

Abstract
Atrial fibrillation (AF) is the most frequent clinical arrhythmia. Atrial fibrosis is an important factor in initiating and maintaining AF. However, the collagen turnover and its regulation in AF has not been completely elucidated. We tested the hypothesis that the extracellular matrix changes are more severe in patients with permanent AF in comparison with those in patients in sinus rhythm (SR). Intraoperative biopsies from the right atrial appendages (RAA) and free walls (RFW) from 24 patients with AF undergoing a mini-Maze procedure and 24 patients in SR were investigated with qualitative and quantitative immunofluorescent and Western blot analyses. As compared with SR, all patients with AF exhibited dysregulations in collagen type I and type III synthesis/degradation. Tissue inhibitors of metalloproteinases (TIMP2) was significantly enhanced only in RAA-AF. As compared with SR, collagen VI, matrix metalloproteinases MMP2, MMP9 and TIMP1 were significantly increased while TIMP3 and TIMP4 remained unchanged in all AF groups. Reversion-inducing cysteine-rich protein with Kazal motifs (RECK), a newly discovered MMPs inhibitor, was elevated in RFW as compared to RAA-AF (P<0.05) and RFW-SR (P<0.05). The level of transforming growth factor (TGF)-β1 was higher in AF than SR. Smad2 and phosphorylated Smad2 showed an elevation in RFW-AF as compared to RFW-SR, RAA-AF, and RAA-SR groups (P<0.05). Conclusions: Atrial fibrosis in AF is characterized by severe alterations in collagen I and III synthesis/degradation associated with disturbed MMP/TIMP systems and increased levels of RECK. TGF-β1 contributes to atrial fibrosis via TGF-β1-Smad pathway by phospho-rylating Smad2. These processes culminate in accumulations of fibrillar and non-fibrillar collagens leading to excessive atrial fibrosis and maintainance of AF.
Keywords: atrial fibrillation, fibrosis, extracellular matrix, metalloproteinases
Introduction
Atrial fibrillation (AF) is the most frequent form of arrhythmia in clinical practice. Among mechanisms of AF, multiple wavelets [1], focal sources [2] and spiral-wave activity or ‘mother rotor’ have been proposed [3, 4]. The development of AF is characterized by structural changes of atrial cardiomyocytes including cellular hypertrophy, disintegration of the contractile apparatus, accumulation of glycogen, myolysis and alterations in connexins expression [5–9]. Moreover, AF and its duration are associated with atrial fibrosis which causes abnormal conduction through the atria and creates itself a substrate for AF especially in the settings of heart failure [9–15].
The major heart collagens are collagen type I and type III that comprise almost 95% of the cardiac extracellular matrix (ECM) and ∼5% of the total collagen constitutes collagen type VI [16, 17]. Since collagens are relatively stiff material with a high tensile strength, even small changes in its concentrations, proportions and quality indicated by cross-linking degree have been demonstrated to induce marked effects on heart functional properties including myocardial stiffness and consequently diastolic and systolic performance [18, 19].
A group of enzymes capable of degrading almost all ECM proteins, matrix metalloproteinases (MMPs) contribute to both normal and pathological tissue remodelling [20–25]. The balance between activated MMPs and tissue inhibitors of metalloproteinases (TIMPs) controls the extent of myocardial ECM remodelling [26, 27] including the atrial tissue. However, recent studies have demonstrated the existence of alternative MMPs inhibitors [28–31], namely reversion-inducing cysteine-rich protein with Kazal motifs (RECK) which is thought to be an important regulator of ECM remodelling [28, 32, 33]. However, RECK expression has not been studied in relation to atrial fibrosis and AF.
Aminoterminal propeptide of type I (PINP) and aminoterminal propeptide of type III (PIIINP) are the markers of newly synthesized non-cross-linked collagen type I and type III, whereas carboxyterminal telopeptide of type I collagen (ICTP) and aminoterminal telopeptide of type III collagen (IIINTP) characterize cross-linked but partly degraded collagens [34]. A recent study showed that the level of circulating ICTP correlates with the type of AF, that is paroxysmal, persistent or permanent AF [35]. However, it must be emphasized, that only direct microscopic examination of the atrial tissue and determination of the atrial protein content can confirm or refute the results of non-invasive methods for assessing the degree of atrial fibrosis and collagen quality, since collagen markers can appear in serum also by liver fibrosis, systemic sclerosis and many bone and joint diseases [36–38]. However, there are no data regarding the markers of collagen synthesis/degradation directly in the atrial myocardium in patients with AF.
Transforming growth factor (TGF-β1) is a known profibrotic cytokine that has been demonstrated to induce selective atrial fibrosis in a transgenic mouse model [39] indicating the critical role of TGF-β1 in the pathogenesis of AF. These data have provided a direct link between atrial fibrosis and AF [14]. We hypothesized therefore that TGF-β1 and its major signalling pathway through Smads (vertebrate sma and mad (mothers against decapentallegic) genes may play a role in the development of atrial fibrosis thereby contributing to the maintenance of AF also in human patients.
From this background and in order to test the hypothesis that the degree of ECM changes and remodelling are more profound and exacerbated in patients with AF than in patients in SR, the present work set out with several interrelated goals. The first was to study collagen VI and expression levels of collagen type I and type III synthesis and degradation markers in patients with AF in comparison with those in patients in SR. The second was to evaluate the levels of MMPs and TIMPs, as well as alternative MMPs inhibitor RECK in AF. A third major goal was to study the role of TGF-β1-Smad signalling pathway in atrial fibrosis and AF.
Material and methods
Patients
The study group included 24 patients with a mean duration of AF ranging from 1 to 20 years who underwent a mini-Maze procedure. The surgical procedure has been detailed described [40]. In brief, longitudinal incision to the right atrium between vena cava superior and vena cava inferior (VCI), T-incision around the VCI, excision of the right atrial appendage (RAA). Excision of the left atrial appendage (LAA), circumcision of the pulmonary veins, incision from the amputation line of the LAA to the pulmonary veins.
The aim of the mini-MAZE was to reduce aortic cross-clamp time, to minimize injury of structures at risk, for example circumflex coronary artery and coronary sinus, but to interrupt the most frequent reentry circuits. To decrease the operative risk and to reduce the cross-clamp time and the time of extracorporal circulation, we modified the procedure and did not perform the following incisions as compared to Cox Maze III: endocardial incision to tricuspid and mitral annulus and incision of interatrial septum. Furthermore, cryoablation was not performed during mini-MAZE.
The AF patients were matched for left ventricular function, right and left atrial sizes with 24 patients in normal sinus rhythm (SR). In addition, given the evidence that cardiopulmonary bypass and the cross-clamp duration modulate the expression and activity of MMPs and TIMPs [41], as well as numerous cytokines implicated in ECM remodelling [42], the study groups were matched for duration of extracorporal circulation and the aortic cross-clamp time. Clinical data are presented in Table 1. The study protocol was approved by the Ethics Committee of the Kerckhoff Clinic, Bad Nauheim, Germany. All patients gave written informed consent and the study conforms to NIH guidelines.
1.
Baseline characteristics of patients with AF and patients in SR
| SR | AF | |
|---|---|---|
| Number | 24 | 24 |
| Gender (male/female) | 16/8 | 14/10 |
| Age (years) | 61.2 ± 9.3 | 64.5 ± 8.2 |
| Duration of AF (years) | - | 4.5 ± 3.8 |
| NYHA class (I-IV) | 2.3 ± 0.4 | 2.5 ± 0.5 |
| Underlying cardiac disease (n) | ||
| CAD | 2 | 1 |
| MVD | 5 | 8 |
| TVD | 2 | 1 |
| ASD | 1 | - |
| DCM | 5 | 2 |
| MVD/TVD | 5 | 2 |
| MVD/CAD | 5 | 2 |
| MVD/TVD/CAD | 2 | 4 |
| ECC time (minutes) | 121.2 ± 29.9 | 131.5 ± 36.1 |
| Cross-clamp time (minutes) | 84.1 ± 19.7 | 93.8 ± 22.4 |
| RA size (mm) | 55.1 ± 8.7 | 57.1 ± 6.9 |
| LA size (mm) | 63.7 ± 10.9 | 64.5 ± 7.4 |
| LF function | ||
| Ejection fraction (%) | 46.4 ± 12.8 | 45.8 ± 9.8 |
| LVESD (mm) | 35.6 ± 11.5 | 33.8 ± 10.3 |
| LVEDD (mm) | 55.4 ± 8.1 | 56.5 ± 7.7 |
| Medication (n) | ||
| Calcium antagonists | 9 | 13 |
| Beta-blockers | 8 | 12 |
| ACE inhibitors | 11 | 13 |
| Digitalis | 9 | 7 |
| Diuretics | 5 | 7 |
ASD, atrial septal defect; AVD, aortic valve disease; CAD, coronary artery disease; DCM, dilated cardiomyopathy; ECC, extracorporal circulation; LA, left atrium; LV, left ventricle; LVEDD, end-diastolic diameter; LVESD, LV end-systolic diameter; MVD, mitral valve disease; TVD, tricuspid valve disease. The data are presented as means ± SD.
