

Abstract
Glucocorticoids (GCs) are provided as co-medication with chemotherapy in breast cancer, albeit several lines of evidence indicate that their use may have diverse effects and in fact may inhibit chemosensitivity. The molecular basis of GC-induced resistance to chemotherapy in breast cancer remains poorly defined. Recent researchers, in an attempt to clarify some aspects of the underlying pathways, provide convincing evidence that GCs induce effects that are dependent upon the glucocorticoid-receptor (GR)-mediated transcriptional regulation of specific genes known to play key roles in cellular/tissue functions, including growth, apoptosis, differentiation, metastasis and cell survival. In this review, we focus on how GC-induced chemoresistance in breast cancer is mediated by the GR, unravelling the molecular interplay of GR signalling with other signalling cascades prevalent in breast cancer. We also include a detailed description of GR structure and function, summarizing data gained during recent years into the mechanism(s) of the cross-talk between the GR and other signalling cascades and secondary messengers, via which GCs exert their pleiotropic effects.
Keywords: glucocorticoid receptor, breast cancer, glucocorticoid resistance, apoptosis, cell survival, cell signalling, chemotherapy resistance
Introduction
Breast cancer is an aggressive, high-mortality cancer and the most common cancer among women in the United States [1]. It is associated with a high incidence of axillary lymph node involvement in patients with breast tumour sizes less than 1 cm [2].
Converging evidence indicates that co-administration of dexamethasone with emetogenic chemotherapy appears to be a suitable antiemetic therapy in cancer patients with solid tumours, including breast cancer [3, 4].
Although glucocorticoids (GCs) are used to support chemotherapy, pre-clinical data (cellular studies in vitro) suggest that GC show diverse and even contradictive effects on chemosensivity in many non-haematological tumour cells from breast, bone, brain, cervix, melanoma and neuroblastoma [5, 6]. Notably, a breast cancer xenograft study has shown that dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumour cell apoptosis [7]. On the other hand, in a recent study, assessing the effects of dexamethasone pre-treatment on the anti-cancer activity of adriamycin in a model of breast cancer, dexamethasone potentiates the anti-tumour activity of adriamycin and may be used as chemosensitizer and chemoprotectant [8]. In view of the controversial data and due to the use of dexamethasone as co-medication in conventional breast cancer treatment modalities, a detailed molecular investigation of the GC signalling in breast cancer is needed urgently.
Extensive evidence suggests that carcinogenesis is a multi-factorial process, characterized by a progressive transformation of normal human cells into highly malignant invasive cancer cells. Many regulatory circuits and signal transduction pathways are disrupted in cancerous cells, including de-regulation of growth factor signalling events, defects in cellular proliferation and/or apoptosis, in differentiation, in invasion and metastasis. Alterations of crucial importance are observed at the molecular level, dictating the expression of multiple genes, including survival genes, onco-genes, oncosuppressor genes as well as genes associated with hypoxia, inflammation and angiogenesis.
In this review, we have summarized some recent discoveries that elucidate the molecular mechanisms via which GCs induce changes in breast-cancer-associated signal transduction pathways, including regulation of growth factors, transcription factors (TFs), mitogen-activated protein kinases (MAPKs), oncogenes, tumour suppressor genes, survival genes, proliferation and apoptosis-related genes and proteins, cytokines and other intracellular secondary messengers. The advent of molecular biology techniques and microarray technology has made it possible to unravel how GCs, via the glucocorticoid receptor (GR), are causally involved in resistance to chemotherapy-induced cell death in breast cancer. We considered essential to include also a detailed description of the structure and functions of the GR, the cornerstone molecule mediating the GCs'cellular effects.
Glucocorticoid receptor structure and mechanism of action
GCs secreted by the adrenal gland in response to various stress signals, exert pleiotropic effects in virtually every organ and tissue of human body, including regulation of metabolism, cell growth, apoptosis and differentiation, inflammation, vascular tone, mood and cognitive function as well as immunosuppressive actions [9].
GCs mediate their effects on target cells through binding to their intracellular receptor, the GR, a member of the nuclear receptor family. The classic GR, today known as GR-α, binds GC whereas a second GR, now called GR-β does not bind GC. Both GR-α and GR-β are products of the same gene, located on chromosome 5, and result from differential splicing through the alternative use of two distinct terminal exons, 9α and 9β respectively. The first 727 amino acids from the N-terminus are identical in the two iso-forms, GR-α being a 777 amino acid protein whereas GR-β isoform contains 742 amino acids. In GR-β the 50 carboxyterminal amino acids of GR-α have been replaced by 15 non-homologous amino acids (encoded by exon 9β) in the C-terminus. GR-α contains three distinct functional domains – the ligand-binding domain (LBD), the DNA-binding domain (DBD) and the N-terminal domain (known as transactivation domain AF-1). The LBD contains also a transactivation domain (AF-2) that is involved in transcriptional activation of target genes [10–12] (Fig. 1).
1.
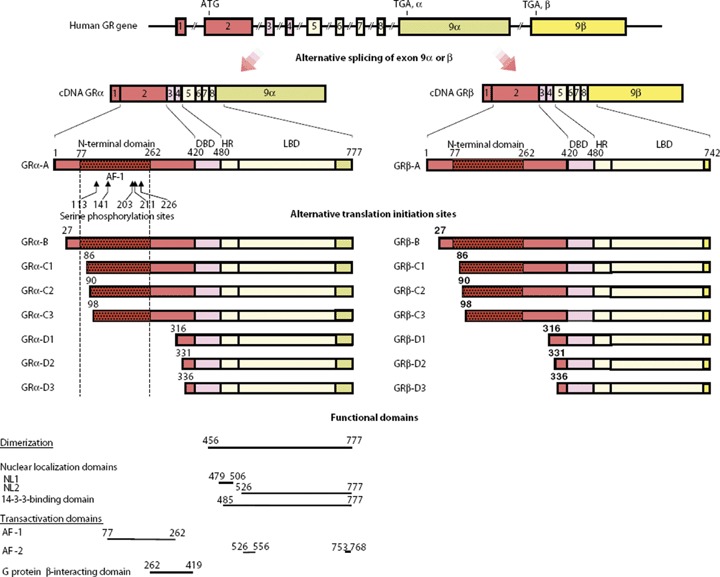
Genomic and complementary DNA, protein structures and functional domains of human GR isoforms. The human GR gene consists of 10 exons. Exon 1 is an untranslated region; exon 2 encodes the N-terminal ‘immunogenic’ domain; exons 3 and 4 encode the DNA-binding domain;and exons 5 through 9 encode the hinge region and the LBD. The GR gene contains two terminal exon 9s (9α and 9β), which are alternatively spliced to produce the classic GR-α (GRα-A) and the non-ligand-binding GRβ-A, which exerts dominant negative effects upon GR-α (GRα-A). C-terminal domains colored as light green and yellow in GRαs and GR-βs show unique portions of their amino acid sequences. GR-α N-terminal translational isoforms expressed from a single GR-α transcript are shown in the middle of the figure. The GR-β transcript may also produce similar N-terminal isoforms from the same start sites as GR-α. AF-1 and -2, activation function 1 and 2; DBD, DNA-binding domain; HR, hinge region; LBD, ligand-binding domain; NL1 and 2, nuclear translocation signal 1 and 2. ‘From G. P. Chrousos, T. Kino. Intracellular glucocorticoid signalling: A formerly simple system turns stochastic. Sci. STKE 2005, pe48. Reprinted with permission from AAAS’.
In the absence of GCs, GR-α resides in the cytoplasm forming a complex with heat shock proteins (HSPs) 90, 70, 50, 20 and other proteins (chaperones) which maintain GR in a conformation suitable for ligand binding. GR-α, after binding to GCs, undergoes conformational changes, dissociates from the HSPs, homodimerizes and translocates into the nucleus where it interacts directly with its specific DNA sequences, the glucocorticoid-response elements (GREs), in the promoter of target genes. The GRa/GRE complex results in stimulation or reduction of the GRE-mediated gene transcription (known as transactivation effect) (Fig. 2).
2.
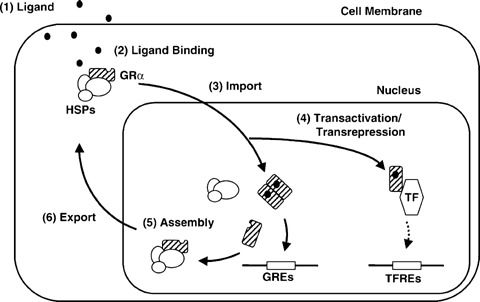
Shuttling of GR-α between the cytoplasm and the nucleus and its transactivating or transrepressive activities. Possible sites of intervention, which may change the activity of GR-α are indicated by numbers. GR: glucocorticoid receptor; GREs: glucocorticoid-responsive elements; TFREs: transcription factor responsive elements; HSP: heat shock proteins; TF: transcription factor. ‘This article (figure) was published in Journal of Steroid Biochem. Mol. Biol. 85, T. Kino, MU De Martino, E. Charmandari, M. Mirani, GP. Chrousos, Tissue glucocorticoid resistance/hypersensitivity syndromes, 457–467, Copyright Elsevier 2003’.
GR-α monomers, activated by glucocorticoids, may also interact with other TFs, such as CRE-binding protein (CREB), signal transducer and activator of transcription 5 (STAT5), activator protein 1 (AP-1) and nuclear factor-kB (NF-κB), via protein-protein interactions, thus influencing indirectly the activity of these TFs on their target genes (known as transre-pression effect) [13–15] (Fig. 2). Many other cellular proteins, the co-activators such as the nuclear receptor co-activator complexes p160, p300/CREB-binding protein (CPB), p300/CBP-associated factor (P/CAF) as well as the co-repressors may interact with GC–GR complex through AF-1 and AF-2 activation functions [16]. AF-2 function is activated upon ligand binding whereas AF-1 function interacts with components of the basic transcriptional machinery. The histone acetyltransferase activity shown by co-activators is of particular importance in chromatin remodelling since it loosens the chromatin structure and facilitates the binding of transcriptional machinery to DNA [17]. The hGR-β is constitutively localized in the nucleus of cells, it does not bind glucocorticoids and is transcriptionally inactive [18, 19]. Recent studies have shown that each GR-α mRNA and GR-β mRNA is translated from at least eight initiation sites into multiple GR-β and possibly GR-β isoforms having variable transcriptional activities with distinct transactivation and transrepression patterns [20, 21] (Fig. 1).
