

Abstract
Foetal cells secrete more growth factors, generate less immune response, grow and proliferate better than adult cells. These characteristics make them desirable for recombinant modification and use in microencapsulated cellular gene therapeutics. We have established a system in vitro to obtain a pure population of primary human foetal myoblasts under several rounds of selection with non-collagen coated plates and identified by desmin staining. These primary myoblasts presented good proliferation ability and better differentiation characteristics in monolayer and after microencapsulation compared to murine myoblast C2C12 cells based on creatine phosphokinase (CPK), major histocompatibility complex (MHC) and multi-nucleated myotubule determination. The lifespan of primary myoblasts was 70 population doublings before entering into senescent state, with a population time of 18–24 hrs. Hence, we have developed a protocol for isolating human foetal primary myoblasts with excellent differentiation potential and robust growth and longevity. They should be useful for cell-based therapy in human clinical applications with microencapsulation technology.
Keywords: primary cell culture, immortalization, cell therapy, gene therapy, microencapsulation, alginate
Introduction
Human foetal tissue research has led to the development of a number of important research and medical advances, particularly in the area of transplantation. Allogenic organ transplantation with foetal tissues rather than adult tissues is attractive because of better survival of the graft. In animal models, when adult allogeneic kidneys are transplanted into an immuno-competent recipient, rejection occurs immediately, causing deterioration of renal architecture and a dense lymphocytic infiltration [1, 2]. In contrast, transplanted foetal kidneys survive extremely well, showing excellent maturation of renal elements and no sign of rejection [3]. Studies with murine foetal kidneys demonstrated that there were decreased expression of MHC [4] and some cell adhesion molecules (CAMs) such as N-CAM, A-CAM, ICAM, VCAM [5]. These, together with a paucity of antigen-presenting cells (APCs) [6, 7], meant that foetal kidneys induced a less effective alloantigen-primed T cells response, leading to reduced immunogenicity. However, other foetal tissues, such as skin, small intestine, pancreas or liver, were rejected soon after transplantation [6, 8, 9]. These results indicated that different foetal organs have variable propensity for rejection. Selective application of foetal tissues provides an ideal candidate for organ replacement therapy.
Foetal cell therapy is another area of intense research. Foetal cells, particularly stem cells, are also clinically valuable for transplantation [8] because they possess properties of intrinsic plasticity, robust growth and proliferation, growth factor production, and reduced antigenicity compared to adult cells. Transplantation of foetal fibroblasts showed none of the clinical or histological signs of acute inflammation or rejection, while demonstrating better survival than adult fibroblasts [10]. Foetal nerve cells are also being examined as grafts for Parkinson's disease [11, 12] and amyotrophic lateral sclerosis (ALS) [13]. As our understanding of stem cell biology improves, we will hopefully learn how to manipulate these cells into differentiating into a desired tissue, expanding the therapeutic options [14]. It has been demonstrated that stem cells can be modified to secrete biochemicals to improve host cell survival or decrease inflammation. Neural stem cells derived from mouse foetus endogenously secrete glia-cell-line-derived neu-rotrophic factor (GDNF) and promote the recovery of dopaminergic neurons in nigral reconstitution of dopaminergic-depleted areas in mouse models of Parkinson's disease. However, the current limited understanding of how stem cells differentiate after transplantation and the tumourigeneic potential of embryonic stem cells are concerns in their clinical therapy.
Our laboratory has developed a novel approach to cell-based therapy by encapsulating genetically modified allogeneic cells secreting therapeutic products. Microcapsules composed of alginate-polylysine-alginate (APA) layers provide a selectively permeable immuno-isolated microenvironment for allo-geneic cells that protects them from host immune mediators. The cells can be engineered to secrete therapeutic proteins for the treatment of a variety of diseases. The cells to be encapsulated should possess several properties such as: robust proliferative potential necessary for gene transfection in vitro; capability to express and secrete the transgene product and the ability to maintain stable transgene expression after encapsulation in vitro and in vivo. Since the development of microencapsulation, many cell lines have been identified that possess these requirements and have successfully been used to deliver therapeutic proteins in different animal models [15]. In general, immortalized cell lines survive better than primary cells and are more feasible for long-term applications, whereas ‘reservoirs’ of primary cells are hard to maintain because of their inability to proliferate indefinitely following genetic modification. Several types of proliferative cell lines derived from fibroblasts 16], myoblasts [17], hybrido-ma cells [18], PC12 [19], BHK [20], 293 cells [21–23], CHO [24] have been used effectively in various types of microcapsules. However, cultured myoblasts possess several advantages over other cell types when used as recombinant gene product delivery vehicles in immuno-isolation devices. They can proliferate continuously, hence are amenable to genetic modification, but they can also differentiate terminally. The capability of myoblasts to withdraw from the cell cycle through differentiation into myotubules circumvents the potential problem of continued proliferation and overcrowding of cells within the microcapsules. This permits maintenance of their long-term viability in vivo and allows for sustained expression and secretion of the therapeutic gene products, thus providing more long-term efficacy for disease treatment. Murine myoblast C2C12 cells have successfully delivered therapeutic proteins, such as human growth factor [17], human factor IX [15] and β-glu-curonidase [25] into corresponding animal models. However, C2C12 is a transformed cell line which causes tumour development in nude mice [26]. Diverse cell types, such as nerve cells, adipocytes and fibroblasts, which can have undesirable or unpredicatable effects on the transplantation, inevitably contaminate cultured primary muscle cells. However, in 1981, Webster & Blau developed a method for the isolation of human myoblasts with high purity [27]. The method was applied in this study to isolate and establish primary human foetal myoblasts for use in microencapsulated therapy. We have compared the characteristics of human foetal myoblast proliferation and differentiation in monolayer and after encapsulation with the C2C12 cells that are widely used in mouse and in vitro experiments.