Tissue preparation
Tissue samples were taken intraoperatively after aortic cross-clamp release (SR groups) and during mini-MAZE (AF groups) from RAA and the right atrial free wall (RFW) and were immediately frozen in liquid nitrogen and stored at 80°C. From each patient in SR, 2–3 tissue samples (20–40 mg wet weight) were divided into two equal parts for immunohistochemical (IHC) and Western blot (WB) analyses. From the patients who underwent a mini-MAZE procedure, more numerous and much larger tissue samples were obtained. However, the size of tissue samples from these patients was adjusted to that of patients in SR.
Immunolabelling and fluorescent microscopy
The tissue samples were mounted in Tissue-TeK® O.C.T.™ (Sakura) and 5 μm thick cryosections were prepared using a Leica CM3050S cryotome. Immunolabelling was performed using protocols as previously described [43]. In brief, cryosections were air dried and fixed for 10 min. in acetone at −20°C, 4% paraformaldehyde or Carnoys’ solution at room temperature. After washing in phosphate buffered saline (PBS) sections were incubated with 1% bovine serum albumin for 30 min. to block nonspecific binding sites. After rinsing in PBS, the samples were incubated overnight with primary antibodies which are listed in Table 2. Polyclonal antibodies to the ICTP, aminoterminal propeptide of type I collagen (PINP), aminoterminal propeptide of type III collagen (PIIINP) and IIINTP were prepared and used as previously described [34, 44]. After incubation with the primary antibodies, the sections were thoroughly washed in PBS and incubated with secondary biotin-conjugated antimouse, anti-rabbit or anti-sheep IgG antibodies (Dianova) for 1 hr followed by incubation with Cy2-conjugated streptavidin. Myofibrills were labelled for F-actin with phalloidin-conjugated with tetramethylrhodamine isothiocyanate (TRITC) or with Alexa 633. Nuclei were stained blue with 4′, 6-diamidino-2-phenylindole (DAPI) (Molecular Probes).
2.
Primary antibodies used for IHC and WB
| Antibody | Source | Catalog Nr./Clone | Company | Dilutions: IHC/WB |
|---|---|---|---|---|
| MMP2 | Mono- (mouse) | IM68/Ab-8 | Calbiochem | —/1:500 |
| MMP2 | Poly- (sheep) | 5980-0211 | Biotrend | 1:100/— |
| MMP9 | Mono- (mouse) | IM72/Ab-8 | Oncogene | —/1:500 |
| MMP9 | Poly- (sheep) | 5980-0911 | Biotrend | 1:100/— |
| TIMP1 | Poly- (rabbit) | T 8187 | Sigma | 1:100/1:1000 |
| TIMP2 | Mono- (mouse) | sc-5539/H-140 | Santa Cruz | 1:100/1:500 |
| TIMP3 | Poly- (rabbit) | sc-30075/H-55 | Santa Cruz | 1:100/1:500 |
| TIMP4 | Poly- (rabbit) | T 8312 | Sigma | 1:100/1:1000 |
| Collagen VI | Poly- (rabbit) | 3080a | Rockland | 1:200/1:4000 |
| RECK | Mono- (mouse) | R97120/28 | BD Biosciences | 1:50/1:300 |
| TGF-β1 | Mono- (mouse) | ab647/2E6 | Abcam | —/1:300 |
| TGF-β1 | Mono- (mouse) | MAB240/9016.2 | R& D | 1:50/— |
| Smad2 | Poly-(rabbit) | 3122/86F7 | Cell Signaling | 1:50/1:500 |
| Phospho -Smad2 | Poly-(rabbit) | 3104/Ser245/250/255 | Cell Signaling | —/1:500 |
| Vimentin-Cy3 | Mono- (mouse) | C9080/V9 | Sigma | 1:300/— |
| Smooth muscle (SM)-α-actin | Mono- (mouse) | 1A4 | Sigma | 1:300/— |
| Actin | Mono- (mouse) | HHF-35 | Sigma | —/1:1000 |
Confocal microscopy
The tissue samples were examined with a Leica TCS NT or Leica SP2 confocal scanning laser microscope. Confocal images were obtained using concomitant multi-channel scanning. Each recorded image was taken using two con-focal detectors for reflected fluorescence and consisted of 1024 × 1024 pixels. Series of confocal optical sections were taken through the depth of the tissue sample at 0.5–1 μm intervals. In order to improve image quality and to obtain a high signal/noise ratio each image from the series was signal-averaged. After data acquisition, the images were transferred to a Silicon Graphics Indy or Octane workstations (Silicon®, Grabsbrunn, Germany) for image restoration and reconstruction using Imaris®, the multi-channel image processing software (Bitplane®, Zürich, Switzerland). The principles of this method have been previously described [9, 45].
Quantitative analysis
For quantitative analysis cryosections from at least two different blocks of RA tissue in each case were used. All samples were immunolabelled simultaneously with identical conditions of fixation and dilutions of primary and secondary antibodies. Sections exposed to PBS instead of primary antibodies served as negative controls. Per each patient, at least 10 randomly selected microscopic fields (size 400 μm x 400 μm) were analysed with a fluorescent Leica DMRB microscope using a 40 PlanApo objective (Leica). Immunolabelled cryosections were studied using image analysis (Leica) and Image J® softwares. Quantification of each protein was performed blindly, having on the screen only one channel showing F-actin labelling. For each protein a specific setting was established and kept constant in all measurements. The area occupied by MMPs, TIMPs and collagen markers was expressed as percentage of specific labelling per tissue area.
Western blot
Frozen tissue was homogenized in lysis buffer and centrifuged for 10 min. RA extracts were loaded onto 4–12% polyacrylamide gel and separated under reducing conditions. Proteins were electrotransferred onto nitrocellulose membrane (Invitrogen) and blocked with 5% non-fat dry milk in Tris-buffered saline Tween (TBST) at 4°C. After washing with TBST proteins were incubated overnight at 4°C with primary antibodies which are listed in Table 2. Bound antibodies were detected by peroxidase-conjugated human antimouse, anti-rabbit or anti-sheep IgG horseradish-peroxidase-conjugated and SuperSignal West-Femto (Pierce) detection system. Quantification of immunoblots was done by scanning on a VersaDoc™ 4000 (Biorad) using ImageQuant software. The densitometric values of the investigated proteins were normalized per densitometric values of the 43 kD band of actin. All WBs were performed minimum in duplicate.
Statistical analysis
All data are presented as means ± S.E.M., if not otherwise indicated. Differences between group means were analysed using the two-way unpaired t-test or the Mann-Whitney U-test for two comparisons. For multiple comparisons we used ANOVA, followed by analysis with the Bonferroni t-test. Differences between groups were considered significant at P<0.05.
Results
Collagen VI
Previously we have shown in a similar group of patients with permanent AF that the amount of collagen type I and III was much higher than that in patients in SR [9]. In this study we have examined whether expression levels of non-fibrillar collagens (collagen VI) in patients with AF differ from those in patients in SR. IHC staining of collagen VI revealed that this protein was located mostly in the endomysium surrounding groups or individual cardiomyocytes and capillaries (Fig. 1A). In the AF group collagen VI was much more abundant and occupied enlarged extracellular spaces surrounding individual cardiomyocytes and vessels (Fig. 1B). In comparison with patients in SR, the quantity of collagen VI was significantly increased in patients with AF both in RA appendages and free walls (Fig. 1C).
1.