Glucocorticoids regulate the expression of a wide array of target genes by both positive and negative regulatory mechanisms. Gene profiling revealed enhancing as well as suppressive actions of dexamethasone on immune cells, 9% of the genes were down-regulated while 12% of genes were up-regulated [22]. The immunosuppressive anti-inflammatory effects of glucocorticoids are mediated via the tran-srepression function of GR whereas the undesirable side effects (e.g. insulin resistance, growth failure, osteoporosis, glucocorticoid resistance) are thought to occur mainly via the activation of gene transcription (transactivation function) [9, 14, 15]. It is now appreciated that a mutual antagonism between cytoplasmic TFs, e.g. NF-κB (which is activated by inflammatory signals) and GR-α (which is activated by endogenous or exogenous GCs) results in opposed functions in regulating inflammation(stimulatory versus inhibitory, respectively). Excess activation of one TF (e.g. NF-κB activation) will shift cellular responses towards insensitivity and/or resistance to cortisol [9, 14, 15]. Conceivably, apart from the cognate ligand, multiple factors may influence the transcriptional activity of GR, such as the TFs AP-1 and NF-κB, thus affecting negatively the GR-mediated transactivation effects and tissue sensitivity to glucocorticoids.
Other examples of the cross-talk between the GR-mediated signalling with other signalling cascades include post-translational modifications, as well as the interaction of GR with other cell-specific intracellular molecules and secondary messengers. Extracellular signals are known to activate a number of GR phosphorylation sites (113, 141, 203, 211 and 226) which are located at the N-terminal domain of GR proteins [21, 23, 24]. For example, the extracellular signal-regulated kinase (ERK), the c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) phosphorylate at serine 211 and 226. Cyclin-dependent kinases (CDKs) phosphorylate at serines 203 and 211. A positive correlation has been observed between the amount of S211 phosphorylation and transactivation activity of GR in stably transfected U-20S cells. In the absence of ligand, GR is phosphorylated at serines 203 or 211 and resides in the cytoplasm. In presence of GCs, GR phosphorylated at S211 translocates into nucleus, but not the GR phosphorylated at 203, indicating that GR-phosphorylation status regulates its sub-cellular localization and its transcriptional activity [25]. Conceivably, the MAPK-mediated signals alter GR phosphorylation status and its transcriptional activity and affect GC-induced effects, thus inhibiting or increasing tissue sensitivity to GCs [26, 27].
The molecular mechanism(s) via which GR-signalling leads to cell growth arrest, apoptosis and differentiation are currently of high research interest. Extensive evidence supports that in haematological malignancies the transactivation as well as the transrepression activity of GR is required for mediating GC-induced apoptosis [28–32]. Moreover, data suggest that GR-induced processes regulate the transition from G1 to S phase, the late G1 and S phase being sensitive to glucocorticoids whereas G2, M and early G1 phase being resistant to GCs [33]. Of note, in S phase GR is phosphorylated (at a low basal level) whereas in mitotic phase (G2/M) GR is hyperphosphorylated suggesting that hyperphos-phorylation of GR might account for the GC resistance during G2M phase [34]. Current studies however, provide data supporting that GR itself is fully functional throughout the entire cell cycle, but GR responsiveness is repressed in mitosis due to chromatin condensation rather than to specific modification of GR [35]. It must be stressed, that in lymphoid cells the p38 MAPK is a key mediator in GC-induced apoptosis associated with a site-specific phosphorylation of the human GR at serine 211 [36]. While GR phosphorylation is associated with cell cycle regulated transcriptional control, protein phosphates (PP5) have been shown to participate as inhibitors of GR-mediated signalling networks leading to growth arrest [37].
In addition to phosphorylation, acetylation and methylation are also important post-translational protein modifications that affect the function of proteins. Although there is not any direct evidence that GR activity may be modified by direct acetylation and methylation, some reports support that acetylation and methylation of GR-accessory proteins may modulate GR function in an indirect fashion and certainly warrants further investigation [21, 38–41]. The ubiquitin-proteasome-mediated degradation pathway regulates also the glucocorticoid signalling system by controlling the degradation rates of GR [42]. The phosphorylation status of GR protein plays an important role in the turnover of the receptor, since mutation of all phosphorylation sites in GR has resulted in increased receptor half-life and abolished down-regulation [24].
The N-terminal domain of GR contains also a novel site that interacts with the β-component of the heterotrimeric quanine nucleotide-binding protein (G protein) complex, resulting in the suppression of GR-induced transactivation of GC responsive genes. Conceivably, G-protein coupled receptor (GPCR)-signals activated by extracellular hormones and other compounds may also influence GR transcriptional activity and tissue sensitivity to glucocorticoids [43].
Finally, a series of studies have documented the presence of GR in mitochondria of various cell lines and tissues and the presence of nucleotide sequences in the human mitochondrial genome, with strong similarities to GREs [44–46]. The potential of GR to bind to putative mitochondrial GREs, has given support to hypothesis that mitochondrial genes may be sites of primary action of glucocorticoids [44]. Furthermore, a role of mitochondrial GR has been suggested in glucocorticoid-induced apoptosis in haematological cells [47].
Cell survival and apoptosis
Among the most important regulatory circuits that are disrupted in breast cancer cells are the defects in cell proliferation and/or apoptosis. Effective anti-cancer agents act by modulating expression of genes controlling apoptosis or cell proliferation. Accumulated data provide evidence that while GCs induce apoptosis in leukaemic cells, lymphocytes and lymphoma cells, they seem to protect against apoptosis in mammary epithelial cells [5, 6].
The advent of microarray technology resulted in the identification of genes and patterns of gene expression associated to apoptosis. Wu et al. performed a large-scale oligonucleotide microarray screen of GR-regulated genes and found that several of the genes induced 30 min. after GR activation encode proteins that function in mammary epithelial cell survival signalling pathways [48]. The majority of these target genes encode signal transduction proteins, e.g. the serum and GC-inducible kinase (SGK-1), the MAPK phosphatase-1 (MKP-1), TFs as well as cell cycle/DNA repair proteins.
SGK-1 is a serine/threonine kinase, a downstream target of the phosphatidylinositol-3-kinase (PI3K) pathway, which mitigates growth factor deprivation-induced apoptosis in mammary epithelial cells [49]. A GRE (SgK GRE) between –1000 and –975 bp has been defined in SgK promoter, the binding of GR to SgK GRE conferring glucocorticoid responsiveness [50]. Glucocorticoids increase MKP-1 gene expression. The promoter of MKP-1 contains three putative GREs while over-expression of MKP-1 has also been associated with increased tumorigenicity in breast cancer [51–54].
Wu et al. identified SGK-1 and MKP-1 as direct transcriptional targets of GR activation involved in survival signalling since (i) endogenous SGK-1 and MKP-1 protein levels were increased in breast cancer cell lines treated with Dex before chemotherapy treatment (paclitaxel and doxorubicin), (ii) ectopic expression of either SGK-1 or MKP-1 inhibited chemotherapy-induced apoptosis in a way similar to Dex pre-treatment, (iii) there was a decrease in Dex-mediated protection from chemotherapy in the presence of either SGK-1 small interfering RNA (siRNA) or MKP-1 siRNA [48]. Taken together, SGK-1 and MKP-1 are direct transcriptional targets of GR activation and contribute, at least in part, to a GR-mediated signalling that ultimately inhibits chemotherapy-induced apoptosis in breast cancer cells.
MAPKs are activated by a diverse array of extra-cellular stimuli and include extracellular signal-regulated kinases (ERK 1/2), c-Jun N-terminal kinase (JNK) and the p38 protein kinase [55, 56]. The MAPK phosphatase (MKP-1), induced by dexamethasone, growth factors and cellular stress, dephosphorylates and inactivates both SAPK/JNK and p38 [57–59].
Seeking for a potential role of MAPK signal pathway in GR-mediated inhibition of apoptosis in breast cancer cells, Wu et al. showed that paclitaxel treatment resulted in MAPK activation and apoptosis of MDA-MB-231 breast cancer cells, and that both processes were inhibited by Dex pre-treatment [60]. They demonstrated that the induction of MKP-1 (its promoter contains three putative GREs) following Dex treatment, resulted in the specific inhibition of paclitaxel-induced ERK 1/2 and JNK activity [51, 60]. In addition, MKP-1 siRNA inhibited GR-mediated ERK and JNK dephosphorylation and cell survival. Finally, a set of the Ets-like TF-1 (ELK-1)-dependent target genes (e.g. tPA) were down regulated 2–24 hrs following GR activation suggesting that glucocorticoid signalling mediates both early (direct) and later (indirect) gene expression alterations leading to cell survival.
Of note, Wu et al. reported that GR-activation mediates further Forkhead TF 3a (FOXO3a) phosphorylation and inactivation in mammary epithelial cells, concomitantly with a reduction in transcriptional target genes of FOXO3a such as IGF-binding protein-3 (IGFBP-3) [61]. In conclusion, the GR-mediated inactivation of genes such as MAPKs, IGFBP-3, tPA and GR-mediated activation of genes such as MAPK phosphatase (MKP-1) contribute to glucocorticoid-mediated mammary epithelial cell survival.
Ultimately, understanding how GCs inhibit cell death may lead to the identification of molecular targets for breast cancer treatment.
Growth factor signalling
De-regulation of growth signalling pathways and acquired autonomy in growth signals are among the main characteristics which dictate malignant mammary cellular growth. One of the most active growth factors in breast glandular cell proliferation is the epidermal growth factor (EGF). It exerts potent growth-promoting effects in mammary epithelium and is involved in the proliferation and migration of breast cancer cells [62–65], processes mediated mainly via binding to the EGF receptor [66, 67]. Glucocorticoids have been shown to increase EGF binding to its receptor accompanied by increases also in EGF-R mRNA levels in human breast cancer cell lines [68].
