

Abstract
CaMKII is a calcium and calmodulin-activated kinase that has been shown to regulate learning and memory in the brain, and contractility in blood vessels. Following Ca activation, CaMKII autophosphorylates, gaining a calcium-independent autonomous activity that reflects a molecular memory of having previously come into contact with calcium. The present study addresses whether the molecular memory properties of CaMKII are involved in the modulation of sustained vascular tone. We demonstrate a history-dependence of α agonist-induced vascular tone and show that CaMKII activation in vascular cells is also history dependent. Autophosphorylation of Thr287, which is classically associated with autonomous activity, does not persist during tone maintenance after transient increases in intracellular calcium levels. However, we have found that another site, Thr305, known from in vitro studies to be inhibitory, is regulated by α agonists in that the inhibitory action is removed, thus leading to a delayed reactivation of CaMKII as measured by Thr287 phosphorylation. By the use of a small molecule CaMKII inhibitor (KN93) as well as a decoy peptide (autoinhibitory peptide; AIP) we show a cause and effect relationship between CaMKII reactivation and sustained vascular tone maintenance. Thus, it appears that a complex interplay between the regulation of Thr305 and Thr287 provides a novel mechanism by which a history-dependence is developed and contributes to a new facet of molecular memory for CaMKII of relevance to vascular tone maintenance.
Keywords: CaMKII, differentiated vascular smooth muscle, contractility, molecular memory, protein kinase, vasospasm
Introduction
Calmodulin-dependent protein kinase II (CaMKII) is a Serine/Threonine kinase expressed as four different isoforms [1, 2], each encoded by a separate gene. The kinase is a large multimer of 12–14 subunits ranging in molecular mass from 50–65 kD. The α and β isoforms are largely restricted to neural tissues whereas the α and β isoforms are more widely distributed. It is thought that the γ isoform predominates in differentiated smooth muscle and that the δ isoform predominates in cultured smooth muscle cells [3–5]. Recent studies ([6–8]) utilizing both pharmacological and molecular knock down approaches have made it clear that CaMKII is a significant regulator of contractility in differentiated vascular smooth muscle (dVSM).
Binding of Ca/CaM activates the individual CaMKII subunits and also results in inter-subunit activation by an autophosphorylation on T287 (CaMKII γ iso-form, T286 in the CaMKII isoform) that causes both an increase in the calmodulin (CaM) binding affinity and the generation of Ca/CaM-independent activity. The ability of CaMKII to continue to have activity in the absence of Ca/CaM or in the presence of low concentrations of Ca/CaM has been referred to as ‘autonomous activity’ or, a ‘molecular memory’, of having previously come into contact with Ca/CaM [9] and, for the brain isoforms (α and β), has been linked to processes such as learning, synaptic plasticity and cognitive processes in the brain [10].
In the current study, we tested the hypothesis that the potential molecular memory function of CaMKII contributes to vascular tone maintenance. The results indicate that classical autonomous activity of CaMKII through T287 phosphorylation alone does not contribute to tone maintenance directly, but that a novel type of regulation at the less studied CaMKII inhibitory phosphorylation site, T305, may indirectly regulate vascular tone through inhibitory effects on CaMKII activation.
Materials and methods
Tissue preparation and contraction measurements
All procedures were approved by the Boston Biomedical Research Institute and Boston University Institutional Animal Care and Use Committees. As previously described [8], the aorta was excised from euthanized ferrets and immersed in oxygenated (95% O2 to 5% CO2) physiological saline (PSS) consisting of 120 mmol/l NaCl, 5.9 mmol/l KCl, 1.2 mmol/l NaH2PO4, 25 mmol/l NaHCO3, 11.5 mmol/l dextrose, 1 mmol/l CaCl2 and 1.4 mmol/l MgCl2 (pH = 7.4). The aorta was carefully dissected to remove all connective tissue and the endothelium. The aorta was cut into circular rings (∼3 mm wide), which were then attached to a force transducer and stretched to ∼1.3 times their resting length to approximate the optimal length for force production (a resting tension of ∼2.5 g). The tissue was allowed to equilibrate at 37°C for 1 hr in oxygenated PSS and then were stimulated for up to 10 min with 51 mmol/l KCl for purposes of normalization. At the end of the experiment the tissues were quick-frozen by immersion into a dry ice-acetone slurry containing 10% trichloroacetic acid (TCA) and 10 mM dithiothreitol (DTT).