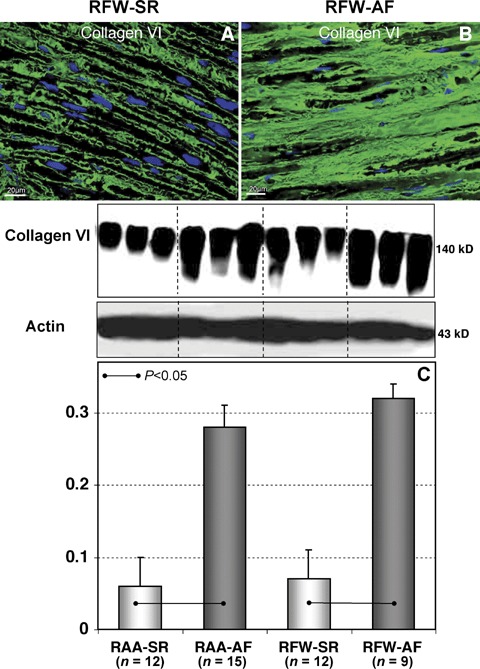
Representative immunofluorescent images of collagen VI labelling in RFW sectioned in longitudinal planes to the long myocyte axis in patients in sinus rhythm (SR) (A) and in patients with AF (B). Nuclei are stained blue with DAPI. (C) A typical Western blot (WB) for collagen VI and quantitative data of WB in different atrial tissues in patients in SR and in patients with atrial fibrillation (AF). The data are expressed as the ratio of densitometric units of collagen VI to those of actin.
Collagen type I and III markers
IHC staining of different collagen markers displayed their plentiful ECM localization as either thicker or finer fibers (Fig. 2A and B). Quantitative IHC and WB analyses showed significant up-regulation of collagen I and collagen III synthesis (PINP and PIIINP markers correspondingly) in both RAA-AF and RFW-AF groups in comparison with RAA-SR and RFW-SR groups (Fig. 2C and E). These changes in patients with AF were accompanied by significant elevations of cross-linked, but partly degraded collagens type I and III (ICTP and IIINTP markers) in contrast to SR groups (Fig. 2D and F).
2.
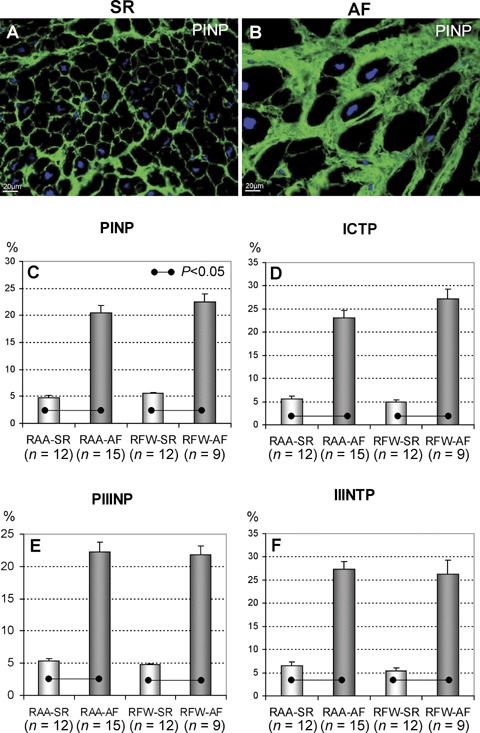
Representative immunofluorescent images of PINP in right atrial appendages (RAA) in patients in SR (A) and in patients with AF (B). Nuclei are stained blue with DAPI. Quantitative IHC data of PINP (C), carboxyterminal telopeptide of type I collagen (ICTP) (D), PIIINP (E) and IIINTP (F). The data are expressed as percent of positive labelling per tissue area.
As compared to controls, the PINP/ICTP ratio was significantly decreased in RFW-AF while the RAA-AF group showed only a tendency of a decreased the PIIINP/IIINTP ratio (Table 3). Similarly, the RFW-AF group showed a trend toward prevalence of collagen III (PIIINP) over collagen I synthesis (PINP).
3.
Ratios between collagen I and collagen III synthesis and degradation
| Ratio | RAA-SR (n= 12) | RAA-AF (n= 15) | RFW-SR (n= 12) | RFW-AF (n= 9) |
|---|---|---|---|---|
| PINP/ICTP | 0.95 ± 0.15 | 1.02 ± 0.15 | 1.32 ± 0.12 | 0.87 ± 0.1* |
| PIIINP/IIINTP | 1.24 ± 0.3 | 0.82 ± 0.14 | 1.02 ± 0.15 | 0.93 ± 0.1 |
| PINP/PIIINP | 1.26 ± 0.52 | 1.17 ± 0.21 | 1.26 ± 0.23 | 1.05 ± 0.06 |
| ICTP/IIINTP | 0.78 ± 0.09 | 0.85 ± 0.06 | 1.04 ± 0.07 | 0.97 ± 0.16 |
- P < 0.05 RFW-AF versus RFW-SR
MMPs and TIMPs
By IHC, MMP2 and MMP9 were localized in the ECM, perivascular regions (Fig. 3A and B) and in interstitial cells, mostly in fibroblasts, as indicated by vimentin staining (data not shown) and myofibroblasts as shown using immunolabelling for SM-α-actin (Fig. 3C and D). As compared with patients in SR (Fig. 3A), MMP2 in AF groups was essentially enhanced and occupied larger ECM spaces (Fig 3B).
3.
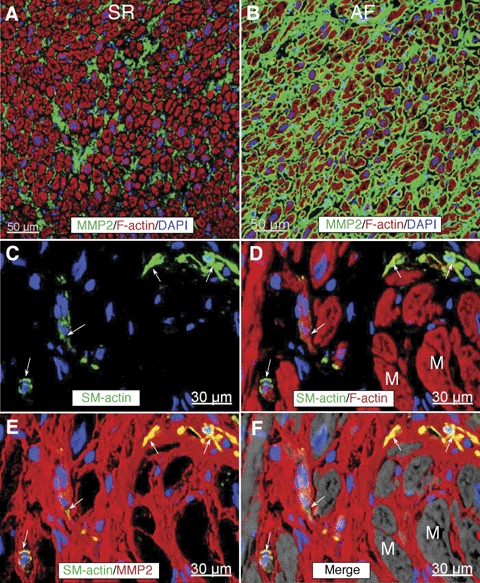
Representative confocal images of matrix metalloproteinases (MMP2) labelling (green) in RFW in a patient in SR (A) and in a patient with AF (B). Nuclei are stained blue with DAPI and f-actin is stained red with phalloidin conjugated with Alexa633.(C and D) are confocal micrographs of a tissue section immunolabelled for SM-α-actin, MMP2 and f-actin from a patient with AF. Arrows indicate interstitial cells which are postive for SM-α-actin. Notice that these cells express MMP2 as shown by yellow colocalization colour. M, myocytes. Nuclei are stained blue with DAPI and f-actin is stained red with phalloidin conjugated with Alexa633.
Expression levels of MMPs were analysed by IHC and WB analysis. Both methods demonstrated almost similar results (Fig. 4). According to quantitative IHC anaysis, MMP2 was significantly increased in RA appendages and free walls in AF groups as compared to SR (Fig. 4A), and demonstrated a tendency to be increased according to WB data (Fig. 4C). Quantification of MMP9 by IHC and WB showed that expression levels of MMP9 in AF groups were significantly higher than in SR groups (Fig. 4B and D).
4.
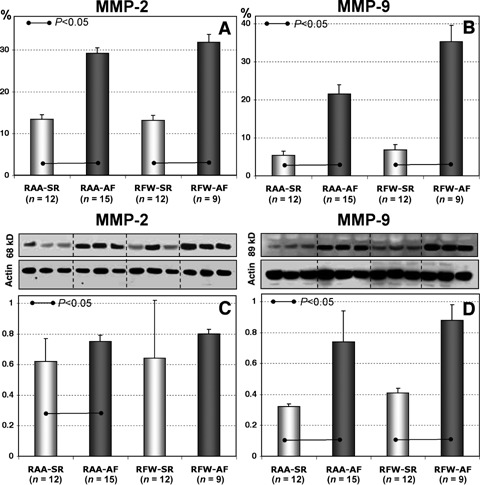
Quantitative IHC data of MMP2 (A) and MMP9 (B). The data are expressed as percent of positive labelling per tissue area. Representative WB for MMP2 (C) and MMP9 (D) and quantitative data of WB in different atrial tissues in patients in SR and in patients with AF. The data are expressed as ratios of either MMP2 or MMP9 expression levels to the actin content.