Research results indicate that EGF stimulates sphingosine kinase (SK-1) activity accompanied by increased mRNA and protein expression of SK-1, effects mediated, mechanistically, via protein kinase C (PKC) and the phosphoinositide 3-kinase. Sphingosine kinase (SK) (SK-1 and SK-2 subtypes) is the key enzyme catalysing the formation of sphingosin 1-phosphate (S1P), a cellular regulator which acts via binding to cell surface G-protein-coupled receptors (S1P receptors) [69] and regulates numerous cellular processes including cell movement and proliferation [70–73]. Data support that SK is activated by various growth and survival factors [74]. Recently, interesting findings by Döll et al. support that dexamethasone treatment inhibits the EGF-induced SK-1 mRNA expression and activity, via a GR-dependent manner, resulting in reduced EGF-induced proliferation and migration [75]. It must be noted that dexamethasone by itself had no significant effect on SK-1 mRNA expression despite the fact that the SK-1 promoter contains a putative GRE (very near to the translation site). Furthermore, transfection with SK-1 siRNA inhibited EGF-induced proliferation, thus supporting that the sphingosine kinase-1 may represent a novel target for breast cancer therapy [75, 76]. The mechanism(s) underlying the lowering effect of dexamethasone on EGF-induced SK-1 expression and the role of GR in this pathway needs to be further elucidated.
Oncogenes
Among the hallmarks of breast cancer is the prevalence of dominant oncogenes which are activated and ultimately lead to cellular growth, proliferation and escape from apoptosis. The protooncogene c-fms encodes the growth factor CSF-1 receptor (CSF-1R). Abnormal expression of CSF-1, CSF-1R are associated with increased disease risk and poor outcome while transfection of c-fms into normal mammary epithelial cells increases cellular invasion and anchorage-independent growth [77]. Injection, on the other hand, of human breast cancer cells expressing high c-fms levels into spleen in a severe immunodeficient mice model of experimental metastasis, has resulted in increased tumorigenicity and metastasis as compared to control breast cancer cells [78].
Glucocorticoids, including dexamethasone, upregulate c-fms in breast cancer cells in vitro, via a GR-dependent pathway [79, 80], associated with an increase in cancer invasiveness [81, 82]. Sequence upstream of both c-fms gene promoters contained potential GREs, while elimination of the GRE closest to the promoter abolished glucocorticoid stimulation. These data point out that GCs regulate the proliferation/differentiation of neoplastic mammary epithelial cells, at least in part, via the regulation of the c-fms gene product, i.e. the CSF-1R. Furthermore, Flick et al. have shown that it is actually a composite GRE that mediates the GCs-induced increase in c-fms protooncogene. It contains overlapping binding sites for AP-1 proteins and the GR. Of note, a reduced basal transcription activity of this promoter and lack of GC stimulation was observed when the AP-1 site was mutated, implicating that AP-1 proteins, GR and associated co-factors regulate the c-fms first promoter expression. Differences in recruitment of the various components seem to be responsible for cell specific repression and activation of c-fms gene in breast carcinoma cell lines [83].
Another example of how GCs modulate downstream intracellular regulators of c-fms, involves the serum-glucose kinase 1 (SGK-1), a serinethreonine kinase known to be associated with survival pathways [49, 84, 85]. Tangir et al., by using an oligonucleotide array representing 16,700 known expressed human genes, analysed the gene expression profile of breast cancer cells exposed to dexamethasone. They reported that SGK-1 was consistently over-expressed in the Dex-exposed cells, an increase correlated with Dex-induced adhesiveness [86]. Such data implicate that SGK-1 may be the mediator of c-fms-induced adhesiveness and related aggressive phenotype of breast cancer cells after exposure to GC [86].
Conceivably, therapeutic approaches targeting GR-mediated gene expression may block the invasiveness, motility and adhesiveness of breast cancer.
Tumour suppressor genes
The tumour suppressor p53 is of paramount importance since it protects the cells from different stress conditions, including DNA damage and hypoxia. P53 mediates its effects via activation of gene transcription and the regulation of genes (stimulation or repression) containing PBS (p53 binding sites) which ultimately lead to growth arrest and apoptosis. Loss of its activity has been associated with various types of human cancer, including breast cancer. The function of p53 depends on its cross-talk with many other cellular proteins and nuclear receptors [87].
The functional interactions between p53 and GR are of crucial importance in different physiological and pathological conditions. The cross-talk between p53 and GR has been shown to lead to mutual inhibition by various mechanisms, either at the level of their cytoplasmic sequestration or at a transcriptional level of regulation. Complementation, however, and synergism between p53 and GR functions have also been reported [88].
The positive or negative p53-GR interactions have been looked in various cellular processes, in particular, however, in growth arrest, proliferation and apoptosis of cells [88]. In breast carcinoma, wild type p53 has been suggested to be cytoplasmic [89], implicating that p53 may be complexed by GR in the cytoplasm, raising the possibility that GR either alone or in concert with other proteins serves as cytoplasmic anchor for p53, thereby regulating its function as a TF [84]. In mammary epithelial cells, apart from the cytoplasmic sequestration, p53 and GR also interact at the transcriptional level for the regulation of SGK. As mentioned previously, SGK is a novel member of the serine/threonine protein kinase family that is transcriptionally regulated by serum and glucocorticoids in both non-tumorigenic and transformed mammary epithelial cells. It functions as a key cell survival component in response to different environmental stress stimuli in non-tumorigenic mammary epithelial cells [85].
Elucidation of the molecular mechanisms involved revealed that p53 stimulates the promoter activity of SGK (four of the p53 binding sites in the SGK promoter are specifically recognized by the p53 protein) [90]. It is also of interest that p53 has been shown to inhibit the binding of GR to the SGK-GREs or a consensus GRE. Conversely, activated GRs have also the potential to suppress the p53 transactivation function but not its transrepression activity, thereby indicating a mutual interference of transactivation functions of p53 and GR, possibly through their direct interaction at the level of transcriptional regulation [50].
It is of importance that Urban et al., in human lung carcinoma cells, identified a functional link for the p53 tumour suppressor protein in dexamethasone-induced growth suppression [91]. They showed that suppression of p53 expression is associated with a decrease in the basal expression of the CDK inhibitor protein p21 (WAF1/Cip1) and a concomitant increase in the rate of cell proliferation. Suppression of p53 blocked the dexamethasone-induced p21 expression and G(1) growth arrest. Dexamethasone treatment was also associated with hyperphosphorylation of p53 at Ser-15, an effect observed when PP5 (a serine/threonine phosphatase known to suppress p53) expression was suppressed. Overall, Urban et al. indicated that the basal expression of p53 plays a functional role in a GR-mediated response, regulating the expression of p21 via a mechanism that is suppressed by PP5 and associated with the phosphorylation of p53 at Ser-15 [91].
In light of above, an excessive activation of either p53 or GR-mediated signalling will shift the balance between apoptotic and survival routes, thus determining the ultimate fate of the cell.
Metastasis suppressor genes
Highly metastatic tumour cells are characterized by reduced expression of metastasis suppressor genes while their re-expression has been shown to inhibit metastasis [92, 93]. Conceivably, among the new approaches in breast cancer therapy is the development of drugs targeting metastasis suppressor gene expression. The nm23 gene family consisted from eight family members is of particular importance, the nm23-H1 playing a key role in human breast cancer [94–96]. Recent studies present evidence that dexamethasone elevates Nm23-H1 and Nm23-H2 expression via a GRE-dependent transcriptional mechanism [97, 98]. Furthermore, methoxyproges-terone acetate (MPA) has been shown to elevate also the Nm23-H1 protein expression, via a GR-mediated mechanism, 3-fold over a 10 nm to 1 μM dose range in breast carcinoma in vitro while reducing the soft agar colonization of metastatic breast cancer cell lines by ∼50%. On this respect, MPA is considered as a first generation lead agent for the elevation of Nm23-H1 metastasis suppressor expression and the inhibition of metastatic colonization.
Metastasis is a multi-step process, mediated by multifunctional gene products that digest basal membranes, interact with extracellular matrix proteins, protect cells from death and promote angiogenesis. Gene profiling studies have shown that among the differentially expressed entities in breast cancer metastasis, were the invasion-, extracellular matrix protein- and tissue remodelling-related genes, cell cycle regulatory genes and other genes. Of note, a 76-gene profiling has represented a strong prognostic factor for the development of metastasis in breast cancer.
The divergence of GC action on metastasis related genes, such as the lowering effect on metastasis suppressor Nm23 gene vis-á-vis the c-fms-induced expression presented in this review, may be partly explained by the fact that ligand-mediated regulation of single target genes may not necessarily converge and may differ from its generalized effects on metastasis which are the reflection of much more complex transcriptional and translational functions. Animal studies and human studies have also shown that receiving GCs had a statistically significant increase in metastasis [99–102].
Transcription factors
The dual action of GCs enhancing or repressing gene activity in a cell type specific manner has been attributed to the cross-talk of GR with other multiple cellular factors such as TFs, other regulatory proteins (co-activators, co-repressors), chief among them the TFs AP-1 and NK-kB.
AP-1 is activated by various stimuli, including mitogens, oncoproteins, thus playing a major role in cellular proliferation, survival and differentiation, invasion, angiogenesis and apoptosis and overall in carcinogenesis [103–106]. AP-1 has been implicated in breast cancer as it has been correlated with tumour grade, cell cycle regulating protein expression, while its inhibition causes blockade of signal transduction pathways and inhibits breast cancer growth [107–111]. In the mouse mammary gland, dexamethasone has been shown to inhibit involution and programmed cell death [112]. Injection of Dex, leading to milk accumulation, has been accompanied by increase in PKA, c-fos, junB and junD mRNA levels as well as increase in AP-1 DNA-binding activity. However, in Dex-treated animals some AP-1 and c-Jun target genes (stromelysin-1 and SGP-2 induced during normal involution) were inhibited. Taken together, the cross-talk between GRs and AP-1 may lead to impairment in AP-1 activity and apoptosis in the post-lactational mammary gland. In addition, it seems that the anti-apoptotic activity of Dex enhances the differentiation related products (such as milk) [112].