Materials and methods
Growth media
Growth medium for primary myoblasts contained Ham's nutrient mixture F-10 with 20% foetal bovine serum (FBS) and basic fibroblast growth factor (bFGF) (1.0 ng/ml) Differentiation medium for myogenesis consisted of Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 2% horse serum. The growth medium for primary fibroblasts was made up of DMEM with 20% FBS. Murine C2C12 myoblasts (ATCC, CRL#1772) were grown in DMEM +10% FBS. All the cultures were grown in the presence of penicillin (100 U/ml) and streptomycin sulfate (100 μ g/ml). The plates used for human foetal myoblasts were coated with 0.01% collagen in 0.1 N acetic acid. Primary myoblasts and fibroblasts were frozen in FBS + 10% DMSO.
Isolation of primary human foetal myoblasts
The thigh muscle sample was obtained from a 9-week foetus from a therapeutic abortion as approved by Institutional Research Ethics Board according to guidelines established by the Canadian Tri-Council Policy Statement on Ethical Conduct for Research Involving Humans. Collected biopsies were stored in cold F-10 medium before delivery to the laboratory. The muscle tissue was washed with cold phosphate buffered saline (PBS) 3–4 times to remove blood, carefully dissected in F-10 medium on ice to remove as much connective tissue as possible, and then minced into tiny pieces. The tissue was then dissociated in TBA (0.11% trypsin, 0.11 mg/ml collage-nase type II, 1% BSA) for 30 min.at 37°C with mixing at intervals of 5 min. A total of 5ml of growth medium was added to triturate with sterilized pipette to break up the clumps. The dissociated cells were filtered through a cell strainer (100 μm, Falcon, Becton Dickinson) to remove the undigested clumps and then centrifuged at 1000 rpm × 10 min. (Allegra 6R Centrifuge, Beckman Coulter) at 4°C. The pellet was suspended in 5 ml complete medium (F-10 + 20% FBS + 1% S/P + 1 ng/ml bFGF) and plated onto a non-collagen-coated plate for incubation overnight at 37°C. The primary fibroblasts were obtained by harvesting the attached cells on these non-coated plates. The medium with unattached cells was transferred into a collagen-coated plate. In the first 2–3 passages, the cells were plated on a non-coated plate for half hour after digestion with 0.25% trypsin before transfer of the unattached cells to a collagen-coated plate for further myoblast isolation. The number of population doublings (PD) at every passage was calculated according to the formula logN/log2, where N is the number of cells at the time of passage divided by the number of cells initially attached after seeding. The cultured cells were considered to have entered the senescent state when their PD time exceeded 24 hrs.
Immunohistochemistry (IHC) staining of desmin
Cells were fixed in 95% methanol at –20°C for 20 min., rinsed in PBS and incubated in blocking solution (1% BSA + 0.5% Triton-100 in PBS) for 30 min. A monoclonal antibody of mouse anti-human desmin (PharMingen, Becton Dickinson Inc., San Jose, CA, USA) was applied at a dilution of 1:1000 in blocking solution for 1 hr at room temperature. The cells were then washed with PBST (PBS with 0.5% Triton-100), and alkaline phosphatase-conjugated goat anti-mouse secondary antibody (Promega, Madison, WI, USA) was applied at 1:1000 in PBST for 1 hr at room temperature. The stain was developed with 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitro blue tetrazoli-um (NBT) (Promega, Madison, WI, USA) according to manufacturer's instructions.