Peptide loading
An inhibitory CaMKII decoy peptide (H2N-FITC-βA-GYGRKKRRQRRR- KKKLRRQEAFDAL -COOH) was synthesized by the Boston Biomedical Research Institute peptide synthesis core using sequence from the autoinhibitory domain of CaMKII (KKKLRRQEAFDAL). An N-terminal FITC tag was added to monitor loading efficiency and a TAT sequence (GYGRKKRRQRRR) was added to facilitate loading. This peptide has been previously characterized [11]. A control peptide (FITC-β A—GYGRKKRRQRRR-EAK-FRLKRAQLKD) was synthesized by randomizing the peptide sequence. Tissues were loaded with the peptides by incubation for three hours at 50 μmol/l (diluted in PSS), with one solution change midway. Uniform cellular loading was confirmed by confocal microscopy of the FITC signal.
Immunoblots
Quick–frozen tissue samples were homogenized and analysed via western blot as previously described [8]. Briefly, the tissue samples were acetone washed and homogenized in a buffer containing 20 mmol/l 3-(N-mor-pholino) propane sulfonic acid (MOPS), 4% SDS, 10% glycerol, 10 mmol/l DTT, 20 mmol/l (β-glycerophosphate, 5.5 μmol/l leupeptin, 5.5 μmol/l pepstatin, 20 kallikrein inhibiting units (KIU) aprotinin, 2 mmol/l Na3VO4, 1 mmol/l NaF, 100 μmol/l ZnCl2, 20 μmol/l 4-(2-Aminoethyl) ben-zenesulfonylfluoride hydrochloride (AEBSF) and 5 mmol/l ethylene glycol tetraacetic acid (EGTA). The samples were protein matched using a modified Lowry assay (DC Protein Assay Kit, BioRad) and then boiled in Laemmli sample buffer. The samples were separated using SDS polyacry-lamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore). Protein matching between samples was confirmed by staining the membrane with napthol blue black and measuring the densitometry of the actin bands. Immunoblotting was performed using enhanced chemiluminescence (Pierce) or the odyssey infrared imaging system (LI-COR Biosciences, Lincoln, Nebraska).
Materials and antibodies
The phosphoT286 (1:500, Millipore, Charlottesville, VA), phosphoT305 (1:10, 000; PhosphoSolutions, Aurora, CO) –specific antibodies were used as primary antibodies. Secondary antibodies conjugated with HRP or infrared dyes (1:1000) were purchased from Molecular Probes (Eugene, Oregon). KN93 and KN92 were purchased from Calbiochem, San Diego, CA.
Statistical analysis
Data were analysed and expressed as means (S.E.unless it is specified otherwise). Data were compared by anova where multiple comparisons were made and an unpaired, two-tailed t-test for force data where only two values were compared.
Results
CaMKII T287 phosphorylation levels are time dependent during α agonist- induced tonic contractions
Ferret aorta tissues display a sustained tonic increase in contractile force in response to α adrenergic agents, such as phenylephrine (PE) (Fig. 1A). In contrast, we have previously published that, for the same cell type, [Ca2+]i levels peak by 30 sec. after the addition of a maximally effective concentration of PE and then to fall to near baseline levels by 30 min. in the continued presence of PE [12]. We tested the hypothesis that CaMKII activation, as measured by autophosphorylation at T287, persists after [Ca2+]i falls to near baseline levels by virtue of the generation of autonomous activity characteristic of CaMKII in vitro. Thus, tissues were exposed to 10−5 M PE for increasing times from 0 to 60 min., quick frozen and probed in immunoblots for phospho-T287 levels.
1.