NF-κB is another significant example that has been shown to be involved in inactivation of apoptosis pathways in mammary epithelium [111, 113, 114]. NF-κB activated by TNF-α results in the induction of anti-apoptotic proteins, while derangements in NF-κB activation or its transcriptional anti-apoptotic products may render cells extremely sensitive to TNF-α-induced apoptosis [115, 116]. TNF-α is a cytokine which stimulates both, cell death pathways but survival pathways also [117]. Depending on signalling activated intracellular adaptors TNF-α may lead to apoptosis or it may activate multiple cell survival intracellular signals such as NF-κB, JNK, p38 and ERK [118]. Ultimately, the balance between pro- and anti-apoptotic regulators, will define the disease evolution, i.e. whether TNF-α will be a tumour necrosis factor or a tumour-promoting factor [119]. Chronically produced endogenous TNF-α by a tumour is considered to enhance tumour development while locally administered high doses of TNF-α elicit a powerful anti-tumoral effect [120]. Notably, NF-κB activation by both, PI3K or Akt, both involved in cell survival pathways, has been shown to suppress TNF-α-induced apoptosis in MCF-7 cells [121]. AKT, a kinase with a strong homology to SGK1, is activated by the PI3-K pathway and is involved in cell survival pathways [122–124].
In human mammary epithelial cells (MCF10A), Dex activation, via a GR-mediated mechanism, resulted in potent survival pathways, independently of the anti-apoptotic PI-3K and Akt/protein kinase B signalling pathways [125]. GCS, in particular Dex, has been shown to completely abrogate the TNF-α-induced apoptosis in MCF-7 cells [126, 127]. Recent data demonstrated that NF-κB, but not PI3K/Akt activation is required for the Dex protective effect against TNF-α-mediated cell death, and correlates with lack of degradation of the anti-apoptotic protein c-IAPI [128, 129]. Concluding, the anti-apoptotic effect of Dex in the TNF-α-induced apoptosis in MCF-7 cells is probably mediated via NF-κB and it is likely that the NF-κB dependent gene expression of anti-apoptotic proteins (c-IAPI) is Dex-dependent possibly through the GC receptor. On this respect, the NF-κB system may be a potential therapeutic target against the GC-dependent form of breast cancer.
Signalling pathways cross-talk
Melatonin, secreted by the pineal gland, elicits various biological responses in many cell types and tissues, including suppression of MCF-7 breast cancer cell growth [130–132]. It mediates its effects via its specific receptors MT1 and MT2, both members of the GPCR superfamily [133, 134]. The MT1 receptor is considered responsible for the melatonin's growth-suppressive action in MCF-7 cells [135]. Cardinal role in the melatonin-mediated breast cancer cell growth inhibition, plays the coupling of MT1 receptor to the Gai2 protein coupled receptor and subsequent suppression of oestrogen receptor alpha (ERα) transcriptional activity and cell proliferation [136–138].
Considerable interest has been generated on recent findings revealing that melatonin modulates not only the transcriptional activity of the oestrogen receptor (ERα) but melatonin has the potential to repress also the dexamethasone-induced activation of GR [139]. To this effect, activation of GPCRs may work as a biological modifier of the transcriptional activity of Dex-induced GR-mediated effects and possibly inhibit the anti-proliferative effects of GCs in breast cancer cells.
Crosstalk with other steroid receptor signalling
Recent as well as earlier evidence points out the extensive cross-talk of GR signal transduction with other steroid receptor-mediated signalling, including progesterone and oestrogen receptor pathways.
Glucocorticoids and progestins bind to receptors, GR and progesterone receptor (PR), respectively, that share many structural and functional similarities. Several lines of evidence indicate that anti-progestins via PR inhibit GR-mediated transcription in breast cancer cells [140, 141]. Glucocorticoid-like effects of progesterone have been shown in some tissues, while progesterone-like effects of glucocorticoids in other tissues have also been demonstrated [142]. Interesting recent findings document the overlapping but distinct profiles of gene expression elicited by glucocorticoids and progestins [143]. Notably, in breast cancer cells expressing PR, dexamethasone and cortisol mimic the effects of progesterone by inducing significant growth inhibition, cell spreading and focal adhesions, effects shown to be mediated by cross-talk with PR [144]. On the contrary, in PR-negative but GR-positive breast cancer cells, dexamethasone induces a small increase of focal adhesions and a small growth stimulation, effects not mediated by progesterone. The role of the cross-talk between GR and PR signalling cascades in breast cancer cells is gradually unravelled, the GR and PR sharing some overlapping activity in mediating focal adhesion but not in regulating cell proliferation [144]. Given that GCs and progestins are used in breast cancer therapeutic modalities, the GC therapy in PR-positive breast cancer may have combined effects of both progestins and GCs. In light of above, it seems necessary to determine the applicability and efficacy of the GC/progestin strategies in breast carcinoma therapeutics. To this effect, further investigation is warranted for definitive answers.
Estradiol has been shown to inhibit GR expression and to induce glucocorticoid resistance in MCF-7 human breast cancer cells [145]. Conversely, the transcription of the oestrogen-induced pS2 gene was partially inhibited by exposing MCF-7 cells to gluco-corticoids [146]. It is of interest that a synergistic action of glucocorticoid and estradiol response elements has also been reported [147].
A recent research approach, in an effort to elucidate the mechanisms participating in the cross-talk between GR and ER signalling, unravels new data on the GR/ER molecular interplay. Researchers engineered an MCF-7/GR cell line and the MMTV-LUC as a model for GR-mediated transcriptional activity [148]. Importantly, the presence of oestrogen agonists (estradiol, genistein), but not antagonists (tamoxifen or raloxifene) inhibited GR-mediated MMTV-LUC transcription and chromatin remodelling. Moreover, oestrogen agonists inhibited glucocorticoid induction of p21 mRNA and protein levels, implicating that the repressive effect applies to other GR-regulated genes and proteins in MCF-7 cells. Notably, oestrogen agonists down regulated GR protein levels via the proteasomal degradation pathway coupled to an increase in p53 and its key regulator protein Mdm2, an E3 ubiquitin ligase shown to target the GR for degradation [148]. In light of above, oestrogen agonists activate a cascade of events leading to p53 stability and shifting the Mdm2 E3 ubiquitin ligase activity towards destruction of the GR in MCF-7 cells. In contrast, ER antagonists played a permissive role in mediating GR activity.
The above data suggest a further layer of complexity in the ER/GR cross-talk, the ubiquitin-proteasome pathway playing crucial role in preventing or facilitating GR-mediated transcriptional activity. It is important to mention that interfering in proteasome function has been shown to impact GR action at multiple levels (e.g. GR turnover, GR transactivation) [149]. Conceivably, an important goal in future therapeutic strategies in breast cancer would be agents targeting ubiquitin-mediated proteasomal regulation of GR.
Cytokines
Earlier as well as recent studies implicate a role of interleukin-6 in breast cancer. Krueger et al. showed that IL-6 inhibits proliferation of breast carcinoma cell lines (ZR-75-1 and T-47D) [150]. Today, accumulated evidence suggests that cytokines have both stimula-tory and inhibitory effects on breast cancer growth, depending on their relative concentrations and the presence of other modulatory factors in the tumour microenvironment. Certain cytokines IL-6 and IL-18 have been considered to act against breast cancer since they showed promising correlations with disease stage and progression [151]. However, a literature search conducted by Knupfer and Preiss, unravels surprising, new data indicating both tumour-promoting and inhibitory effects of IL-6 in in vitro experiments of breast tumour cells and breast tumour tissues [152]. Considering patients' serum IL-6 levels, data support IL-6 to be a negative prognosticator in breast tumours. Furthermore, a recent study by Tunon et al. aimed to characterize the role of tumour necrosis factor-alpha (TNF-α) and its receptors (TNFRI and TNFRII) in human benign breast lesions and tumours [153]. Their data support that the percentage of samples positive for TNF-α and TNFRll was higher in in situ carcinoma than in benign breast diseases and TNFRII and TNF-α was even higher in infiltrating tumours. TNFRI was similarly expressed in all groups studied. TNF-α, mtp53, p21 and IL-6 expression were associated with increasing malignancy. The researchers suggested that TNF-α might be an important factor in breast cancer promotion as its proliferation and survival effects seems to be enhanced through the increased expression of TNFRII.
A vast amount of data describe the different levels of interaction between glucocorticoids and cytokines in immune system, the molecular cross-talk between GR and TFs as well as the posttranslational modifications playing a pivotal role for the final biological action of cytokines [154] (Table 1). However, there is scant information about the cytokine/GR interactions in breast cancer. In view of the use of GCs in breast cancer and the crucial role of cytokines, in particular IL-6 and TNF-α, in breast cancer outcome, the molecular mechanism(s) underlying their interaction warrants further investigation.
1.
Main transcription factors (TFs) involved in glucocorticoid regulation of cytokine gene expression and action.
| Cytokine | Main transcription factors affected by glucocorticoids |
|---|---|
| IL-1 | AP-1, NF-κB |
| IL-2 | AP-1/NFAT, NF-κB |
| IL-4 | NFAT, STAT-6 |
| IL-5 | GATA-3, AP-1/NFAT, NF-κB |
| IL-6 | AP-1, NF-κB |
| IL-12 | STAT-4 |
| IFN-γ | AP-1/CREB/ATF, T-bet |
| TNF-α | AP-1, NF-κB |
11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2): conversion of cortisol to cortisone
Endogenous glucocorticoids may have a high impact in controlling breast cancer growth. The 11β-HSD2 enzyme plays a pivotal role in converting endogenous cortisol into its inactive metabolite, cortisone. Interestingly, Lipka et al. demonstrated that enzyme over-expression led to an increase in MCF-7 breast cancer cellular growth of about 120%, implicating a possible therapeutic role for 11βHSD2 inhibitors in the treatment of breast cancer [155].