Microencapsulation of cells
Cells were encapsulated as described previously [28]. Briefly, the cell concentration was 2.0 × 106/ml 1.5% Keltone potassium alginate (Kelco, Chicago, IL, USA). The alginate/cell suspension was then extruded through a 27-gauge blunt-end hypodermic needle (Vita Needle, MA, USA) into a beaker containing cold 1.1% calcium chloride solution to form gelled microcapsules of approximately 500 μm in diameter, and then the microcapsules were subjected to a series of washes to form microcapsules laminated with poly-L-lysine and alginate.
Release of encapsulated cells
Microcapsules were transferred into 50-ml conical tubes and washed four times with phosphate-buffered saline (PBS). Sodium citrate (30 ml, 0.055 M) was added and the tube was placed on ice on a rocking table for 10 min. The microcapsules were drawn into a syringe and forced through a 27-gauge needle to release the encapsulated cells. The cells were then washed with PBS and spun at 1500 rpm at 4°C for 10 min. The cell pellet was washed and centrifuged as before. This preparation was used for CPK activity assay and for cytocentrifugation.
Determination of viable cell number per capsule
The number of viable cells from a known number of micro-capsules was determined with AlamarBlue reagent (AccuMed In, Westlake, OH). Five hundred microlitres of growth medium were added into each well in a 24-well plate, 100 μ L suspended capsules or a serial dilution of a known number of C2C12 or primary myoblast cells (as determined by a Coulter Z1 particle counter, Coulter Corporation, Mississauga, Ontario) were loaded into corresponding wells, and then 50 μL AlamarBlue reagent was applied to each well. After 4-hrs incubation at 37°C and 5% CO2, 100 μL medium from each well was transferred to a clean 96-well (Nunc) plate and read with fluorescent reader (GENios, TECAN Boston, MA) at an excitation wavelength of 535 nm and an emission wavelength of 595 nm. Cell number per well is determined with reference to the corresponding standards. Capsules were then counted in each well to estimate the number of viable cells per capsule.
Determination of myoblasts differentiation
Creatine phosphokinase activity (CPK).
The cell pellet or the cells released from the microcapsules were suspended in 0.5 ml PBS for sonication to release intracellular proteins. CPK activity was analysed using the CK-MPR1 kit (Roche Diagnostics Inc., Indianapolis, IN). The change in absorbance (at 340 nm) at 1 min. interval was used to calculate the CPK activity (U/(g protein) according to the manufacturer's instruction. Protein was determined with the Bradford assay according to manufacturer's instruction (BioRad, Mississauga, ON).
Myosin heavy chain immuno-stain.
Unencapsulated cells from monolayer or cells released from microcapsules were spun onto microscope slides by cytocentrifugation. The slides were then incubated at 37° C in PBST (PBS with 0.05% Tween-20) with 5% non-fat dry milk (NFDM) for 30 min., followed by a 60 min. incubation at RT in a 1:10 dilution of MF20 monoclonal antibody against myosin heavy chain (ATTC CRL-2039, Rockville, MD) in blocking buffer, and then a final 60 min. RT incubation with a 1:1000 dilution of alkaline phosphatase-conjugated goat anti-mouse IgG antiserum (Promega, Madison, WI) in blocking buffer. The slides were then treated with substrate NBT/BCIP (GibcoBRL, Galthersburg, MD) to develop the stain for alkaline phosphatase activity according to manufacturer's instruction.
Myogenic index.
Cells deposited on microscope slides were stained with Giemsa and examined under the light microscope. Cells with more than three nuclei were scored as multi-nucleated myotubes. Myogenic index = number of multi-nucleate cells/total number of cells in a visual field examined under a 10× objective lense. At least 300 cells were counted for each sample.
Results
Enrichment of myoblasts from primary cultures
Primary cultures of human foetal skeletal muscle consist of a mixture of myoblasts and fibroblasts. To enrich for myoblasts, we exploited their differences in propensity to attach to non-collagen coated plates [27]. Primary fibroblasts attached to the plates sturdily within 30 min., while myoblasts remained unattached even after overnight incubation. It is important also to separate these two populations at the first few passages of the primary cultures. Hence, after incubating the initial cell pellet isolated from the tissues on un-coated plates at the first passage, we transferred only the floating cells into collagen-coated plates the next day. In the following 2–4 passages, only the floating cells were transferred into coated plates after trypsinization and 30 min.incubation.