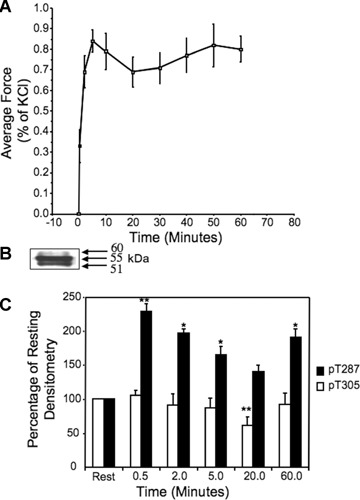
Time course of contractile response and CaMKII phosphorylation after addition of phenylephrine (10−5 M) to aortic rings.(A) Isometric force, normalized as a percentage of the force response to a 51 mM KCl PSS challenge performed at the beginning of each experiment n= 5 separate animals. (B) Typical immunostain of aortic homogenate for T287 phosphorylation illustrating the three groups of bands described in text as paralleling known CaMKII γ variants. The sample was quick frozen after 10 min.exposure to 51mM KCl PSS.(C) Time course of phosphorylation of CaMKII γ, 55kD band, at the T287 site (dark bars) and at the T305 site (clear bars).*, P<0.05 versus rest samples; *, P<0.05 versus rest samples **, P<0.01 versus rest samples, (data analysed before normalization to resting). n= 5–12 separate animals.
We have previously published that this tissue expresses 6 different γ CaMKII variants [5]. As expected, staining whole cell homogenates quick frozen after contractile stimulation with an anti-phospho-T287 antibody gave multiple bands (Fig. 1B). We were able to resolve three groups of bands on a consistent basis with both KCl PSS and PE stimulation, groups at approximately 60kD, 55kD and 51kD, probably representing variants G1&G2, B & J and C1 & C2 variants, respectively, based on gels with purified recombinant proteins [5]. The top band was difficult to quantitate because of interference from the pronounced 55kD band, but the quantitative time course for the 51 kD and 55 kD bands were indistinguishable in response to PE and only data for the 55kD band are shown here.
The average data for the time course of T287 phosphorylation of the 55kD band are shown in Fig. 1C, dark bars. Phosphorylation reaches a peak at 30 sec., then falls to a minimum by 20 min., mimicking transient increases in [Ca2+]i levels in this cell type [12]. Unexpected, however, was a subsequent increase in T287 phosphorylation at 60 min.(Fig. 1C).
CaMKII T305 is regulated in dVSM in response to α agonists
CaMKII T305 phosphorylation is reported, from in vitro studies, to be inhibitory with respect to catalytic activity of CaMKII since it blocks the binding of Ca/CaM [2]. In vitro phosphorylation at T305 has been described as increasing only when Ca is removed from solutions after CaMKII is initially activated and T287 has undergone autophosphorylation in the presence of Ca/CaM [13–16]. Whether this site is phosphorylated in vivo in dVSMCs has not been described. We monitored T305 phosphorylation by immunoblot of aortic samples quick frozen during activation with 10−5 M PE. From 0.5 to 20 min., a time when [Ca2+]i [12] and T287 phosphorylation (Fig. 1C) decrease, T305 also decreases but in this case the levels fall below baseline, indicating that this site is phosphorylated under resting conditions (Fig. 1C). This decrease removes an inhibitory influence from the kinase and may explain the subsequent increase in autophosphorylation of T287.
α agonist-induced contractility is inhibited by KN93
The question arises as to whether the increased activation of CaMKII, as monitored by the increased T287 phosphorylation and decreased T305 phosphorylation contributes to the simultaneous sustained increase in contractility. Thus, the effect of a small molecule inhibitor of CaMKII, KN93, on tissues was compared to sham treated tissues or tissues treated with an inactive congener of KN93, KN92. As is shown in Figure 2, KN93 causes a significant decrease in T287 (Fig. 2A) as well as T305 (Fig. 2B) phosphorylation and steady-state contractile force (Fig. 2C) at the 60 min. time-point, compared to the PE treated tissues. In contrast, the KN92 treated tissues were not different from the PE treated tissues in any of these parameters (Fig. 2). No effect on contractility was seen at 20 min.(data not shown).