GR expression in human breast tissues
Because of the many reports claiming a tumorigenic effect of GC-GR-mediated signalling in mammary cells, the expression of GR has been assessed in many breast cancer cell lines and breast cancer tissue. Data support a relatively high percentage of GR in primary human breast cancers [156–160]. Significant alterations of GR expression has been seen in breast cancer stroma, particularly in 2 and 3 tumour grades [161]. However, the presence of many cell types in tumour tissue lysates and the absence of data regarding the expression of GR in non-tumour cells in whole tissue has raised many questions about the potential role of GR in mammary tumorigenesis.
On this respect, Lien et al. investigated GR expression in situ in 400 human breast tissue samples, comprising normal tissue and a range of benign, pre-invasive, and invasive lesions, using immunohistochemical assays [162]. GR was expressed in myoepithelium but not in luminal epithelium suggesting perhaps a pathological role for the myoepithelium in mediating the effects of glucocorticoid hormones in the breast. Quantitative RT-PCR and Western blot analysis showed that GR-α, not GR-β was the predominant GR isoform in the breast. In addition, a pathogenetic role for GR has been implicated since there was a strong GR expression in metaplastic carcinomas and malignant tumours but a lack of GR expression in cancer cells of non-metaplastic carcinomas.
Given the importance of myoepithelium in giving rise to basal/triple negative breast cancer (i.e. ER-negative, PR-negative and HER2-negative), the role of GR expression in triple negative breast cancer awaits determination [163].
Non-genomic glucocorticoid effects
Accumulating evidence demonstrates that rapid GC actions, which occur within seconds, are mediated via interactions with cellular membranes, via membrane bound GRs and cytosolic GRs [164]. Because GR has been shown to be transiently attached through the G-protein β subunit to the inner surface of the plasma membrane, this interaction may also explain some non-genomic actions of glucocorticoids observed at the plasma membrane [43, 165].
Clinically relevant concentrations of GCs have shown effects within 16 seconds, on bioenergetics, on calcium and sodium concentration, on adenylate cyclase (AC)-/protein kinase A (PKA)-dependent inhibition of chloride ion secretion and on JNK/c-Jun signalling cascade [44, 164]. The release of Src from the multi-protein complex which follows the binding of dexamethasone to the cytosolic GR, is implicated in the rapid GC effects. GCs have also been shown to activate endothelial nitric oxide synthase in a non-genomic manner mediated by PI3K and Akt phosphorylation [164]. Taken together, targeting molecules involved in the non-genomic GC effects such as JNK, AC, PKA, PI3K/Akt, which are also important in breast cancer cell signalling, may represent a very promising therapeutic approach.
Concluding remarks
Apoptosis plays a key role in mammary gland remodelling and development. Although many of the molecular mechanisms regulating apoptosis are not yet understood, a considerable amount of data have shown that many regulatory molecules and signalling pathways trigger apoptosis during normal mammary gland development and remodelling. These include members of the bcl-2 family, namely the proapoptotic molecules such as Bcl-xs, Bad, Bax, Bid and the anti-apoptotic molecules such as Bcl-2, Bcl-XL. Key elements of the death receptor pathway such as FasL and downstream factors such as capsases 1, 3, 7, 8, 9 and caspase-10, cytochrome C are also crucial in the apoptotic process. Growth factors (EGF, IGF-I, IGF-II, TGF-B-3), IGF-binding proteins (IGFBP-5), cytokines (TNF-α, IL-6), kinases (PKA, PKB/Akt), integrins, matrix metalloproteinases (MMPs), oncosuppressor proteins (p53), CDK inhibitors (p21) have also a critical role in controlling apoptosis. Other routes involved in the normal mammary gland-related apoptotic processes include those mediated by TFs such as AP-1, NF-κB, Stat3, Stat5 [111]. Glucocorticoids have also been shown to inactivate apoptosis pathways during normal mammary gland development [111].
Taken together, multiple pathways and a large number of molecules contribute to apoptosis in normal mammary epithelial cells, however, little is known about changes in the apoptosis-related pathways during progression to breast cancer.
In this review we provide the current knowledge of the known so far molecular mechanisms underlying the inhibitory effects of GCs on apoptosis-related pathways in breast cancer and in normal mammary epithelial cells. However, in spite of the important clinical significance of the GC-induced inactivation of apoptosis in the cancerous mammary epithelial cells, many of the key elements in the underlying anti-apoptotic mechanisms remain largely unknown. To this effect, it will be of crucial importance to determine the impact of GCs in the members of Bcl-2 family (proapoptotic and anti-apoptotic proteins). Other putative target molecules could be the caspases, the metalloproteinases, as well as the impact of GCs on mitochondrial membrane potential and cytochrome C release. Given the importance of GR phosphorylation (i) in regulating a host of receptor functions such as transcriptional activation, stability and recycling, (ii) in coordinating crucial processes such as growth and apoptosis, the role of phosphorylated GR in breast cancer warrants also further investigation. Elucidation of the interactions of GR with other signalling molecules crucial in breast cancer aetiopathology such as the oestrogen receptor, p53, NF-κB and cytokines will be valuable in breast cancer management. Recent data demonstrating that translocation of GR to the mitochondria is associated with apoptosis along with findings showing the localization of GR within the mitochondria in some cell types, implicate that a role of mitochondrial GR in breast cancer awaits further characterization. Conceivably, much work needs to be done to fully explore the GC/GR-induced alterations in the various pathways associated with breast cancer. The use of gene and/or protein expression profiling by microarray technology in elucidating the underlying mechanisms will provide us valuable information about the ‘signature’ of GR-regulated genes that might be helpful in the design of future therapeutic strategies.
Acknowledgments
Limitations of space preclude extensive citation of the literature; we apologize to those whose work is not mentioned herein.
References
- 1.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J. Clin. 53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Carter CL, Allen C, Henson DE. Relation of tumor size, lymph node status and survival in 24,740 breast cancer cases. Cancer. 1989;63:181–7. doi: 10.1002/1097-0142(19890101)63:1<181::aid-cncr2820630129>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 3.Herr I, Gassler N, Friess H, Büchler MW. Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids. Apoptosis. 2007;12:271–91. doi: 10.1007/s10495-006-0624-5. [DOI] [PubMed] [Google Scholar]
- 4.Grote T, Hajdenberg J, Cartmell A, Ferguson S, Ginkel A, Charu V. Combination therapy for chemotherapy-induced nausea and vomiting in patients receiving moderately emetogenic chemotherapy: palonoseetron, dexamethasone and aprepitant. J Support Oncol. 2006;4:403–8. [PubMed] [Google Scholar]
- 5.Lu YS, Lien HC, Yeh PY, Yeh KH, Kuo ML, Kuo SH, Cheng AL. Effects of glucocorticoids on the growth and chemosensitivity of carcinoma cells are heterogeneous and require high concentration of functional glucocorticoid receptors. World J Gastroenterol. 2005;11:6373–80. doi: 10.3748/wjg.v11.i40.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang C, Beckermann B, Kallifatidis G, Liu Z, Rittgen W, Edler L, Buchler P, Debatin KM, Buchler MW, Friess H, Herr I. Corticosteroids induce chemotherapy resistance in the majority of tumour cells from bone, brain, breast, cervix, melanoma and neuroblastoma. Int J Oncol. 2006;29:1295–301. [PubMed] [Google Scholar]
- 7.Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol Ther. 2006;5:933–40. doi: 10.4161/cbt.5.8.2875. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Wang Y, Rayburn ER, Hill DL, Rinehart JJ, Zhang R. Dexamethasone as a chemosensitizer for breast cancer chemotherapy:potentiation of the anti-tumor activity of adriamycin, modulation of cytokine expression and pharmacokinetics. Int J Oncol. 2007;30:947–53. [PubMed] [Google Scholar]
- 9.Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Science's stke. 2005;2005:pe48. doi: 10.1126/stke.3042005pe48. [DOI] [PubMed] [Google Scholar]
- 10.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–41. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bamberger CM, Bamberger AM, DeCastro M, Chrousos GP. Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–41. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- 13.Kino T, Chrousos GP. Tissue-specific glucocorticoid resistance-hypersensitivity syndromes: multifactorial states of clinical importance. J Allergy Clin Immunol. 2002;109:609–13. doi: 10.1067/mai.2002.123708. [DOI] [PubMed] [Google Scholar]
- 14.De Bosscher K, Van den Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 15.Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol. 2003;85:457–67. doi: 10.1016/s0960-0760(03)00218-8. [DOI] [PubMed] [Google Scholar]
- 16.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators:cellular and molecular biology. Endocr Rev. 1999;20:321–44. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 17.Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 1998;12:1638–51. doi: 10.1101/gad.12.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakley RH, Jewell CM, Yudt MR, Bofetiado JA, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274:27857–77. doi: 10.1074/jbc.274.39.27857. [DOI] [PubMed] [Google Scholar]
- 19.Oakley RH, Webster JC, Sar M, Parker CR, Jr, Cidlowski JA. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology. 1997;138:5028–38. doi: 10.1210/endo.138.11.5501. [DOI] [PubMed] [Google Scholar]
