-0432.CCR-06-0368. [DOI] [PubMed] [Google Scholar]
- 105.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA. 2007;104:1777–82. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Price ND, Reed JL, Palsson BO. Genomescale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol. 2004;2:886–97. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 107.Mulquiney PJ, Kuchel PW. Modelling metabolism with Mathematica detailed examples including erythrocyte metabolism. 1. Boca Raton, Fla.: CRC Press; 2003. [Google Scholar]
- 108.Price ND, Schellenberger J, Palsson BO. Uniform sampling of steady-state flux spaces: means to design experiments and to interpret enzymopathies. Biophys J. 2004;87:2172–86. doi: 10.1529/biophysj.104.043000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thiele I, Price ND, Vo TD, Palsson BO. Candidate metabolic network states in human mitochondria. Impact of diabetes, ischemia, and diet. J Biol Chem. 2005;280:11683–95. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 110.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 111.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 112.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–61. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 113.Christopher R, Dhiman A, Fox J, Gendelman R, Haberitcher T, Kagle D, Spizz G, Khalil IG, Hill C. Data-driven computer simulation of human cancer cell. Ann N Y Acad Sci. 2004;1020:132–53. doi: 10.1196/annals.1310.014. [DOI] [PubMed] [Google Scholar]
- 114.Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, Thorsson V. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hayete B, Gardner TS, Collins JJ. Size matters: network inference tackles the genome scale. Mol Syst Biol. 2007;3:77. doi: 10.1038/msb4100118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–90. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 117.Kumar N, Hendriks BS, Janes KA, De Graaf D, Lauffenburger DA. Applying computational modeling to drug discovery and development. Drug Discov Today. 2006;11:806–11. doi: 10.1016/j.drudis.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 118.Hood L, Perlmutter RM. The impact of systems approaches on biological problems in drug discovery. Nat Biotechnol. 2004;22:1215–7. doi: 10.1038/nbt1004-1215. [DOI] [PubMed] [Google Scholar]