2.
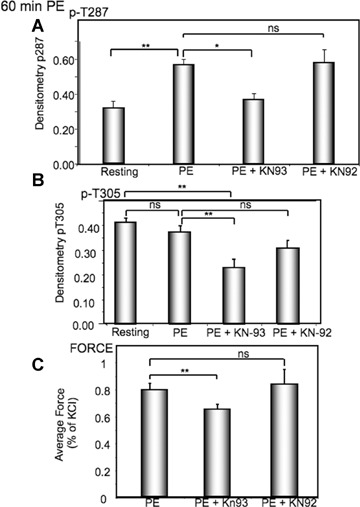
CaMKII inhibitor, KN93, inhibits CaMKII phospho-rylation and tonic force in response to PE.(A) Effect of KN93 versus an inactive congener, KN92 on PE(10−5 M)-induced increases in T287 phosphorylation at the late, 60 min.time-point.(B) Effect of KN93 versus that of an inactive congener, KN92 on PE-induced changes in T305 phosphorylation.(C) Effect of KN93 versus that of an inactive congener, KN92 on PE-induced increases in contractile force at the late, 60 min time point. In all cases, n= 6–10 separate animals. *, P<0.05; **P<0.01 compared to rest; NS, not significantly different.
CaMKII T287 phosphorylation is history dependent
We have made the observation that the amplitude of the force response to a 20 min. exposure to 10−5 M PE is history dependent. When given as a single 20 min.challenge, the contraction to a maximally effective (10−5 M) concentration of PE averages about 65% of the response to steady state challenges to KCl PSS performed at the beginning of the experiment. However, if the same 20 min.exposure to 10−5 M PE occurs as part of a concentration- response protocol, force increases to about 96% of the response to the KCl PSS challenge (Fig. 3C).
3.
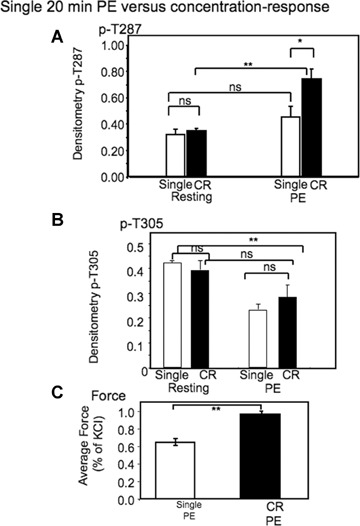
History dependence of steady state phosphorylation and force responses to a maximally effective concentration of PE.(A) Comparison of T287 phosphorylation levels after 20 min.exposure to 10−5 M PE either as a single challenge or after exposure to increasing concentrations of PE (CR) from 10−8 to 10−5 M).(B) Comparison of T305 phosphorylation after 20 min exposure to 10−5 M PE either as a single challenge or after exposure to increasing concentrations of PE (CR) from 10−8 to 10−5.(C) Comparison of contractile force levels after a 20 min.exposure to PE either as a single challenge or after exposure to increasing concentrations of PE (CR) from 10−8 to 10−5 M.*, P<0.05; **, P<0.01. NS, not significantly different. In all cases n= 5–21 animals.
The increase in contractility could be due to additivity with other mechanisms, such as myosin light chain kinase activation or caldesmon phosphorylation which might occur at lower concentrations during the concentration-response protocol. However, when CaMKII T287 phosphorylation was measured in tissues quick frozen during the same protocol, T287 phosphorylation levels were found to be significantly increased by the concentration response protocol even though both samples were frozen after 20 min. of exposure to 10−5 M PE (Fig. 3A). T305 phosphorylation levels were also measured in this protocol (Fig. 3B). In this case no significant difference was detectable between the single exposure and the concentration response protocol for this phosphorylation site, suggesting a difference in the kinetics of phosphorylation/de-phosphorylation T287 versus T305.