- 20.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Duma D, Jewell CM, Cidlowsky JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Molec Biol. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Galon J, Franchimont D, Hiroi N, Frey G, Boetner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 23.Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann NY Acad Sci. 2004;1024:86–101. doi: 10.1196/annals.1321.007. [DOI] [PubMed] [Google Scholar]
- 24.Hoeck W, Groner B. Hormone-dependent phoshorylation of the glucocorticoid receptor occurs mainly in the amino-terminal transactivation domain. J Biol Chem. 1990;265:5403–8. [PubMed] [Google Scholar]
- 25.Wang Z, Frederick J, Garabedian MJ. Deciphering the phoshorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277:26573–80. doi: 10.1074/jbc.M110530200. [DOI] [PubMed] [Google Scholar]
- 26.Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3947–54. doi: 10.1128/mcb.17.7.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogatsky I, Logan SK, Garabedian MJ. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA. 1998;95:2050–5. doi: 10.1073/pnas.95.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helmberg A, Auphan N, Caelles C, Karin M. Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J. 1995;14:452–60. doi: 10.1002/j.1460-2075.1995.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramdas J, Harmon JM. Glucocorticoid-induced apoptosis and regulation of NF-kappaB activity in human leukemic T cells. Endocrinology. 1998;139:3813–21. doi: 10.1210/endo.139.9.6180. [DOI] [PubMed] [Google Scholar]
- 30.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Transcriptional control of steroid-regulated apop-tosis in murine thymoma cells. Mol Endocrinol. 1996;10:967–78. doi: 10.1210/mend.10.8.8843413. [DOI] [PubMed] [Google Scholar]
- 31.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schütz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–41. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 32.Frankfurt O, Rosen ST. Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol. 2004;16:553–63. doi: 10.1097/01.cco.0000142072.22226.09. [DOI] [PubMed] [Google Scholar]
- 33.Hsu SC, DeFranco DB. Selectivity of cell cycle regulation of glucocorticoid receptor function. J Biol Biochem. 1995;270:3359–64. doi: 10.1074/jbc.270.7.3359. [DOI] [PubMed] [Google Scholar]
- 34.Hu JM, Bodwell JE, Munck A. Cell cycle-dependent glucocorticoid receptor phosphorylation and activity. Mol Endocrinol. 1994;8:1709–13. doi: 10.1210/mend.8.12.7708059. [DOI] [PubMed] [Google Scholar]
- 35.Abel GA, Wochnik GM, Rüegg J, Rouyer A, Holsboer F, Rein T. Activity of the GR in G2 and mitosis. Mol Endocrinol. 2002;16:1352–66. doi: 10.1210/mend.16.6.0842. [DOI] [PubMed] [Google Scholar]
- 36.Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB. p38 mitogen-activated protein kinase (MAPK) is a key mediator in gluco-corticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phoshorylation of the human glucocorticoid receptor at serine 211. Molec Endocrinol. 2005;19:1569–83. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 37.Zuo Z, Urban G, Scammell JG, Dean NM, McLean TK, Aragon I, Honkanen RE. Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry. 1999:38. doi: 10.1021/bi990842e. [DOI] [PubMed] [Google Scholar]
- 38.Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280:33792–9. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- 39.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft Do, Pratt WB, Yao TP. HDAC6 regulates hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 41.Aoyagi S, Archer TK. Modulating molecular chaperone hsp90 functions through reversible acetylation. Trends Cell Biol. 2005;15:565–7. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–21. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 43.Kino T, Tiulpakov A, Ichijo T, Cheng L, Kozasa T, Chrousos GP. G protein beta interacts with the glucocorticoid receptor and suppresses its transcriptional activity in the nucleus. J Cell Biol. 2005;169:885–96. doi: 10.1083/jcb.200409150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheller K, Seibel P, Sekeris CE. Glucocorticoid and thyroid hormone receptors in mitochondria of animal cells. Int Rev Cytol. 2003;222:1–61. doi: 10.1016/s0074-7696(02)22011-2. [DOI] [PubMed] [Google Scholar]
- 45.Moutsatsou P, Psarra AG, Tsiapara A, Paraskevakou H, Davaris P, Sekeris CE. Localization of the glucocorticoid receptor in rat brain mitochondria. Arch Biochem Biophys. 2001;386:69–78. doi: 10.1006/abbi.2000.2162. [DOI] [PubMed] [Google Scholar]
- 46.Koufali M-M, Moutsatsou P, Sekeris CE, Breen KC. The dynamic localization of the glucocorticoid receptor in rat C6 glioma cell mitochondria. Mol Cell Endocrinol. 2003;209:51–60. doi: 10.1016/j.mce.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J Exp Med. 2006;203:189–201. doi: 10.1084/jem.20050433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 49.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649–54. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- 50.Maiyar AC, Phu PT, Huang AJ, Firestone GL. Repression of glucocorticoid receptor transactivation and DNA binding of a glucococrticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumor suppressor protein. Mol Endocrinol. 1997;11:312–29. doi: 10.1210/mend.11.3.9893. [DOI] [PubMed] [Google Scholar]
- 51.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–16. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003;191:229–37. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 53.Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–93. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 54.Noguchi T, Metz R, Chen L, Mattei MG, Carrasco D, Bravo R. Structure, mapping and expression of erp, a growth factor-inducible gene encoding a non-transmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol Cell Biol. 1993;13:5195–205. doi: 10.1128/mcb.13.9.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–6. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 56.Davis RJ. MAPKs: new JNK expands the group. Trends Biochem Sci. 1994;19:470–3. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Gorospe M, Yang C, Holbrook NJ. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun N-terminal kinase activity and AP-1-dependent gene activation. J Biol Chem. 1995;270:8377–80. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- 58.Imasato A, Desbois-Mouthon C, Han J, Kai H, Cato AC, Akira S, Li JD. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of toll-like receptor 2. J Biol Chem. 2002;277:47444–50. doi: 10.1074/jbc.M208140200. [DOI] [PubMed] [Google Scholar]
- 59.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–11. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu W, Pew T, Zou M, Pang D, Conzen SD. Glucocorticoid receptor-induced MAPK phos-phatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem. 2005;280:4117–24. doi: 10.1074/jbc.M411200200. [DOI] [PubMed] [Google Scholar]
- 61.Wu W, Zou M, Brickley DR, Pew T, Conzen SD. Glucocorticoid receptor activation signals through forkhead transcription factor 3a in breast cancer cells. Mol Endocrinol. 2006;20:2303–14. doi: 10.1210/me.2006-0131. [DOI] [PubMed] [Google Scholar]
- 62.Earp HS, Dawson TL, Li X, Yu H. Heterodimerization and functional interaction between EGF receptor family members:a new signaling paradigm with implications for breast cancer research. Breast Cancer Res Treat. 1995;35:115–32. doi: 10.1007/BF00694752. [DOI] [PubMed] [Google Scholar]
- 63.De Cupis A, Favoni RE. Oestrogen/growth factor cross-talk in breast carcinoma: a specific target for novel antioestrogens. Trends Pharmacol Sci. 1997;18:245–51. doi: 10.1016/s0165-6147(97)01083-3. [DOI] [PubMed] [Google Scholar]
- 64.Murphy LJ, Sutherland RL, Stead B, Murphy LC, Lazarus L. Progestin regulation of epidermal growth factor receptor in human mammary carcinoma cells. Cancer Res. 1986;46:728–34. [PubMed] [Google Scholar]
- 65.Dickson RB, Lippman ME. Growth factors in breast cancer. Endocr Rev. 1995;8:559–89. doi: 10.1210/edrv-16-5-559. [DOI] [PubMed] [Google Scholar]
- 66.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–43. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 67.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–72. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 68.Ewing TM, Murphy LJ, Ng ML, Pang GY, Lee CS, Watts CK, Sutherland RL. Regulation of epidermal growth factor receptor by progestins and glucocorticoids in human breast cancer cell lines. Int J Cancer. 1989;44:744–52. doi: 10.1002/ijc.2910440432. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez T, Hla T. Structural and functional characteristics of SIP receptors. J Cell Biochem. 2004;92:913–22. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 70.Hla T. Physiological and pathological actions of sphingosine-1-phosphate. Semin Cell Dev Biol. 2004;15:513–20. doi: 10.1016/j.semcdb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 71.Huwiler A, Pfeilschifter J. Altering the sphingosine-1-phosphate/Ceramide balance: a promising approach for tumor therapy? Curr Pharmaceut Des. 2006;35:4625–35. doi: 10.2174/138161206779010422. [DOI] [PubMed] [Google Scholar]
- 72.Liu H, Chakravarty D, Maceyka M, Milstien S, Spiegel S. Sphingosine kinases: a novel family of lipid kinases. Prog Nucleic Acid Res Mol Biol. 2002;71:493–511. doi: 10.1016/s0079-6603(02)71049-0. [DOI] [PubMed] [Google Scholar]
- 73.Van Brocklyn JR, Lee MJ, Menzelev R, Olivera A, Edshall L, Cuvillier O, Thomas DM, Coopman PJ, Thangada S, Liu CH, Hla T, Spiegel S. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol. 1998;142:229–40. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiegel S, Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem. 2002;277:25851–4. doi: 10.1074/jbc.R200007200. [DOI] [PubMed] [Google Scholar]