To assess the success of separating the myoblasts from the fibroblasts primary cultures, we stained for desmin which is an intermediate filament protein and a biomarker of myogenic cells [29]. Figure 1B shows that the primary fibroblasts had a low level (approximate 5%) of desmin positive cells, indicating that the fibroblasts were likely contaminated with some myoblasts during the separating process. However, the primary myoblasts cultured from the same initial tissue sample demonstrated very clear and positive desmin staining throughout (Fig. 1C). In fact, when compared to the mouse C2C12 myoblasts as the positive control, the levels of desmin positive cells were similar (Fig. 1D). Hence, the protocol to isolate primary human foetal myoblasts from muscle biopsies was successful.
1.

Myogenic enrichment examined by desmin staining. (A) Fibroblasts at passage 2 under phase contrast microscopy; (B) The same fibroblasts at passage 2 under light transmission microscopy; (C) C2C12 cell line; (D) Human primary myoblasts at passage 5. All the cells were grown for 24 hrs, fixed with 95% methanol and stained with anti-desmin antibody. (Magnification: × 160)
Differentiation characteristics of human foetal primary myoblasts
The differentiation potential of the primary myoblasts was assessed by CPK activity. Increased activity is characteristic of the onset of differentiation after changing from growth medium to a differentiation-inducing medium. As shown in Figure 2, over a period of 9 days in differentiation medium, the primary fibroblasts maintained only a baseline level of CPK activity. However, both primary myoblasts and C2C12 cells showed initially escalating levels that eventually decreased back to the baseline. While both cultures showed elevated CPK levels compared to the fibrob-lasts, the human myoblasts activity peaked early at day 3 whereas the C2C12 myoblasts showed the highest plateau from days 4–6. Furthermore, the specific CPK activity in the human myoblasts was greater than twofold higher than that achieved by the C2C12 myoblasts. The difference in the pattern of CPK activity indicated that the primary myoblasts differentiated earlier and more acutely than the C2C12.
2.

Creatine phosphokinase (CPK) activity in differentiation medium. The cells were cultured in growth medium for 24 hrs and switched into differentiation medium. The cells were trypsinized and pelleted by centrifugation at different time points. The pellets were suspended in phosphate buffered saline (PBS) and sonicated to release the cytoplasm. The CPK was assayed and standardized by protein concentration.
Since MHC is a marker for the well-differentiated state of myotubes, this was used to assess the differentiation potential of the primary cell isolates.Figure 3A confirmed the non-myogenic nature of the primary fibroblasts, showing no MHC expression even after 9 days in culture with the differentiation medium. For the positive control, the mouse myoblast C2C12 cells, there was clear though sparse MHC expression (Fig. 3B) after 9 days in the differentiation medium. But during the process of differentiation, many cells died and detached, demonstrating poor survival. However, the primary human myoblasts showed a much more robust state of differentiation (Figs 3C–D). They started to differentiate as early as 2 days after culturing in the differentiation medium, with high expression of MHC and multi-nucleated myotubes clearly evident by that time (Fig. 3C). This well-differentiated state in a densely populated culture of multi-nucleated cells was maintained even at day 12 after differentiation, as shown in Figure 3D. If the differentiation medium was changed regularly, this state could be maintained up to 30 days (data not shown).
3.

Expression of major histocom-patibility complex (MHC). The cells were cultured in growth medium for 24 hrs and switched into differentiation medium. At different time points, the cells were fixed with 95% methanol and stained with anti-MHC antibody. (A) Human foetal primary fibroblasts with no positive stain; (B) C2C12 cells were differentiated into myotubes, but with lots of dead cells; (C) Human foetal primary myoblasts demonstrated high expression of MHC and multi-nucleated myotubes at day 2 with few dead cells; (D) human foetal primary myoblasts remained in well-differentiation state at day 12 with less dead cells. (Magnification: × 160)
The extent of differentiation into multi-nucleated myotubes could be visualized after Giemsa staining as shown in Figure 4. After 9 days in differentiation medium, the multi-nucleated cells were readily observed in the primary human myoblasts (Fig. 4C), whereas none could be observed among the primary fibroblasts (Fig. 4A) and few even in the C2C12 control (Fig. 4B).
4.