The interstitial localization of TIMP1, TIMP2, TIMP3 and TIMP4 was almost similar and varied between groups only in the percent of area occupied by positive staining (data not shown). By WB, the content of TIMP3 and TIMP4 in RA appendages and free walls showed no differences between AF and SR groups (Fig. 5C and D). In contrast, TIMP1 in patients with AF as compared with patients in SR was up-regulated in both, RA appendages and free walls (Fig. 5A), while TIMP2 was enhanced only in the RAA-AF group (Fig. 5B). The amount of TIMP2 was almost unchanged in RFW-AF and RFW-SR groups. Importantly, in patients in SR there was a significant difference in TIMP2 between RA free walls and appendages. Together, these data indicate that a regional heterogeneity in TIMP2 expression exists in different structures forming the RA atrium.
5.
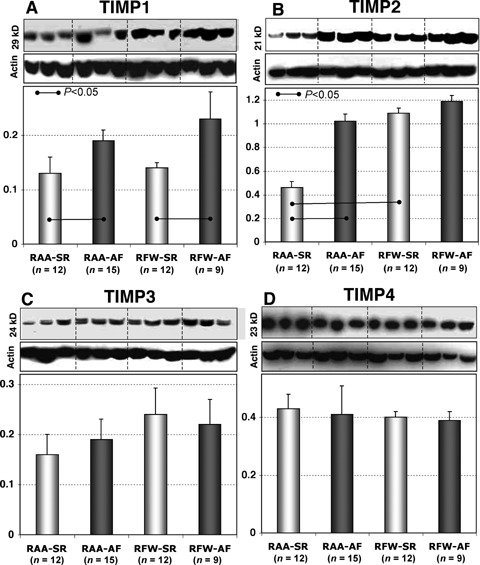
Representative WB and quantitative data of TIMP1 (A), TIMP2 (B), TIMP3 (C) and TIMP4 (D) in different atrial tissues from patients in SR and in patients with AF.
RECK
By IHC, RECK expression was found in patients of both AF and SR groups. The localization of RECK was confined mainly to interstitial cells or diffusely as a punctate labelling in the ECM (Fig. 6A and B). We did not find visually apparent differences in RECK expression in AF and SR groups except the observation that in SR groups RECK-positive staining was found in isolated ECM cells; whereas in AF groups more often diffuse positive spots or cell accumulations in the interstitium were observed. WB analysis revealed a significant increase in RECK expression levels only in RFW-AF group as compared to RAA-AF, RAA-SR and RFW-SR groups, respectively (Fig. 6C).
6.
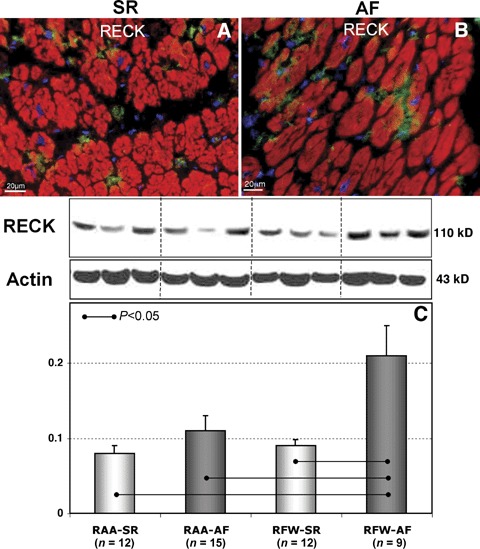
Representative immunofluorescent images of Reversion- inducing cysteinerich protein with Kazal motifs (RECK) labelling in RAA in patients in SR (A) and in patients with AF (B). Nuclei are stained blue with DAPI. Typical WB and quantitative WB data for RECK in different atrial tissues in patients in SR and in patients with AF (C).
TGF-β1 and Smads
TGF-β1 labelling was confined mainly to the interstitial cells (Fig. 7A and B). The number of TGF-β1-positive cells was much higher in patients with AF than in patients in SR. Double labelling for TGF-β1 and vimentin showed that TGF-β1 is localized in (myo)fibroblasts (Fig. 7C and D) and macrophages (data not shown). The level of TGF-β1 was significantly up-regulated in both RAA-AF (P<0.05) and RFW-AF (P<0.01) as compared to RAA-SR and RFW-SR groups (Fig. 7E).
7.
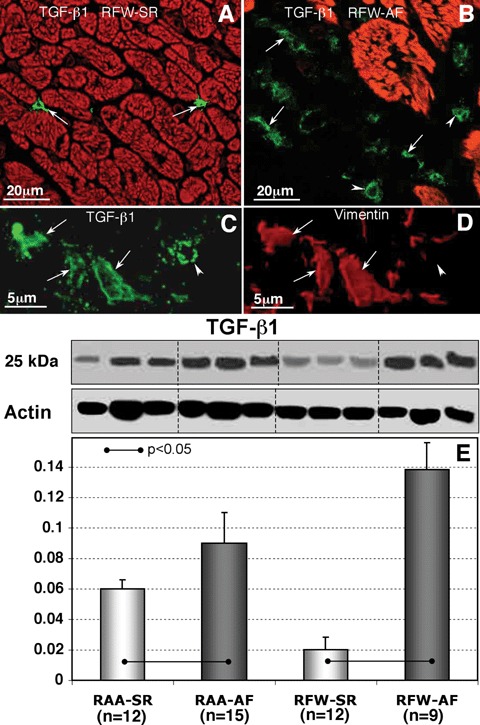
Confocal images showing transforming growth factor (TGF)-β1 expression in RFW-SR (A) and RRFW-AF (B). Myocytes are stained red with phalloidin-conjugated with tetramethyl-rhodamine isothiocyanate (TRITC). (C and D) are high-magnications confocal micrographs of a tissue section immunolabelled for TGF-β1 and vimentin from a patient with AF. In (A)–(D) arrows indicate spindle, fibroblast-like cells and arrowheads denote round monocyte- macrophage-like cells. A representative WB for TGF-β1 and its quantity in different atrial tissues from patients in SR and in patients with AF (E).
By IHC, Smad2-positive staining was observed in atrial cardiomyocytes at intercalated disks (Fig. 8A and B). In addition, Smad2-positive signal was found in fibroblast-like cells with long processes situated between cardiac cells (Fig. 8A and B). By WB, Smad2 and phosphorylated Smad2 showed a significant elevation in RFW-AF group in comparison with RFW-SR and RAA-AF (Fig. 8C and D).
8.
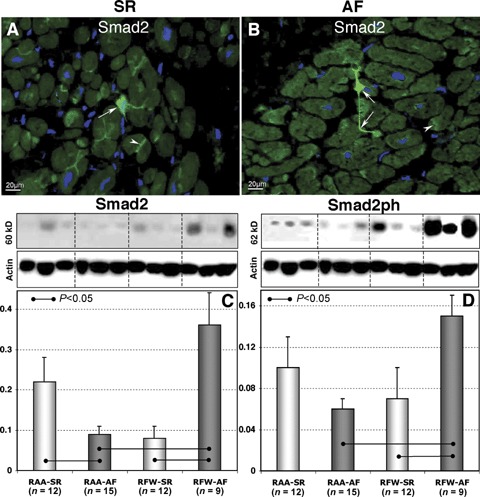
Representative immunofluorescent images of Smad2 labelling in RA appendages in patients in SR (A) and in patients with AF (B). Nuclei are stained blue with DAPI. Arrows denote fibroblast-like cells and arrowheads indicate intercalated disk-like structures. Typical WB and quantitative WB data of Smad2 (C) and Smad2ph (D) in different atrial tissues in patients in SR and in patients with AF.
Discussion
Atrial ECM remodelling and AF
There are controversial opinions whether structural changes in the atria are related to AF or due to underlying diseases [46–50]. In the present study, we tested the hypothesis and show that ECM changes and remodelling are more profound and exacerbated in patients with AF than in patients in SR. Recent evidence in support of this hypothesis has emerged from the observations that rapidly-paced atrial myocytes profoundly alter ECM-gene expression [51]. In addition, experimental results from TGF-β1 transgenic mice have clearly shown that atrial fibrosis alone is sufficient to create a substrate for AF [39]. Taken together, our observations and recently published data strongly suggest that AF per se may cause ECM remodelling in previously normal hearts or may aggravate ECM remodelling due to underlying heart diseases and degree of atrial dilation.
Previous (immuno)histochemical studies have convincingly demonstrated that atrial fibrosis consisting of the fibrillar collagen type I and III is a typical feature of AF, especially its permanent form. The present study has documented also an abundant collagen VI in patients with AF. All these data were recently confirmed at the level of gene expression in experimental models of AF, as well as in human patients with AF [52, 53]. These studies demonstrated dysregulation of ECM genes associated with collagen production and among them eight collagen genes and fibronectin.