- 75.Döll F, Pfeilschifter J, Huwiler A. The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochimica et Biophysica Acta. 2005;72:81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 76.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–9. [PubMed] [Google Scholar]
- 77.Sapi E, Flick MB, Rodov S, Gilmore-Hebert M, Kelley M, Rockwell S, Kacinski BM. Independent regulation of invasion and anchorange-independent growth by different autophosphorylation sites of the macrophage colony-stimulating factor 1 receptor. Cancer Res. 1996;56:5704–12. [PubMed] [Google Scholar]
- 78.Toy EP, Bonafe N, Savlu A, Zeiss C, Zheng W, Flick M, Chambers SK. Correlation of tumor phenotype with c-fms proto-oncogene expression in an in vitro intraperitoneal model for experimental human breast cancer metastasis. Clin Exp Met. 2005;22:1–9. doi: 10.1007/s10585-005-0718-4. [DOI] [PubMed] [Google Scholar]
- 79.Chambers SK, Wang Y, Gilmore-Hebert M, Kacinski BM. Post-transcriptional regulation of c-fms proto-oncogene expression by dexamethasone and of CSF-1 in human breasts carcinomas in vitro. Steroids. 1994;59:514–22. doi: 10.1016/0039-128x(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 80.Sapi E, Flick MB, Gilmore-Hebert M, Rodov S, Kacinski BM. Transcriptional regulation of the c-fms (CSF-1R) proto-oncogene in human breast carcinoma cells by glucocorticoids. Oncogene. 1995;10:529–42. [PubMed] [Google Scholar]
- 81.Filderman AE, Bruckner A, Kacinski BM, Deng N, Remold HG. Macrophage colony-stimulating factor (CSF-1) enhances invasiveness in CSF-1 receptor-positive carcinoma cell lines. Cancer Res. 1992;52:3661–6. [PubMed] [Google Scholar]
- 82.Toy EP, Chambers JT, Kacinski BM, Flick MB, Chambers SK. The activated macrophage colony-stimulating factor (CSF-1) receptor as a predictor of poor outcome in advanced epithelial ovarian carcinoma. Gynecol Oncol. 2001;80:194–200. doi: 10.1006/gyno.2000.6070. [DOI] [PubMed] [Google Scholar]
- 83.Flick MB, Sapi E, Kacinski BM. Hormonal regulation of the c-fms proto-oncogene in breast cancer cells is mediated by a composite glucocorticoid response element. J Cell Biochem. 2002;85:10–23. [PubMed] [Google Scholar]
- 84.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–65. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leong ML, Maiyar AC, Kim B, O'Keeffe BA, Firestone GL. Expression of the serum- and gluco-corticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–82. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 86.Tangir J, Bonafe N, Gilmore-Hebert M, Henegariu O, Chambers SK. SGK1, a potential regulator of c-fms related breast cancer aggressiveness. Clin Exp Metast. 2004;21:477–83. doi: 10.1007/s10585-004-4226-8. [DOI] [PubMed] [Google Scholar]
- 87.Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13:293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- 88.Sengupta S, Wasylyk B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen and estrogen receptors. Ann NY Acad Sci. 2004;1024:54–71. doi: 10.1196/annals.1321.005. [DOI] [PubMed] [Google Scholar]
- 89.Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci USA. 1992;89:7262–6. doi: 10.1073/pnas.89.15.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maiyar AC, Huang AJ, Phu PT, Cha HH, Firestone GL. P53 stimulates promoter activity of the sgk serum/glucocorticoid-inducible serine/theonine protein kinase gene in rodent mammary epithelial cells. J Biol Chem. 1996;271:12414–22. doi: 10.1074/jbc.271.21.12414. [DOI] [PubMed] [Google Scholar]
- 91.Urban G, Golden T, Aragon IV, Cowsert L, Cooper SR, Dean NM, Honkanen RE. Identification of a functional link for the p53 tumor suppressor protein in dexamethasone-induced growth suppression. J Biol Chem. 2003;278:9747–53. doi: 10.1074/jbc.M210993200. [DOI] [PubMed] [Google Scholar]
- 92.Yoshida BA, Sokoloff MM, Welch DR, Rinker-Schaeffer CW. Metastasis-suppressor genes: a review and perspective on an emerging field. J Natl Cancer Inst. 2000;92:1717–30. doi: 10.1093/jnci/92.21.1717. [DOI] [PubMed] [Google Scholar]
- 93.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Cancer Res. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 94.Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO. The human Nm23/nucleoside dihos-phate kinases. J Bioenerg Biomembr. 2000;32:247–58. doi: 10.1023/a:1005584929050. [DOI] [PubMed] [Google Scholar]
- 95.MacDonald NJ, Freije JM, Stracke ML, Manrow RE, Steeg PS. Site directed mutagenesis of nm23-H1:mutation of proline 96 or serine 120 abrogates its motility inhibitory activity upon transfection into human breast carcinoma cells. J Biol Chem. 1996;271:25107–16. doi: 10.1074/jbc.271.41.25107. [DOI] [PubMed] [Google Scholar]
- 96.Bemis LT, Schedin P. Reproductive state of rat mammary gland stroma modulates human breast cancer cell migration and invasion. Cancer Res. 2000;60:3414–8. [PubMed] [Google Scholar]
- 97.Ouatas T, Clare SE, Hartsough MT, DeLaRosa A, Steeg PS. MMTV-associated transcription factor binding sites increase NM23-H1 metastasis suppressor gene expression in humar breast carcinoma cell lines. Clin Exp Metastasis. 2002;19:35–42. doi: 10.1023/a:1013897022827. [DOI] [PubMed] [Google Scholar]
- 98.Ouatas T, Halverson D, Steeg PS. Dexamethasone and medroxyprogesterone acetate elevate Nm23-H1 metastasis suppressor gene expression in metastatic human breast cacinoma cells: new uses for old compounds. Clin Cancer Res. 2003;9:3763–72. [PubMed] [Google Scholar]
- 99.Hao X, Sun B, Hu L, Lähdesmäki H, Dunmire V, Feng Y, Zhang SW, Wang H, Wu C, Wang H, Fuller GN, Symmans WF, Shmulevich I, Zhang W. Differential gene and protein expression in primary breast malignancies and their lymph node metastases as revealed by combined cDNA microarray and tissue microarray analysis. Cancer. 2004;100:1110–22. doi: 10.1002/cncr.20095. [DOI] [PubMed] [Google Scholar]
- 100.Herr I, Buchler MW. New in vivo results support concerns about harmful effects of cortisone drugs in the tretment of breast cancer. Cancer Biol Ther. 2006;5:941–2. doi: 10.4161/cbt.5.8.3285. [DOI] [PubMed] [Google Scholar]
- 101.Herr I, Marme A. Glucocorticoids and progression of breast cancer. Cancer Biol Ther. 2005;4:1415–6. doi: 10.4161/cbt.4.12.2354. [DOI] [PubMed] [Google Scholar]
- 102.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 103.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–6. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 104.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 105.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–6. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 106.Van Dam H, Castellazzi M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene. 2001;20:2453–64. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- 107.Liu Y, Ludes-Meyers J, Zhang Y, Munoz-Medellin D, Kim HT, Lu C, Ge G, Schiff R, Hilsenbeck SG, Osborne CK, Brown PH. Inhibition of AP-1 transcription factor causes blockade of multiple signal transduction pathways and inhibits breast cancer growth. Oncogene. 2002;21:7680–9. doi: 10.1038/sj.onc.1205883. [DOI] [PubMed] [Google Scholar]
- 108.Milde-Langosch K, Bamberger AM, Methner C, Rieck G, Loning T. Expression of cell cycle-regulatory proteins rb, p16/MTS1, p27/KIP1, p21/WAF1, cyclin D1 and cyclin E in breast cancer: correlations with expression of activating protein-1 family members. Int J Cancer. 2000;87:468–72. [PubMed] [Google Scholar]
- 109.Milde-Langosch K, Kappes H, Riethdorf S, Loning T, Bamberger AM. FosB is highly expressed in normal mammary epithelia, but down-regulated in poorly differentiated breast carcinomas. Breast Cancer Res Treat. 2003;77:265–75. doi: 10.1023/a:1021887100216. [DOI] [PubMed] [Google Scholar]
- 110.Bamberger AM, Methner C, Lisboa BW, Stadtler C, Schulte HM, Loning T, Milde-Langosch K. Expression pattern of the AP-1 family in breast cancer: association of fosB expression with a well-differentiated, receptor-positive tumor phenotype. Int J Cancer. 1999;84:533–8. doi: 10.1002/(sici)1097-0215(19991022)84:5<533::aid-ijc16>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 111.Green KA, Streuli CH. Apoptosis regulation in the mammary gland. Cell Mol Life Sci. 2004;61:1867–83. doi: 10.1007/s00018-004-3366-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng Z, Marti A, Jehn B, Altermatt HJ, Chicaiza G, Jaggi R. Glucocorticoid and progesterone inhibit involution and programmed cell death in the mouse mammary gland. J Cell Biol. 1995;131:1095–103. doi: 10.1083/jcb.131.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brantley DM, Yull FE, Muraoka RS, Hicks DJ, Cook CM, Kerr LD. Dynamic expression and activity of NF-kappaB during post-natal mammary gland morphogenesis. Mech Dev. 2000;97:149–55. doi: 10.1016/s0925-4773(00)00405-6. [DOI] [PubMed] [Google Scholar]
- 114.Haffner MC, Berlato C, Doppler W. Exploiting our knowledge of NF-kappaB signaling for the treatment of mammary cancer. J Mammary Gland Biol Neoplasia. 2006;11:63–3. doi: 10.1007/s10911-006-9013-5. [DOI] [PubMed] [Google Scholar]
- 115.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kapaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAPI and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 116.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–9. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 117.Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15:24–7. doi: 10.1038/sj.cr.7290259. [DOI] [PubMed] [Google Scholar]
- 118.Gupta S. A decision between life and death during TNF-alpha-induced signaling. J Clin Immunol. 2002;22:185–94. doi: 10.1023/a:1016089607548. [DOI] [PubMed] [Google Scholar]
- 119.Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13:135–41. doi: 10.1016/s1359-6101(01)00020-x. [DOI] [PubMed] [Google Scholar]
- 120.Lejeune FJ, Ruegg C, Lienard D. Clinical applications of TNF-alpha in cancer. Curr Opin Immunol. 1998;10:573–80. doi: 10.1016/s0952-7915(98)80226-4. [DOI] [PubMed] [Google Scholar]
- 121.Burow ME, Weldon CB, Melnik LI, Duong BN, Collins-Burow BM, Beckman BS, McLachlan JA. P13-K/AKT regulation of NF-kappaB signaling events in suppression of TNF-induced apoptosis. Biochem Biophys Res Commun. 2000;271:342–5. doi: 10.1006/bbrc.2000.2626. [DOI] [PubMed] [Google Scholar]