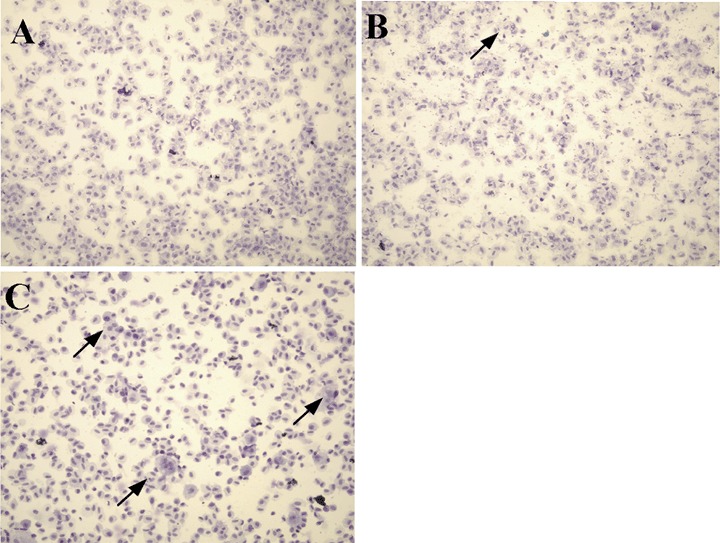
Multi-nucleated cells stained by Giemsa.(A) Human foetal primary fibroblasts; (B) C2C12 cells; (C) Human foetal primary myoblasts. The cells were cultured in growth medium for 24 hrs and transferred into differentiation medium. At day 9, the cells were trypsinized and centrifuged onto slides. The cells on the slides were fixed in 95% methanol and stained in Giemsa solution.(Magnification: × 160)
Differentiation properties of encapsulated human primary foetal myoblasts
In order to apply human primary foetal myoblasts to microencapsulation, we compared cell proliferation and differentiation of the human myoblasts and C2C12 cells after microencapsulation. The encapsulated human myoblasts and C2C12 cells were cultured in growth media for 2 weeks and transferred into differentiation media. In growth media, C2C12 cells formed small aggregates, while human primary cells did not. The cell number per capsule increased from 172 to 351 in 2 weeks in encapsulated C2C12 cells; 168–263 in encapsulated primary myoblasts. During the proliferation, we observed a few C2C12 cells started to escape from the microcapsules and adhere to the culture plate after 2 weeks, while the plate for primary myoblasts was still clear after 6 weeks. In addition, we did not see any bursting of capsules during our experiments, even at high levels of proliferation. After transfer to differentiation media, the cells were sampled for proliferation on days 0, 1, 5, 8, 11, 14 after the media change. Both types of cells demonstrated a similar pattern of viability, and the cell number per capsule decreased over the following 2 weeks (Fig. 5A).
5.

Comparison of growth and differentiation of encapsulated foetal human myoblasts and C2C12 cells. Encapsulated cells were grown in growth media for 2 weeks and transferred to differentiation media and sampled at different time points for growth (A), CPK activity (B), MHC expression (C) and myogenic index (D).
The differentiation status in differentiation media was analysed by measuring the activity of CPK, MHC expression and myogenic index. Human primary myoblasts showed higher CPK activity compared to C2C12 cells from day 0. CPK activity in primary myoblasts increased and reached the highest at day 5 and then decreased after that. However, C2C12 cells had increasing CPK for the duration of the experiment, indicating a slower differentiation than primary myoblasts (Fig. 5B).
A similar trend was seen with another differentiation marker, MHC. MHC expression was remarkably higher in primary myoblasts than C2C12 cells and increased over the experiment (Fig. 5C). In addition to MHC, the percentage of multi-nucleate cells (myo-genic index) was determined. Compared to C2C12 cells, which demonstrated a slight increase of cell fusion over 2 weeks, primary myoblasts showed dramatically superior differentiation (Fig. 5D). Even at day 22, MHC and myogenic index continued to increase in primary myoblasts.
Lifespan of primary human foetal myoblasts
The lifespan of the human foetal myoblasts was ascertained under a clearly defined experimental protocol. The cells were sub-cultured at a dilution ratio of 1:4 every 2 days to maintain their myoblastic status. This limited the confluency to less than 80–90% to avoid initiation of differentiation and withdrawal from the cell cycle. We observed that the population doubling time was 18–19 hrs before passage 20, corresponding to 45 PDs. During the following 20–32 passages (70 PDs), the PD time increased to about 24 hrs. After that point, the cells divided much more slowly, with a PD time of 30 hrs at passage 35 and 48 hrs at passage 38. Using our definition of senescence as a PD time of 24 hrs, the maximal PDs of human foetal primary myoblasts was 70.
Discussion
The cells selected for microencapsulation must be able to survive with good viability in the limited space of the capsules, yet have good growth characteristics for in vitro manipulation. Myoblasts are capable of robust proliferation and terminal differentiation into multi-nucleated myotubes, which makes them ideally suited to microencapsulation. Murine myoblast C2C12 cells have played a critical role in microencapsulation experiments in mice. However, because C2C12 cells are a transformed cell line, there was concern about their tumourigenicity, and it was reported that they could indeed generate tumours in nude mice [26]. They are also immunogenic: encapsulated human Factor IX (hFIX)-secreting C2C12 cells were implanted into mice and hFIX could be detected in the mouse plasma up to 14 days until the appearance of anti-hFIX antibodies [15]. When the C2C12 cells were replaced by mouse foetal myoblast G8 cells which also were engineered to secret hFIX in microcapsules, there was no detectable anti-hFIX antibody until day 60 (the end of the experiment) while maintaining stable hFIX biological activity. These results indicated that human foetal myoblasts could be of great importance in extending the technology of microencapsulation into clinical trials.