Time and history-dependent CaMKII activation contributes to increased contractility in dVSM
To determine if the history-dependent increases in CaMKII T287 phosphorylation and contractile force are related in a cause-and-effect manner, we determined the effect of the small molecule CaMKII inhibitor, KN93 in the concentration-response protocol. As is shown in Fig. 4A, KN93, but not the inactive congener KN92, significantly decreased both T287 phosphorylation (left panel) and contractile force (right panel) relative to the vehicle-treated control.
4.
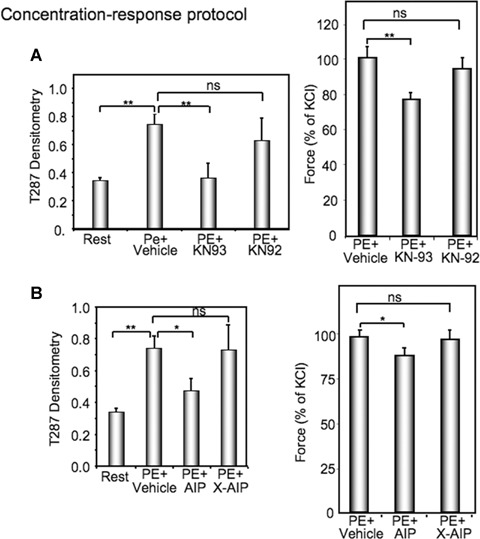
History dependent increases in CaMKII phospho-rylation and force are inhibited by two different CaMKII inhibitors. (A) Comparison of the effect of KN93 versus the inactive congener KN92 on T287 CaMKII phosphorylation (left panel) or on contractile force (right panel) in resting aortic tissues or those exposed to a concentration response protocol ending with 20 min in 10−5 M PE. (B), Comparison of the effect of AIP peptide versus a scrambled version of AIP (X-AIP) on T287 CaMKII phosphorylation (left panel) or on contractile force (right panel) in resting aortic tissues or those exposed to a concentration response protocol ending with 20 min.in 10−5 M PE.*, P<0.05; **P<0.01; NS, not significantly different. n= 3–10.
Because of the possibility of non-specific actions of KN93, we further used a second approach to inhibit CaMKII. We synthesized a previously described [17] decoy peptide containing the autoin-hibitory sequence from the CaMKII regulatory domain, commonly called AIP. As can be seen in Fig. 4B, this peptide similarly inhibited both PE-induced increases in CaMKII T287 phosphorylation (left panel) and contractile force (right panel), further supporting a cause-and-effect relationship between the history-dependent increases in CaMKII activity and contractility.
Discussion
The present study is the first to report that T305 phosphorylation of CaMKII γ is regulated in dVSM. This site has been studied almost exclusively in purified proteins in vitro where it has been shown that T305 is phosphorylated only after: (1) T286 (CaMKII α site corresponding to T287 in CaMKII γ) phosphorylation renders the kinase autonomously active and then (2) the removal of Ca/CaM with EGTA, since bound Ca/CaM protects the phosphorylation site [2]. Very recently T305 phosphorylation of CaMKII α has been shown to be at high levels in postsynaptic densities fractionated from brain slices [18]. Also, in drosophila, a protein, dCASK or Camguk, has been identified and shown to stimulate CaMKII to phosphorylate T306 (drosophila α CaMKII site corresponding to T305 in mammals) under conditions of low-synapse activity and, presumably low [Ca2+]i [19]. Since T305 blocks Ca/CaM binding it will also block activation of the kinase and consequent further T287 phosphorylation. It is said to decrease the ‘gain’ of the system in the synapse. We found that T305 levels are significantly elevated in unstimulated, ‘resting’ tissues. This is expected from the low [Ca2+]i levels in unstimulated tissues and may produce a decreased ‘gain’ with respect to contractility in un-stimulated tissues.