Several experimental models demonstrated that atrial fibrosis is a substrate that promotes and maintains AF [11, 54]. An increased amount of fibrotic tissue which correlated to connexin 43 expression has been shown in the atria of patients with AF in comparison to patients in SR [9, 55]. The increase of fibrosis may cause an abnormal conduction through the atria. Fibrosis disturbs anisotropic conduction and thereby induces slowing of electrical conduction velocities, allowing for the generation of re-entry circuits [10].
The cardiac interstitium is comprised mainly of collagen with small amounts of fibronectin, laminin and elastin. The main myocardial fibrillar collagens are collagen type I, III and VI [16, 17, 56–60]. They consist of three peptide chains being cross-linked during the maturation and therefore are very stable and degradation-resistant structures. Type I collagen represents 50–80% of total cardiac collagen followed by collagen III (10–45%) and type VI (5%) [17]. Type I collagen is the major collagen which has been shown to play a crucial role in maintaining structural and functional integrity of the heart.
Collagen VI is a glycoprotein composed of three distinct polypeptide chains (α1, α2 and α3), each of them is encoded by separated genes [61, 62]. Morphological studies suggest that collagen type VI may act as a connecting unit linking different ECM components. Distribution of collagen VI is not prominent in the epicardium or perimysium, but is abundant in the finer connective tissue septa (endomysium) and around small groups of cardiomyocytes. Collagen VI has been shown to play an important role in the development of fibrosis in the failing heart [63–65]. Myocardial basement membranes are connected to fibronectin and/or collagen V and VI. These three ECM components are, in turn, connected to collagen type III that is connected to collagen type I. Collagen type I is more prevalent in the thicker connective tissue structures whereas collagen type VI and fibronectin are more prevalent in the more delicate ECM framework. Type I collagen has larger diameters of collagen fibres and higher stiffness as compared to collagen type III [66]. It must be mentioned that stiffness and passive mechanical properties of the heart are determined not only by the collagen amount, relative proportions of the types of collagen, the diameter of collagen fibres but to a greater extent also by collagen cross-linking [66, 67]. Cross-linking is a process of collagen fibres covalent linking to one another that results in increased stiffness and resistance to degradation. Collagen type I has a much higher degree of cross-linking than collagen type III and correspondingly higher stiffness.
As already mentioned, PINP and PIIINP are markers of newly synthesized non-cross-linked collagen I and III whereas ICTP and IIINTP characterize cross-linked but partly degraded collagens [34]. We revealed that atria of patients with AF were characterized by abundant synthesis of collagen type I and type III as reflected by PINP and PIIINP markers indicating severe ECM remodelling (Fig. 2). In addition, according to data presented in Table 3, the atria in patients with AF demonstrated local heterogeneity and in particular the predominance of collagen I degradation over its synthesis and a trend in prevalence of collagen III over collagen I synthesis in the RFW-AF group. The RAA-AF group displayed only a tendency of a decreased PIIINP/IIINTP ratio. These data indicate chamber-specific pecularities in the collagen synthesis/degradation balance.
In parallel to overexpression of newly synthesized collagens we observed in atrial ECM of patients with AF a considerable up-regulation of defective, partly degraded collagen type I and type III as indicated by ICTP and IIINTP markers correspondingly. Moreover, the RFW-AF group was characterized by a shift in collagen I balance towards its degradation. This intensive atrial ECM remodelling undoubtedly could have been caused by excessive activities of extracellular proteolytic enzymes – MMPs. We found that MMP2 and MMP9 were increased in RA appendages and free walls in patients with AF in comparison to patients in SR (Fig. 4).
TIMPs are the major inhibitors of MMPs. The level of TIMP1 was enhanced whereas TIMP3 and TIMP4 remained unchanged in both AF groups as compared to SR groups. As compared with patients in SR, TIMP2, which is a specific inhibitor of MMP2, was increased only in fibrillating appendages and showed only a tendency to be upregulated in fibrillating RA free walls. Nevertheless, in AF, there were no significant differences in the TIMP2 content between RA appendages and free walls (Fig. 5).
RECK as a regulator of ECM integrity
Although it is widely established that TIMPs are MMP inhibitors, other proteins have also inhibitory activities against some of the MMPs, including domains of netrins, the procollagen C-terminal proteinase enhancer, tissue factor pathway inhibitor and RECK. Their (patho) physiological significance is still not clear [28–31]. RECK is a 110 kD glycoprotein that contains a serine-proteinase-inhibitor-like domain, two domains that contain the epidermal growth factor-like repeats, and a C-terminal domain encodes a glycosylphosphatidilinositol modification that anchors RECK to the plasma membrane. RECK was first isolated as a transformation suppressor gene by expression cloning in a mouse fibroblast cell line transformed by an activated RAS oncogene [68]. RECK regulates at least MMP-2, MMP-9 and a soluble form of MT1-MMP [28, 68], but its activity against serine proteinases has not been studied yet. RECK is widely expressed in human tissues, however, nothing is known about its expression in the heart. We observed RECK-staining in atrial ECM of all groups. In atria from patients in SR we found isolated interstitial cells. In contrast, in patients with AF we revealed numerous RECK-positive cells or accumulation of RECK-positive material in the interstitium. Although the inhibitory activity of RECK is relatively weak [28], RECK is a cell-associated and membrane-anchored inhibitor and therefore it is plausible to speculate that it functions in a much more specific and directed way. The up-regulation of RECK in free walls of patients with AF in comparison with SR is most probably a compensatory mechanism in the settings of increased collagen degradation. It is established that RECK acts by protecting ECM components (especially the fibrillar collagens) from degradation and thereby maintaining the integrity of ECM [28]. In addition RECK has been shown to be a regulator of angiogenesis [69]. Although both AF groups were characterized by similar MMP/TIMP disbalance and alterations in collagen metabolism, the difference observed in RECK level between free walls and appendages in patients with AF indicates that RECK expression/regulation may be chamber-specific. Alternatively RECK might have other functions that have not yet been elucidated.
TGF-β1 and its Smad pathway
There are at least three interrelated pathways involved in atrial fibrosis: renin-angiotensin system, oxidative stress pathways and TGF-β1 [15, 70–72]. TGF-β1 is a known pro-fibrotic agent and its enhanced expression has been shown to increase myocardial fibrosis. In a transgenic mouse model with the overexpression of active TGF-β1 atrial interstitial fibrosis was observed but the cellular electro-physiology of atrial cardiomyocytes and ventricles was not affected. This study indicates that atria are more susceptible to TGF-β1 influences than ventricles and that atrial fibrosis alone is sufficient to create a substrate for AF implying that TGF-β1 plays an important role in atrial fibrogenesis [39]. Changes in genes regulating TGF-β1 function and signalling were observed in patients with permanent AF and in canine models of AF [52, 53]. TGF-β1-induced fibrosis results in an inhomogeneous environment for electrical propagation, which impedes anisotropic or linear conduction leading to arrhythmia development. In patients with AF an increased level of TGF-β1 was found in serum and it was down-regulated after defibrillation therapy [73]. In our study, TGF-β1 up-regulation was shown in atria of all AF groups as compared to SR controls (Fig. 7). This corresponds with excessive atrial ECM collagen synthesis observed in patients with AF (Figs. 1 and 2).
Signalling by TGF-β1 occurs through type I and type II receptors. The activated type I kinase propagates the signal inside the cell through the phosphorylation of receptor-regulated Smads. Smad proteins are classically divided into three groups: receptor-activated Smads (Smad1, 2, 3, 5 and 8), co-mediator Smads (Smad 4 and Smad10) and inhibitory Smads (Smad 6 and Smad7) [74]. It has been postulated that the effect of TGF 1 in the heart is primarily mediated through Smad2 phosphorylation [75–78]. The TGF-β1-Smad pathway appears to be involved in the activation of collagen-gene promoter sites, increasing DNA translation of collagen I. In the present work, enhanced levels of Smad2 and phosphorylated Smad2 was found in free walls of patients with AF. These data suggest involvement of the TGF-β1-induced atrial fibrosis. However, it is not clear why in fibrillating atria TGF-β1 was up-regulated in both, free walls and appendages, while Smad2 and phosphorylated Smad2 were increased only in RA appendages. It can be supposed that TGF-β1 signalling in appendages is postponed or another alternative pathway for TGF-β1-induced fibrosis might be involved.