The escape of implanted cells from the microcapsules is always a concern. This phenomenon is highly dependent on the initial cell density in the capsules, the cell type and their proliferation features. We initiated the cell density at 2×106 cell/ml alginate which is a relatively low concentration. With this density, there is a low chance of cells protruding from the initial alginate bead and remaining exposed after poly-L-lysine and then alginate coating of the capsules. This should reduce the possibility of cells migrating from or being released by the capsules. Cell type and proliferation status are other important factors. When we encapsulated C2C12 cells at the same density, we noticed that the cells started to ‘leak’from the capsules at 2–4 weeks and appear on the bottom of the tissue culture plates, while canine epithelial Madin–Derby Canine Kidney (MDCK) cells showed the same tendency at 4–6 weeks but at a much reduced level. The encapsulated primary myoblasts, however, had no detectable cell leakage. No ruptured capsules were seen for all of the above cell types even after 2 months, so this ‘leaking’ may represent the relative propensity of a cell type to migrate out of the capsule during proliferation.
A major concern in using primary cells is their limited lifespan and hence their restricted availability, whether they are used for experimental or clinical uses. So far, there are no human primary cells with a long lifespan available even from commercial sources. Because of the superior proliferative ability of foetal cells, we expected primary human foetal cells would have a more extended lifespan and robust growth. We compared the characteristics of human foetal myoblasts with C2C12 cells to determine their suitability for future therapeutic applications. We found that the PD time is 18–24 hrs within 70 PDs, thus demonstrating their excellent proliferation potential.Figures 3C–D demonstrated the foetal myoblasts began to differentiate at day 2, and, in contrast to the C2C12 cells, there was very little cell death, even at day 12 of differentiation. Combined with other differentiation parameters such as CPK (Fig. 2), these data show that the primary human foetal myoblasts differentiated better than C2C12 cells, and have more robust growth properties after differentiation. This makes them good candidates for microencapsulation.
After encapsulation, the overall viability of both C2C12 and human foetal myoblasts in differentiation media was decreased when compared to the growth media. The viability decrease in differentiation media for encapsulated cells probably represents restriction of growth factors and nutrients in this media so that, when the additional reduction of these media constituents occurs because of the diffusion gradient into the microcapsule, the viability of the cells is affected. The viability was decreased to 50% for both cell types, but human foetal myoblasts still showed the superior growth and differentiation features compared to C2C12 cells as was seen in monolayer culture. CPK is an enzyme that is up-regulated at the early stage of myoblast differentiation. CPK activity in encapsulated primary myoblasts reached the highest at day 5 while encapsulated C2C12 cell CPK activity increased gradually until day 14 which indicated a delayed process of differentiation. Further evidence of superior differentiation was seen with MHC and myotube assessment. The levels in primary myoblasts were significantly higher on all days. We also observed that the encapsulated primary myoblasts were still dispersed inside the microcapsule compared to the clumping seen in C2C12 cells after encapsulation. In our previous study, we reported there were small clumps formed after encapsulation which probably promoted the differentiation of encapsulated C2C12 [30]. However, the mechanism why the scattered primary myoblasts presented superior differentiation characteristics is not clear.
An important issue in isolating primary cell lines is the purity of the cell population. When used for cell-based therapy, such as transplantation or immuno-isolation with microencapsulation, the immune responses from the recipients are a crucial consideration. Different cells possess different capacity to present antigen to T cells of the recipients. Fibroblasts are very effective in antigen-presentation, and hence undesirable, but they are the major contamination of myoblasts during the separation process. However, by using the simple but efficient multi-round selections with collagen-coated plates, it was demonstrated that a highly purified population of myoblasts could be obtained.
In conclusion, primary human foetal myoblasts have better proliferation and differentiation when compared to C2C12 cells with or without encapsulation, thus providing a useful tool for gene therapy with microencapsulation.
Acknowledgments
We would like to thank Dr. L. Yoshida for her help in collecting biopsies.