The fact that the resting levels of T305 phosphorylation are significantly higher than those at the 20 min.time point after PE addition suggests that some factor is promoting T305 phosphorylation in the absence of stimulation and also in the absence of detectable T287 phosphorylation and activation of CaMKII activity. Similarly, from theory, based on in vitro work with purified proteins, the falling [Ca2+]I level after the initial [Ca2+]i transient would be expected to promote increasing T305 levels but this is not seen. It is not known if there is a mammalian equivalent of Camguk, but our data suggest that a similar regulatory protein or a kinase other than CaMKII may cause the observed but unexpected pattern of regulation of inhibitory T305 in dVSM.
In parallel, there is also a significant decrease in T287 phosphorylation. Presumably, the occurrence of the autonomous, Ca-independent CaMKII catalytic activity is prevented in vivo because of the presence of phosphatases in the cell that dephosphorylate T287 and terminate catalytic activity. The action of these phosphatases presumably also explains the fall in T305 autophosphorylation levels at this time. This would remove an inhibitory influence on the kinase by un-blocking the Ca/CaM binding site. It is of interest that apparently this site is blocked in the resting state, keeping the kinase in the off state. During the late time-points of the PE response at a time when [Ca2+]I is at near basal levels, the change in the ratio of the T287 to T305 phosphorylation could tend to activate CaMKII again and perhaps explain the subsequent increase seen at late time points in T287 phosphorylation.
We had expected to see evidence for autonomous CaMKII γ activity in dVSM after Ca transients, but we did not. After an agonist-induced [Ca2+]I transient, our results demonstrate that T287 is phosphorylated but that as [Ca2+]I falls, T287 phosphorylation also falls. Presumably this is because cellular phosphatases dephosphorylate T287 before autonomous activity can occur. Rokolya and Singer [6] have demonstrated the presence of autonomous CaMKII activity in homogenates of carotid artery frozen under conditions of high [Ca2+]I, and subsequently treated with EGTA to remove Ca during the assay. However, this assay is performed with the use of phosphatase inhibitors, consistent with the concept that different results are obtained in vivo because of intracellular phosphatase activity.
Thus, a classic example of autonomous activity of CaMKII was not observed; however, in vivo there seems to be a delicate balance between activating versus inhibiting factors (Fig. 5) that allow the unexpected late increase in T287 phosphorylation and, presumably, CaMKII activity. The decrease in the phosphorylation level of the inhibitory T305 site seen in the presence of PE, which must involve a phosphatase as well, appears to allow the subsequent increase in T287 phosphorylation and CaMKII activity at the 60 min. time-point, and could also be considered a reflection of the ‘memory’ properties of CaMKII. It is of note that if PE is removed at the 60 min.time point the muscle does relax. Thus, the cells require the presence of PE to ‘constantly remind’ CaMKII to stay active. This presumably reflects the requirement for PE-triggered signalling pathways that regulate the kinase/phosphatase balance. This late surge in T287 activity is relevant functionally as well since, as we have shown here, it contributes to tone maintenance in the presence of an α agonist. This potential memory property may play a significant role in sustaining vascular tone and could potentially represent a novel mechanism by which vasospasm is maintained.
5.
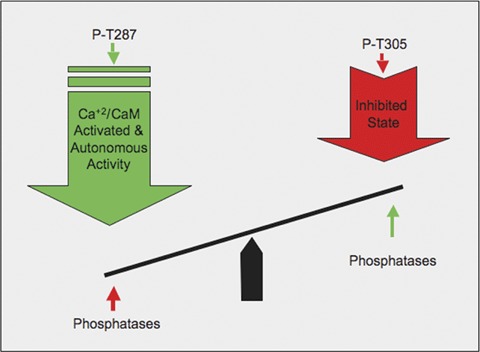
Model of complex interactions between activating phosphorylation at T287, inhibitory phosphorylation at T305 and dephosphorylation by phosphatases leading to a net level of CaMKII phosphorylation.
Acknowledgments
Funding was provided by NIH grants HL31704, HL80003 and HL86655 to K.G.M. and HL46716 and HL076130 to FWS.