Once phosphorylated, Smad2 creates a complex with Smad3 and Smad4 followed by translocation of this complex into the nucleus [79]. In vitro studies have demonstrated translocation of Smad2 into the nuclei of fibroblasts as a result of phosphorylation [80, 81]. We detected Smad2-positive fibroblasts in the atria of AF and SR patients (Fig. 8). Given that only a small fraction of fibroblast-like cells were positive for Smad-2 which does not concur with a much higher number of fibroblasts in the atrial tissue, it is tempting to speculate that these cells represent the interstitial Cajal-like cells which have recently been characterized ultrastructurally and immunohisto-chemically in both, ventricular and atrial tissues [82–84]. In addition, we revealed positive staining of Smad at intercalated disks of atrial cardiomyocytes. These results indicate, albeit indirectly, that the transmission of TGF-β1-Smad signals between fibroblasts and/or Cajal-like cells and cardiomyocytes occurs by direct interactions between these cells as recently reported [83, 85]. It should be emphasized that the expression levels of Smad2 and phosphorylated Smad2 were much higher in AF groups, especially in the RA free walls than that in the SR groups indicating an important role of TGF-β1-Smad in inducing atrial fibrosis.
Clinical perspective
Our findings may have rather prognostic significance. According to data of atrial tissue analysis, any of the collagen I and collagen III markers (PINP, ICTP, IIINTP, PIIINP) can be used for non-invasive estimation of fibrosis in the absence of concomitant non-cardiac diseases. As an anti-fibrotic therapy the blockade of renin-angiotensin-aldosterone system with angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers can reduce the incidence and recurrence of AF. This assumption has recently been strenghthen by the observations that inhibition of the angiotensin-converting enzyme results in decreased MMP9 levels implying that angiotensin-converting enzyme inhibitors may therapeutically be used to target MMP9 function [86].
The progression of fibrosis is induced and accompanied by MMP dysregulation. RECK as a new alternative inhibitor of MMPs has promising prognostic and may have a potential therapeutic utility. In comparison with the well-known TIMPs, RECK, as a MMP-inhibitor has some advantages. At first RECK not only limits MMPs action on different levels but also protects ECM components from degradation and stimulates angiogenesis. In addition, RECK is a membrane-associated inhibitor and acts in a more specific way. The majority of clinical trials using TIMPs analogues were disappointing with lack of efficacy and some side effects. We can suppose that RECK (or its chemical mimetic) which structure differs dramatically from TIMPs could be used as possible therapeutic options in AF treatment.
Study limitations
The findings of the present study should be interpreted in light of certain limitations. As study groups were characterized by different underlying cardiac diseases and the duration of AF was in a wide range (from 1 to 20 years) our results should be interpreted with some caution in terms of the high-interindividual variability in the studied parameters. Another limitation of the present study is that AF is mainly initiated in the left atrium. As for ethical reasons only RA tissues were used, more studies are certainly needed to elucidate whether there are regional heterogeneities in ECM remodelling in different atrial chambers. It should also be emphasized that most of the patients included in the present study had different degrees of heart failure. Several experimental and clinical studies have clearly shown that heart failure and atrial dilatation per se induce atrial ECM remodellingthereby contributing to the development of AF [13, 53, 87]. Nevertheless, our data demonstrating severe alterations in integrity and regulation of the atrial ECM in patients with AF regardless the presence or absence of heart failure provide further support to the ‘mounting evidence that fibrosis generates a major mechanism for AF’as Spach wrote in a recent editorial paper [88].
Conclusions
Taken together, the results of the present study demonstrate a more intensive qualitative and quantitative remodelling of the atrial ECM in patients with AF than in patients in SR. This process is characterized by excessive accumulation of collagens type I, III and VI due to disturbed collagen metabolism and dysregulated MMP/TIMP systems and increases in expression levels of the alternative MMP inhibitor – RECK. Atrial fibrosis is a TGF-β1-dependent process acting via TGF-β1-Smad pathway. Thus, this study indicates the presence of significant changes in atrial collagen synthesis/degradation leading to essential changes of the atrial tissue integrity culminating in excessive atrial fibrosis and maintainance of AF.
Acknowledgments
We thank Carmen Büttner for her expert technical assistance. This study was supported by grants from the Max-Planck-Gesellschaft, München, Germany, for co-operation with the Kerckhoff Clinic, Bad Nauheim (Forschungsprojekt PFOR-403 to S.K., and Forschungsprojekt PFOR-404 to V.P and S.K.)
References
- 1.Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. 1964;67:200–20. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- 2.Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–66. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 3.Mandapati R, Skanes A, Chen J, Berenfeld O, Jalife J. Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation. 2000;101:194–9. doi: 10.1161/01.cir.101.2.194. [DOI] [PubMed] [Google Scholar]
- 4.Skanes AC, Mandapati R, Berenfeld O, Davidenko JM, Jalife J. Spatiotemporal periodicity during atrial fibrillation in the isolated sheep heart. Circulation. 1998;98:1236–48. doi: 10.1161/01.cir.98.12.1236. [DOI] [PubMed] [Google Scholar]
- 5.Everett THt, Li H, Mangrum JM, McRury ID, Mitchell MA, Redick JA, Haines DE. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation. 2000;102:1454–60. doi: 10.1161/01.cir.102.12.1454. [DOI] [PubMed] [Google Scholar]
- 6.Ausma J, Wijffels M, Thone F, Wouters L, Allessie M, Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96:3157–63. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- 7.Ausma J, Litjens N, Lenders MH, Duimel H, Mast F, Wouters L, Ramaekers F, Allessie M, Borgers M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J Mol Cell Cardiol. 2001;33:2083–94. doi: 10.1006/jmcc.2001.1472. [DOI] [PubMed] [Google Scholar]
- 8.Ausma J, Wijffels M, Van Eys G, Koide M, Ramaekers F, Allessie M, Borgers M. Dedifferentiation of atrial cardiomyocytes as a result of chronic atrial fibrillation. Am J Pathol. 1997;151:985–97. [PMC free article] [PubMed] [Google Scholar]
- 9.Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–79. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- 10.Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ Res. 1986;58:356–71. doi: 10.1161/01.res.58.3.356. [DOI] [PubMed] [Google Scholar]
- 11.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Cui G, Esmailian F, Plunkett M, Marelli D, Ardehali A, Odim J, Laks H, Sen L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation. 2004;109:363–8. doi: 10.1161/01.CIR.0000109495.02213.52. [DOI] [PubMed] [Google Scholar]
- 13.Boixel C, Fontaine V, Rucker-Martin C, Milliez P, Louedec L, Michel JB, Jacob MP, Hatem SN. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metallo-proteinases in the rat. J Am Coll Cardiol. 2003;42:336–44. doi: 10.1016/s0735-1097(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 14.Hanna N, Cardin S, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc Res. 2004;63:236–44. doi: 10.1016/j.cardiores.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 15.Boldt A, Wetzel U, Lauschke J, Weigl J, Gummert J, Hindricks G, Kottkamp H, Dhein S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004;90:400–5. doi: 10.1136/hrt.2003.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mays PK, Bishop JE, Laurent GJ. Age-related changes in the proportion of types I and III collagen. Mech Ageing Dev. 1988;45:203–12. doi: 10.1016/0047-6374(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 17.Bashey RI, Martinez-Hernandez A, Jimenez SA. Isolation, characterization, and localization of cardiac collagen type VI. Associations with other extracellular matrix components. Circ Res. 1992;70:1006–17. doi: 10.1161/01.res.70.5.1006. [DOI] [PubMed] [Google Scholar]