References
- 1.Emmrich F, Keitel R, Sorger K, Otto U, Staffa G, Klotzer B. Quantitative alterations of immune serum globulin concentrations in pigs transplanted with a renal allograft. Exp Pathol. 1977;14:334–9. doi: 10.1016/s0014-4908(77)80053-7. [DOI] [PubMed] [Google Scholar]
- 2.Husberg BS. The influence of cyclophosphamide on the rejection of allogenic rat kidney transplants. An in vitro study of cell-bound and antibody-mediated immunity. Clin Exp Immunol. 1972;10:697–711. [PMC free article] [PubMed] [Google Scholar]
- 3.Foglia RP, DiPreta J, Statter MB, Donahoe PK. Fetal allograft survival in immunocompetent recipients is age dependent and organ specific. Ann Surg. 1986;204:402–10. doi: 10.1097/00000658-198610000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Statter MB, Fahrner KJ, Barksdale EM, Jr, Parks DE, Flavell RA, Donahoe PK. Correlation of fetal kidney and testis congenic graft survival with reduced major histocompatibility complex burden. Transplantation. 1989;47:651–60. doi: 10.1097/00007890-198904000-00017. [DOI] [PubMed] [Google Scholar]
- 5.Nouwen EJ, Dauwe S, Van Der Biest I, De Broe ME. Stage- and segment-specific expression of cell-adhesion molecules N-CAM, A-CAM, and L-CAM in the kidney. Kidney Int. 1993;44:147–58. doi: 10.1038/ki.1993.225. [DOI] [PubMed] [Google Scholar]
- 6.Guymer RH, Mandel TE. A comparison of corneal, pancreas, and skin grafts in mice. A study of the determinants of tissue immunogenicity. Transplantation. 1994;57:1251–62. doi: 10.1097/00007890-199404270-00019. [DOI] [PubMed] [Google Scholar]
- 7.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 8.Deutsch AA, Arensman R, Levey R, Folkman J. The effect of antilymphocyte serum on fetal rat intestine transplanted as free subcutaneous homografts. J Pediatr Surg. 1974;9:29–34. doi: 10.1016/0022-3468(74)90006-2. [DOI] [PubMed] [Google Scholar]
- 9.Deutsch AA, Reiss R. Aortogastric fistula: an unusual complication of the thoracic portion of the stomach. Arch Surg. 1978;113:537. doi: 10.1001/archsurg.1978.01370160195036. [DOI] [PubMed] [Google Scholar]
- 10.Hebda PA, Dohar JE. Transplanted fetal fibroblasts: survival and distribution over time in normal adult dermis compared with autogenic, allogenic, and xenogenic adult fibroblasts. Otolaryngol Head Neck Surg. 1999;121:245–51. doi: 10.1016/S0194-5998(99)70179-8. [DOI] [PubMed] [Google Scholar]
- 11.Sladek JR, Jr, Gash DM. Nerve-cell grafting in Parkinson's disease. J Neurosurg. 1988;68:337–51. doi: 10.3171/jns.1988.68.3.0337. [DOI] [PubMed] [Google Scholar]
- 12.Ourednik J, Ourednik V, Lynch WP, Schachner M, Snyder EY. Neural stem cells display an inherent mechanism for rescuing dysfunctional neurons. Nat Biotechnol. 2002;20:1103–10. doi: 10.1038/nbt750. [DOI] [PubMed] [Google Scholar]
- 13.Svendsen CN, Langston JW. Stem cells for Parkinson disease and ALS: replacement or protection? Nat Med. 2004;10:224–5. doi: 10.1038/nm0304-224. [DOI] [PubMed] [Google Scholar]
- 14.Watorek E, Klinger M. Stem cells in nephrology: present status and future. Arch Immunol Ther Exp. 2006;54:45–50. doi: 10.1007/s00005-006-0004-4. [DOI] [PubMed] [Google Scholar]
- 15.Hortelano G, Al-Hendy A, Ofosu FA, Chang PL. Delivery of human factor IX in mice by encapsulated recombinant myoblasts: a novel approach towards allogeneic gene therapy of hemophilia B. Blood. 1996;87:5095–103. [PubMed] [Google Scholar]
- 16.Chang PL, Shen N, Westcott AJ. Delivery of recombinant gene products with microencapsulated cells in vivo. Hum Gene Ther. 1993;4:433–40. doi: 10.1089/hum.1993.4.4-433. [DOI] [PubMed] [Google Scholar]
- 17.Al-Hendy A, Hortelano G, Tannenbaum GS, Chang PL. Correction of the growth defect in dwarf mice with nonautologous microencapsulated myoblasts–an alternate approach to somatic gene therapy. Hum Gene Ther. 1995;6:165–75. doi: 10.1089/hum.1995.6.2-165. [DOI] [PubMed] [Google Scholar]