References
- 1.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hudmon A, Schulman H. Neuronal Ca2+/calmod-ulin-dependent protein kinase II:The role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 3.Singer HA, Benscoter HA, Schworer CM. Novel Ca2+/calmodulin-dependent protein kinase II gamma-subunit variants expressed in vascular smooth muscle, brain, and cardiomyocytes. J Biol Chem. 1997;272:9393–400. doi: 10.1074/jbc.272.14.9393. [DOI] [PubMed] [Google Scholar]
- 4.Singer HA, Abraham ST, Schworer CM. Calcium/calmodulin-dependent protein kinase II. In: Bárány M, editor. Biochemistry of Smooth Muscle Contraction. San Diego.: Academic Press, Inc; 1996. pp. 143–53. [Google Scholar]
- 5.Gangopadhyay SS, Barber AL, Gallant C, Grabarek Z, Smith JL, Morgan KG. Differential functional properties of calmodulin-dependent protein kinase IIgamma variants isolated from smooth muscle. Biochem J. 2003;372:347–57. doi: 10.1042/BJ20030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rokolya A, Singer HA. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol. 2000;278:C537–45. doi: 10.1152/ajpcell.2000.278.3.C537. [DOI] [PubMed] [Google Scholar]
- 7.Kim I, Je HD, Gallant C, Zhan Q, Riper DV, Badwey JA, Singer HA, Morgan KG. Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. J. Physiol. 2000;526:367–74. doi: 10.1111/j.1469-7793.2000.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marganski WA, Gangopadhyay SS, Je HD, Morgan KG. Targeting of a novel Ca+2/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005;97:541–9. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- 9.Fink CC, Meyer T. Molecular mechanisms of CaMKII activation in neuronal plasticity. Curr Opin Neurobiol. 2002;12:293–9. doi: 10.1016/s0959-4388(02)00327-6. [DOI] [PubMed] [Google Scholar]
- 10.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–3. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 11.Hudmon A, Kim SA, Kolb SJ, Stoops JK, Waxham MN. Light scattering and transmission electron microscopy studies reveal a mechanism for calcium/calmodulin-dependent protein kinase II self-association. J Neurochem. 2001;76:1364–75. doi: 10.1046/j.1471-4159.2001.00119.x. [DOI] [PubMed] [Google Scholar]
- 12.Jiang MJ, Morgan KG. Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflug. Archiv. 1989;413:637–43. doi: 10.1007/BF00581814. [DOI] [PubMed] [Google Scholar]
- 13.Colbran RJ, Soderling TR. Calcium/calmodulin-independent autophosphorylation sites of calcium/calmodulin-dependent protein kinase II. Studies on the effect of phosphorylation of threonine 305/306 and serine 314 on calmodulin binding using synthetic peptides. J Biol Chem. 1990;265:11213–9. [PubMed] [Google Scholar]
- 14.Hanson PI, Kapiloff MS, Lou LL, Rosenfeld MG, Schulman H. Expression of a multifunctional Ca2+/calmodulin-dependent protein kinase and mutational analysis of its autoregulation. Neuron. 1989;3:59–70. doi: 10.1016/0896-6273(89)90115-3. [DOI] [PubMed] [Google Scholar]
- 15.Lu CS, Hodge JJ, Mehren J, Sun XX, Griffith LC. Regulation of the Ca2+/CaM-responsive pool of CaMKII by scaffold-dependent autophosphorylation. Neuron. 2003;40:1185–97. doi: 10.1016/s0896-6273(03)00786-4. [DOI] [PubMed] [Google Scholar]
- 16.Patton BL, Miller SG, Kennedy MB. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem. 1990;265:11204–12. [PubMed] [Google Scholar]
- 17.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–12. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- 18.Davies KD, Alvestad RM, Coultrap SJ, Browning MD. alphaCaMKII autophosphorylation levels differ depending on subcellular localization. Brain Res. 2007;1158:39–49. doi: 10.1016/j.brainres.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodge JJ, Mullasseril P, Griffith LC. Activity-dependent gating of CaMKII autonomous activity by Drosophila CASK. Neuron. 2006;51:327–37. doi: 10.1016/j.neuron.2006.06.020. [DOI] [PubMed] [Google Scholar]