- 18.Robinson TF, Geraci MA, Sonnenblick EH, Factor SM. Coiled perimysial fibers of papillary muscle in rat heart: morphology, distribution, and changes in configuration. Circ Res. 1988;63:577–92. doi: 10.1161/01.res.63.3.577. [DOI] [PubMed] [Google Scholar]
- 19.Baicu S, Stroud JD, Livesay VA, Hapke E, Holder JR, Spinale FG, Zile MR. Changes in extracellular collagen matrix alter myocardial systolic performance. Am J Physiol. 2003;284:H122–32. doi: 10.1152/ajpheart.00233.2002. [DOI] [PubMed] [Google Scholar]
- 20.Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–85. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pistol G, Matache C, Calugaru A, Stavaru C, Tanaseanu S, Ionescu R, Dumitrache S, Srefanescu M. Roles of CD147 on T lymphocytes activation and MMP-9 secretion in systemic lupus erythematosus. J Cell Mol Med. 2007;11:339–48. doi: 10.1111/j.1582-4934.2007.00022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moshal KS, Tyagi N, Moss V, Henderson B, Steed M, Ovechkin A, Aru GB, Tyagi SC. Early induction of matrix metalloproteinase-9 transduces signaling in human heart end stage failure. J Cell Mol Med. 2005;9:704–13. doi: 10.1111/j.1582-4934.2005.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med. 2007;11:88–95. doi: 10.1111/j.1582-4934.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jinga DC, Blidaru A, Condrea I, Ardeleanu C, Dragomir C, Szegli G, Stefanescu M, Matache C. MMP-9 and MMP-2 gelatinases and TIMP-1 and TIMP-2 inhibitors in breast cancer: correlations with prognostic factors. J Cell Mol Med. 2006;10:499–510. doi: 10.1111/j.1582-4934.2006.tb00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anghelina M, Moldovan L, Zabuawala T, Ostrowscki MC, Moldovan NL. A subpopulation of peritoneal macrophages form capillary-like lumens and branching patterns in vitro. J Cell Mol Med. 2006;10:708–15. doi: 10.1111/j.1582-4934.2006.tb00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–30. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 27.Spinale FG, Coker ML, Bond BR, Zellner JL. Myocardial matrix degradation and metallopro-teinase activation in the failing heart: a potential therapeutic target. Cardiovasc Res. 2000;46:225–38. doi: 10.1016/s0008-6363(99)00431-9. [DOI] [PubMed] [Google Scholar]
- 28.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa S, Itoh Y, Seiki M, Itohara S, Takahashi C, Noda M. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 29.Herman MP, Sukhova GK, Kisiel W, Foster D, Kehry MR, Libby P, Schonbeck U. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix met-alloproteinases with implications for atherosclerosis. J Clin Invest. 2001;107:1117–26. doi: 10.1172/JCI10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mott JD, Thomas CL, Rosenbach MT, Takahara K, Greenspan DS, Banda MJ. Post-translational proteolytic processing of procollagen C-terminal proteinase enhancer releases a metalloproteinase inhibitor. J Biol Chem. 2000;275:1384–90. doi: 10.1074/jbc.275.2.1384. [DOI] [PubMed] [Google Scholar]
- 31.Welm B, Mott J, Werb Z. Developmental biology: vasculogenesis is a wreck without RECK. Curr Biol. 2002;12:R209–11. doi: 10.1016/s0960-9822(02)00752-2. [DOI] [PubMed] [Google Scholar]
- 32.Noda M, Oh J, Takahashi R, Kondo S, Kitayama H, Takahashi C. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metastasis Rev. 2003;22:167–75. doi: 10.1023/a:1023043315031. [DOI] [PubMed] [Google Scholar]
- 33.Echizenya M, Kondo S, Takahashi R, Oh J, Kawashima S, Kitayama H, Takahashi C, Noda M. The membrane-anchored MMP-regulator RECK is a target of myogenic regulatory factors. Oncogene. 2005;24:5850–7. doi: 10.1038/sj.onc.1208733. [DOI] [PubMed] [Google Scholar]
- 34.Bode MK, Soini Y, Melkko J, Satta J, Risteli L, Risteli J. Increased amount of type III pN-collagen in human abdominal aortic aneurysms: evidence for impaired type III collagen fibrillogenesis. J Vasc Surg. 2000;32:1201–17. doi: 10.1067/mva.2000.109743. [DOI] [PubMed] [Google Scholar]
- 35.Tziakas DN, Chalikias GK, Papanas N, Stakos DA, Chatzikyriakou SV, Maltezos E. Circulating levels of collagen type I degradation marker depend on the type of atrial fibrillation. Europace. 2007;9:589–96. doi: 10.1093/europace/eum072. [DOI] [PubMed] [Google Scholar]
- 36.Koivisto H, Hietala J, Niemela O. An inverse relationship between markers of fibrogenesis and collagen degradation in patients with or without alcoholic liver disease. Am J Gastroenterol. 2007;102:773–9. doi: 10.1111/j.1572-0241.2006.01036.x. [DOI] [PubMed] [Google Scholar]
- 37.Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, Coppock J, Hoogen F, Herrick A, Mayes MD, Veale D, Haas J, Ledbetter S, Korn JH, Black CM, Seibold JR. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–33. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- 38.Donescu OS, Battie MC, Videman T, Risteli J, Eyre D. The predictive role of bone turnover markers for BMD in middle-aged men. Aging Male. 2006;9:97–102. doi: 10.1080/13685530600708631. [DOI] [PubMed] [Google Scholar]
- 39.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–9. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 40.Szalay ZA, Skwara W, Pitschner HF, Faude I, Klovekorn WP, Bauer EP. Midterm results after the Mini-Maze procedure. Eur J Cardiothorac Surg. 1999;16:306–11. doi: 10.1016/s1010-7940(99)00208-0. [DOI] [PubMed] [Google Scholar]
- 41.Lalu MM, Pasini E, Schulze CJ, Ferrari-Vivaldi M, Ferrari-Vivaldi G, Bachetti T, Schulz R. Ischaemiareperfusion injury activates matrix metalloproteinases in the human heart. Eur Heart J. 2005;26:27–35. doi: 10.1093/eurheartj/ehi007. [DOI] [PubMed] [Google Scholar]
- 42.Ruel M, Bianchi C, Khan TA, Xu S, Liddicoat JR, Voisine P, Araujo E, Lyon H, Kohane IS, Libermann TA, Sellke FW. Gene expression profile after cardiopulmonary bypass and cardioplegic arrest. J Thorac Cardiovasc Surg. 2003;126:1521–30. doi: 10.1016/s0022-5223(03)00969-3. [DOI] [PubMed] [Google Scholar]
- 43.Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure-overloaded human myocardium during heart failure progression. J Am Coll Cardiol. 2004;44:1609–18. doi: 10.1016/j.jacc.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 44.Kajantie E, Dunkel L, Risteli J, Pohjavuori M, Andersson S. Markers of type I and III collagen turnover as indicators of growth velocity in very low birth weight infants. J Clin Endocrinol Metab. 2001;86:4299–306. doi: 10.1210/jcem.86.9.7869. [DOI] [PubMed] [Google Scholar]
- 45.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–36. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Ausma J, Wijffels M, Thone F, Allessie M, Borgers M. Structural changes of atrial fibrillation due to sustained atrial fibrillation. Circulation. 1997;96:3157–63. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- 47.Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–4. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- 48.Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, Schoendube F, Hanrath P, Allessie M. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation. 2001;103:691–8. doi: 10.1161/01.cir.103.5.691. [DOI] [PubMed] [Google Scholar]
- 49.Shirani J, Aleddini J. Structural remodeling of the atrial appendage in patients with chronic non-valvular atrial fibrillation: implications for thrombus formation, systemic embolism, and assessment by transesophageal echocardiography. Cardiovasc Pathol. 2000;9:95–101. doi: 10.1016/s1054-8807(00)00030-2. [DOI] [PubMed] [Google Scholar]
- 50.Schotten U, Neuberger HR, Allessie M. The role of atrial dilatation in the domestication of atrial fibrillation. Prog Biophys Mol Biol. 2003;82:151–62. doi: 10.1016/s0079-6107(03)00012-9. [DOI] [PubMed] [Google Scholar]
- 51.Burstein B, Qi X-Y, Yeh Y-H, Calderone A, Nattel S. Atrial cardiomyocyte tachycardia alters cardiac fibroblast function: a novel consideration in atrial remodeling. Cardiovasc Res. 2007;76:442–52. doi: 10.1016/j.cardiores.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos L, Gebauer M, Steinmeyer K, Bleich M, Kääb S, Hinterseer M, Kartmann H, Kreuzer E, Dugas M, Steinbeck G, Nabauer M. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96:1022–9. doi: 10.1161/01.RES.0000165480.82737.33. [DOI] [PubMed] [Google Scholar]