- 18.Orive G, Hernandez RM, Gascon AR, Igartua M, Rojas A, Pedraz JL. Microencapsulation of an anti-VE-cadherin antibody secreting 1B5 hybridoma cells. Biotechnol Bioeng. 2001;76:285–94. doi: 10.1002/bit.10050. [DOI] [PubMed] [Google Scholar]
- 19.Aebischer P, Goddard M, Signore AP, Timpson RL. Functional recovery in hemiparkinsonian primates transplanted with polymer-encapsulated PC12 cells. Exp Neurol. 1994;126:151–8. doi: 10.1006/exnr.1994.1053. [DOI] [PubMed] [Google Scholar]
- 20.Joki T, Machluf M, Atala A, Zhu J, Seyfried NT, Dunn IF, Abe T, Carroll RS, Black PM. Continuous release of endostatin from microencapsulated engineered cells for tumor therapy. Nat Biotechnol. 2001;19:35–9. doi: 10.1038/83481. [DOI] [PubMed] [Google Scholar]
- 21.Lohr M, Bago ZT, Bergmeister H, Ceijna M, Freund M, Gelbmann W, Gunzburg WH, Jesnowski R, Hain J, Hauenstein K, Henninger W, Hoffmeyer A, Karle P, Kroger JC, Kundt G, Liebe S, Losert U, Muller P, Probst A, Puschel K, Renner M, Renz R, Saller R, Salmons B, Schuh M, Schwendenwein I, Rombs K, Wagner T, Walter I. Cell therapy using microencapsulated 293 cells transfected with a gene construct expressing CYP2B1, an ifosfamide converting enzyme, instilled intra-arterially in patients with advanced-stage pancreatic carcinoma: a phase I/II study. J Mol Med. 1999;77:393–8. doi: 10.1007/s001090050366. [DOI] [PubMed] [Google Scholar]
- 22.Lohr M, Hoffmeyer A, Kroger J, Freund M, Hain J, Holle A, Karle P, Knofel WT, Liebe S, Muller P, Nizze H, Renner M, Saller RM, Wagner T, Hauenstein K, Gunzburg WH, Salmons B. Microencapsulated cell-mediated treatment of inoperable pancreatic carcinoma. Lancet. 2001;357:1591–2. doi: 10.1016/s0140-6736(00)04749-8. [DOI] [PubMed] [Google Scholar]
- 23.Lohr M, Hummel F, Faulmann G, Ringel J, Saller R, Hain J, Gunzburg WH, Salmons B. Microencapsulated, CYP2B1-transfected cells activating ifosfamide at the site of the tumor: the magic bullets of the 21st century. Cancer Chemother Pharmacol. 2002;49:S21–4. doi: 10.1007/s00280-002-0448-0. [DOI] [PubMed] [Google Scholar]
- 24.Dawson RM, Broughton RL, Stevenson WT, Sefton MV. Microencapsulation of CHO cells in a hydroxyethyl methacrylate-methyl methacrylate copolymer. Biomaterials. 1987;8:360–6. doi: 10.1016/0142-9612(87)90006-8. [DOI] [PubMed] [Google Scholar]
- 25.Ross CJ, Bastedo L, Maier SA, Sands MS, Chang PL. Treatment of a lysosomal storage disease, mucopolysaccharidosis VII, with microencapsulated recombinant cells. Hum Gene Ther. 2000;11:2117–27. doi: 10.1089/104303400750001426. [DOI] [PubMed] [Google Scholar]
- 26.Hortelano G, Wang L, Xu N, Ofosu FA. Sustained and therapeutic delivery of factor IX in nude haemophilia B mice by encapsulated C2C12 myoblasts: concurrent tumourigenesis. Haemophilia. 2001;7:207–14. doi: 10.1046/j.1365-2516.2001.00492.x. [DOI] [PubMed] [Google Scholar]
- 27.Blau HM, Webster C. Isolation and characterization of human muscle cells. Proc Natl Acad Sci USA. 1981;78:5623–7. doi: 10.1073/pnas.78.9.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li AA, MacDonald NC, Chang PL. Effect of growth factors and extracellular matrix materials on the proliferation and differentiation of microencapsulated myoblasts. J Biomater Sci Polym Ed. 2003;14:533–49. doi: 10.1163/15685620360674236. [DOI] [PubMed] [Google Scholar]
- 29.Kaufman SJ, Foster RF. Replicating myoblasts express a muscle-specific phenotype. Proc Natl Acad Sci USA. 1988;85:9606–10. doi: 10.1073/pnas.85.24.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li AA, Shen F, Zhang T, Cirone P, Potter M, Chang PL. Enhancement of myoblast microencapsulation for gene therapy. J Biomed Mater Res B Appl Biomater. 2006;77:296–306. doi: 10.1002/jbm.b.30342. [DOI] [PubMed] [Google Scholar]