- 122.Downward J. Mechanisms and consequence of activation of protein kinase B/Akt. Curr Opin Biol. 1998;10:262–7. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 123.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 124.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requirs the Akt serine-threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 125.Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2002;60:867–72. [PubMed] [Google Scholar]
- 126.Pagliacci MC, Fumi G, Migliorati G, Grignani F, Riccardi C, Nicoletti I. Cytostatic and cytotoxic effects of tumor necrosis factor alpha on MCF-7 human breast tumor cells are differently inhibited by glucocorticoid hormones. Lymphokine Cytokine Res. 1993;12:439–47. [PubMed] [Google Scholar]
- 127.Messmer UK, Pereda-Fernandez C, Manderscheid M, Pfeilschifter J. Dexamethasone inhibits TNF-alpha-induced apoptosis and IAP protein downregulation in MCF-7 cells. Br J Pharmacol. 2001;133:467–76. doi: 10.1038/sj.bjp.0704093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Machuca C, Mendoza-Milla C, Cordova E, Mejia S, Covarrubias L, Ventura J, Zentella A. Dexamethasone protection from TNF-alpha-induced cell death in MCF-7 levels requires NF-kappaB and is independent from AKT. BMC Cell Biol. 2006;7:9–20. doi: 10.1186/1471-2121-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, Guzman JR, Korgaonkar CK, Donner DB. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappaB activation. J Biol Chem. 2004;279:1615–20. doi: 10.1074/jbc.M306976200. [DOI] [PubMed] [Google Scholar]
- 130.Hill SM, Blask DE. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988;48:6121–6. [PubMed] [Google Scholar]
- 131.Blask DE, Hill SM, Orstead KM, Massa JS. Inhibitory effects of the pineal hormone melatonin and underfeeding during the promotional phase of 7,12-dimethylbenzanthracene-(DMBA)-induced mammary tumorigenesis. J Neural Transm. 1986;67:125–38. doi: 10.1007/BF01243365. [DOI] [PubMed] [Google Scholar]
- 132.Blask DE, Hill SM. Effects of melatonin on cancer: studies on MCF-7 human breast cancer cells in culture. J Neural Transm Suppl. 1986;21:433–49. [PubMed] [Google Scholar]
- 133.Ebisawa T, Karne S, Lerner MR, Reppert SM. Expression cloning of a high-affinity melatonin receptor from Xenopus dermal melanophors. Proc Natl Acad Sci USA. 1994;91:6133–7. doi: 10.1073/pnas.91.13.6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Reppert SM, Godson C, Mahle CD, Weaver DR, Gusella JF. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci USA. 1995;92:8734–8. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yuan L, Collins AR, Dai J, Dubocovich ML, Hill SM. MT1 melatonin receptor overexpression enhances the growth suppressive effect of melatonin in human breast cancer cells. Mol Cell Endocrinol. 2002;192:147–56. doi: 10.1016/s0303-7207(02)00029-1. [DOI] [PubMed] [Google Scholar]
- 136.Brydon I, Roka F, Petit L, De Coppet P, Tissot M, Barrett P, Morgan PJ, Nanoff C, Strosberg AD, Jockers R. Dual signaling of human Mel1a melatonin receptors via Gi2, Gi3 and G9/11 proteins. Mol Endocrinol. 1999;13:2025–38. doi: 10.1210/mend.13.12.0390. [DOI] [PubMed] [Google Scholar]
- 137.Ram PT, Kiefer T, Silverman M, Song Y, Brown GM, Hill SM. Estrogen receptor transactivation in MCF-7 breast cancer cells by melatonin and growth factor. Mol Cell Endocrinol. 1998;141:53–64. doi: 10.1016/s0303-7207(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 138.Kiefer TL, Yuan L, Ram PT, Hill SM. Melatonin inhibits estrogen receptor transactivation and cAMP levels in MCF-7 human breast cancer cells. Breast Cancer Res Treat. 2002;71:37–45. doi: 10.1023/a:1013301408464. [DOI] [PubMed] [Google Scholar]
- 139.Kiefer T, Lai L, Yuan L, Dong C, Burow ME, Hill SM. Differential regulation of estrogen receptor alpha, glucocorticoid receptor and retinoic acid receptor alpha transcriptional activity by melatonin is mediated via different G proteins. J Pineal Res. 2005;38:231–9. doi: 10.1111/j.1600-079X.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 140.Fryer CJ, Nordeen SK, Archer TK. Antiprogestins mediate differential effects on glucocorticoid receptor remodeling of chromatin structure. J Biol Chem. 1998;273:1175–83. doi: 10.1074/jbc.273.2.1175. [DOI] [PubMed] [Google Scholar]
- 141.Fryer CJ, Kinyamu HK, Rogatsky I, Garabedian MJ, Archer TK. Selective activation of the glucocorticoid receptor by steroid antagonists in human breast cancer and osteosarcoma cells. J Biol Chem. 2000;275:17771–7. doi: 10.1074/jbc.M908729199. [DOI] [PubMed] [Google Scholar]
- 142.Wan Y, Nordeen SK. Overlapping but distinct profiles of gene expression elicited by glucocorticoids and progestins. Recent Prog Horm Res. 2003;58:199–226. doi: 10.1210/rp.58.1.199. [DOI] [PubMed] [Google Scholar]
- 143.Wan Y, Nordeen SK. Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol. 2002;16:1204–14. doi: 10.1210/mend.16.6.0848. [DOI] [PubMed] [Google Scholar]
- 144.Leo JC, Guo C, Woon CT, AW SE, Lin VC. Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology. 2004;145:1314–21. doi: 10.1210/en.2003-0732. [DOI] [PubMed] [Google Scholar]
- 145.Krishnan AV, Swami S, Feldman D. Estradiol inhibits glucocorticoid receptor expression and induces glucocorticoid resistance in MCF-7 human breast cancer cells. J Steroid Biochem Mol Biol. 2001;77:29–37. doi: 10.1016/s0960-0760(01)00030-9. [DOI] [PubMed] [Google Scholar]
- 146.Meyer ME, Gronemeyer H, Turcotte B, Bocquel MT, Tasset D, Chambon P. Steroid hormone receptors compete for factors that mediate their enhancer function. Cell. 1989;57:433–42. doi: 10.1016/0092-8674(89)90918-5. [DOI] [PubMed] [Google Scholar]
- 147.Ankenbauer W, Strahle U, Schutz G. Synergistic action of glucocorticoid and estradiol responsive elements. Proc Natl Acad Sci USA. 1988;85:7526–30. doi: 10.1073/pnas.85.20.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kinyamu HK, Archer TK. Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in Mdm2 protein expression. Mol Cell Biol. 2003;23:5867–81. doi: 10.1128/MCB.23.16.5867-5881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Deroo B, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasonal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22:4113–23. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Krueger J, Ray A, Tamm I, Sehgai PB. Expression and function of interleukin-6 in epithelial cells. J Cell Biochem. 1991;45:327–34. doi: 10.1002/jcb.240450404. [DOI] [PubMed] [Google Scholar]
- 151.Rao VS, Dyer CE, Jameel JK, Drew PJ, Greenman J. Potential prognostic and therapeutic roles for cytokines in breast cancer (Review) Oncol Reg. 2006;15:179–85. doi: 10.3892/or.15.1.179. [DOI] [PubMed] [Google Scholar]
- 152.Knupfer H, Preiss R. Significance of interleukin-6 (IL-6) in breast cancer (review) Breast Cancer Res Treat. 2007;102:129–35. doi: 10.1007/s10549-006-9328-3. [DOI] [PubMed] [Google Scholar]
- 153.Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-alpha and its receptors in human benign breast lesions and tumors (in situ and infiltrative) Cancer Sci. 2006;97:1044–9. doi: 10.1111/j.1349-7006.2006.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liberman AC, Druker J, Perone MJ, Arzt E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine and Growth Factor Rev. 2007;18:45–56. doi: 10.1016/j.cytogfr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 155.Lipka C, Mankertz J, Fromm M, Lubbert H, Buhler H, Kuhn W, Ragosch V, Hundertmark S. Impairment of the antiproliferative effect of glucocorticosteroids by 11beta-hydroxysteroid dehydrogenase type 2 overexpression in MCF-7 breast cancer cells. Horm Metab Res. 2004;36:437–44. doi: 10.1055/s-2004-825724. [DOI] [PubMed] [Google Scholar]
- 156.Fazekas AG, MacFarlane JK. Macromolecular binding of glucocorticoids in human mammary carcinoma. Cancer Res. 1977;37:640–5. [PubMed] [Google Scholar]
- 157.Ioannidis C, Papamichail M, Agnanti N, Garas J, Tsawdaroglou N, Sekeris CE. The detection of glucocorticoid receptors in breast cancer by immunocytochemical and biochemical methods. Int J Cancer. 1982;29:147–52. doi: 10.1002/ijc.2910290206. [DOI] [PubMed] [Google Scholar]
- 158.Allegra JC, Lippman ME, Thompson EB, Simon R, Barlock A, Green L, Huff KK, Do HM, Aitken SC. Distribution, frequency and quantitative analysis of estrogen, progesterone, androgen and glucocorticoid receptors in human breast cancer. Cancer Res. 1979;39:1447–54. [PubMed] [Google Scholar]
- 159.Liu SH, Otal-Brun M, Webb TE. Glucocorticoid receptors in human tumors. Cancer Lett. 1980;10:269–75. doi: 10.1016/0304-3835(80)90080-4. [DOI] [PubMed] [Google Scholar]
- 160.Smith RA, Lea RA, Curran JE, Weinstein SR, Griffiths LR. Expression of glucocorticoid and progesterone nuclear receptor genes in archival breast cancer tissue. Breast Cancer Res. 2003;5:R9–12. doi: 10.1186/bcr556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Smith RA, Lea RA, Weinstein SR, Griffiths LR. Progesterone, glucocorticoid, but not estrogen receptor mRNA is altered in breast cancer stroma. Cancer Lett. 2007;255:77–84. doi: 10.1016/j.canlet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 162.Lien HC, Lu YS, Cheng AL, Chang WC, Jeng YM, Kuo YH, Huang CS, Chang KJ, Yao YT. Differential expression of glucocorticoid receptor in human breast tissues and related neoplasms. J Pathol. 2006;209:317–27. doi: 10.1002/path.1982. [DOI] [PubMed] [Google Scholar]
- 163.Bryan BB, Schnitt SJ, Collins LC. Ductal carcinoma in situ with basal-like phenotype: a possible precursor to invasive basal-like breast cancer. Mod Pathol. 2006;19:617–21. doi: 10.1038/modpathol.3800570. [DOI] [PubMed] [Google Scholar]
- 164.Song IH, Buttgereit F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol. 2006;246:142–6. doi: 10.1016/j.mce.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 165.Tasker JG, Di S, Malcher-Lopes R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549–56. doi: 10.1210/en.2006-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]