- 53.Cardin S, Libby S, Pelletier P, Le Bouter S, Shiroshita-Takeshita A, Le Meur N, Léger J, Demolombe S, Ponton S, Glass L, Nattel S. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res. 2007;100:425–33. doi: 10.1161/01.RES.0000258428.09589.1a. [DOI] [PubMed] [Google Scholar]
- 54.Cha T-J, Ehrlich JR, Zhang L, Shi Y-F, Tardif J-C, Leung TK, Nattel S. Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation. 2004;109:412–8. doi: 10.1161/01.CIR.0000109501.47603.0C. [DOI] [PubMed] [Google Scholar]
- 55.Luo MH, Li YS, Yang KP. Fibrosis of collagen I and remodeling of connexin 43 in atrial myocardium of patients with atrial fibrillation. Cardiology. 2007;107:248–53. doi: 10.1159/000095501. [DOI] [PubMed] [Google Scholar]
- 56.Medugorac I. Characterization of intramuscular collagen in mammalian left ventricle. Basic Res Cardiol. 1982;77:589–98. doi: 10.1007/BF01908312. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee D, Sen S. Collagen phenotypes during development and regression of myocardial hypertrophy in spontaneously hypertensive rats. Circ Res. 1990;67:1474–80. doi: 10.1161/01.res.67.6.1474. [DOI] [PubMed] [Google Scholar]
- 58.Mukherjee D, Sen S. Alteration of collagen phenotypes in ischemic cardiomyopathy. J Clin Invest. 1991;88:1141–6. doi: 10.1172/JCI115414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metallo-proteinase inhibition. Circulation. 2003;107:618–25. doi: 10.1161/01.cir.0000046449.36178.00. [DOI] [PubMed] [Google Scholar]
- 60.Weber KT, Janicki JS, Shroff SG, Pick R, Chen RM, Bashey RI. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res. 1988;62:757–65. doi: 10.1161/01.res.62.4.757. [DOI] [PubMed] [Google Scholar]
- 61.Trueb B, Schaeren-Wiemers N, Schreier T, Winterhalter KH. Molecular cloning of chicken type VI collagen. Primary structure of the subunit alpha 2(VI)-pepsin. J Biol Chem. 1989;264:136–40. [PubMed] [Google Scholar]
- 62.Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, Timpl R. Amino acid sequence of the triple-helical domain of human collagen type VI. J Biol Chem. 1988;263:18601–16. [PubMed] [Google Scholar]
- 63.Yoshikane H, Honda M, Goto Y, Morioka S, Ooshima A, Moriyama K. Collagen in dilated cardiomyopathy–scanning electron microscopic and immunohistochemical observations. Jpn Circ J. 1992;56:899–910. doi: 10.1253/jcj.56.899. [DOI] [PubMed] [Google Scholar]
- 64.Mollnau H, Munkel B, Schaper J. Collagen VI in the extracellular matrix of normal and failing human myocardium. Herz. 1995;20:89–94. [PubMed] [Google Scholar]
- 65.Kitamura M, Shimizu M, Ino H, Okeie K, Yamaguchi M, Funjno N, Mabuchi H, Nakanishi I. Collagen remodeling and cardiac dysfunction in patients with hypertrophic cardiomyopathy: the significance of type III and VI collagens. Clin Cardiol. 2001;24:325–9. doi: 10.1002/clc.4960240413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pearlman ES, Weber KT, Janicki JS, Pietra GG, Fishman AP. Muscle fiber orientation and connective tissue content in the hypertrophied human heart. Lab Invest. 1982;46:158–64. [PubMed] [Google Scholar]
- 67.Badenhorst D, Maseko M, Tsotetsi OJ, Naidoo A, Brooksbank R, Norton GR, Woodiwiss AJ. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc Res. 2003;57:632–41. doi: 10.1016/s0008-6363(02)00733-2. [DOI] [PubMed] [Google Scholar]
- 68.Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, Noda M. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci USA. 1998;95:13221–6. doi: 10.1073/pnas.95.22.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoon SO, Park SJ, Yun CH, Chung AS. Roles of matrix metalloproteinases in tumor metastasis and angiogenesis. J Biochem Mol Biol. 2003;36:128–37. doi: 10.5483/bmbrep.2003.36.1.128. [DOI] [PubMed] [Google Scholar]
- 70.Shiroshita-Takeshita A, Schram G, Lavoie J, Nattel S. Effect of simvastatin and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia remodeling in dogs. Circulation. 2004;110:2313–9. doi: 10.1161/01.CIR.0000145163.56529.D1. [DOI] [PubMed] [Google Scholar]
- 71.Nattel S. Aldosterone antagonism and atrial fibrillation: time for clinical assessment. Eur Heart J. 2005;26:2079–80. doi: 10.1093/eurheartj/ehi477. [DOI] [PubMed] [Google Scholar]
- 72.Ehrlich JR, Hohnloser SH, Nattel S. Role of angiotensin system and effects of its inhibition in atrial fibrillation: clinical and experimental evidence. Eur Heart J. 2006;27:512–8. doi: 10.1093/eurheartj/ehi668. [DOI] [PubMed] [Google Scholar]
- 73.Seko Y, Nishimura H, Takahashi N, Ashida T, Nagai R. Serum levels of vascular endothelial growth factor and transforming growth factor-beta1 in patients with atrial fibrillation undergoing defibrillation therapy. Jpn Heart J. 2000;41:27–32. doi: 10.1536/jhj.41.27. [DOI] [PubMed] [Google Scholar]
- 74.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 75.Pokharel S, Rasoul S, Roks AJ, Van Leeuwen RE, Van Luyn MJ, Deelman LE, Smits JF, Carretero O, Van Gilst WH, Pinto YM. N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–61. doi: 10.1161/01.hyp.0000025880.56816.fa. [DOI] [PubMed] [Google Scholar]
- 76.Dixon IM, Hao J, Reid NL, Roth JC. Effect of chronic AT(1) receptor blockade on cardiac Smad overexpression in hereditary cardiomyopathic hamsters. Cardiovasc Res. 2000;46:286–97. doi: 10.1016/s0008-6363(00)00035-3. [DOI] [PubMed] [Google Scholar]
- 77.Hao J, Wang B, Jones SC, Jassal DS, Dixon IM. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol. 2000;279:H3020–30. doi: 10.1152/ajpheart.2000.279.6.H3020. [DOI] [PubMed] [Google Scholar]
- 78.Araujo-Jorge TC, Waghabi MC, Hasslocher-Moreno AM, Xavier SS, Higuchi Mde L, Keramidas M, Bailly S, Feige JJ. Implication of transforming growth factor-beta1 in Chagas disease myocardiopathy. J Infect Dis. 2002;186:1823–8. doi: 10.1086/345882. [DOI] [PubMed] [Google Scholar]
- 79.Greene RM, Nugent P, Mukhopadhyay P, Warner DR, Pisano MM. Intracellular dynamics of Smad-mediated TGFbeta signaling. J Cell Physiol. 2003;197:261–71. doi: 10.1002/jcp.10355. [DOI] [PubMed] [Google Scholar]
- 80.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 81.Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells. 2002;7:1191–204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- 82.Hinescu ME, Gherghiceanu M, Mandache E, Ciontea SM, Popescu LM. Interstitial Cajal-like cells (ICLC) in atrial myocardium: ultrastructural and immunohistochemical characterization. J Cell Mol Med. 2006;10:243–57. doi: 10.1111/j.1582-4934.2006.tb00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mandache E, Popescu LM, Gherghiceanu M. Myocardial interstitial Cajal-like cells (ICLC) and their nanostructural relationships with intercalated discs: shed vesicles as intermediates. J Cell Mol Med. 2007;11:1175–84. doi: 10.1111/j.1582-4934.2007.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Popescu LM, Gherghiceanu M, Hinescu ME, Cretoiu D, Ceafalan L, Regalia T, Popescu AC, Ardeleanu C, Mandache E. Insights into the interstitium of ventricular myocardium: interstitial Cajal-like cells (ICLC) J Cell Mol Med. 2006;10:429–58. doi: 10.1111/j.1582-4934.2006.tb00410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–25. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 86.Yamamoto D, Takai S, Jin S, Tanaka K, Miyazaki M. Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP-9. J Mol Cell Cardiol. 2007;43:670–6. doi: 10.1016/j.yjmcc.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Mukherjee R, Herron AR, Lowry AS, Stroud RE, Stroud MR, Wharton JM, Ikonomidis JS, Crumbley AJ, 3rd, Spinale FG, Gold MR. Selective induction of matrix metalloproteinases and tissue inhibitor of metalloproteinases in atrial and ventricular myocardium in patients with atrial fibrillation. Am J Cardiol. 2006;97:532–7. doi: 10.1016/j.amjcard.2005.08.073. [DOI] [PubMed] [Google Scholar]
- 88.Spach MS. Mounting evidence that fibrosis generates a major mechanism for atrial fibrillation. Circ Res. 2007;1001:839–47. doi: 10.1161/CIRCRESAHA.107.163956. [DOI] [PubMed] [Google Scholar]