

Abstract
The cardiac ankyrin repeat kinase (CARK) gene, also named TNNI3K for its interaction with cardiac troponin I, is both a unique expression and heart-enriched gene. To understand the mechanisms of CARK gene expression and regulation, we first cloned the full-length mRNA sequence and mapped the transcription start site of the mouse CARK gene and characterized its promoter regions. Two transcriptional isoforms of the CARK gene were identified in mouse heart tissue. Truncation analysis of the CARK promoter identified a minimal 151 bp region that has strong basal transcription activity. Mutational analysis revealed five conserved cis-acting elements in this 151-bp long minimal promoter. Mutational and loss-of-functional analysis and co-transfection studies indicated that MEF2 binding region is the most critical cis-acting element in the CARK promoter, and CARK transcription level can be down-regulated by MEF2C antisense. Binding to the MEF2 sites by Mef2c protein was confirmed by electrophoretic mobility shift assay and competition and supershift electrophoretic mobility shift assays.
Keywords: TNNI3K, CARK, promoter, MEF2
Introduction
The heart is one of the earliest organs that form and function during embryogenesis. Its importance for life is highlighted by the scores of lethal phenotypes in mice carrying null mutations in genes critical for cardiac development. Many of these genes encode transcription factors that help specify cardiomyocyte identity or enhance cardiomyocyte survival during development of the heart [1]. Chief among these cardiac-restricted transcription factors are members of several gene families including homeobox, GATA, MADS (MCM1, agamous, deficiens, serum response factor [srf]), bHLH (basic helix-loop-helix) genes [2–5]. These and other families of transcription factors coordinate programs of cardiac-restricted transcripts that, in turn, encode proteins necessary to carry out the unique functions of cardiac muscle.
A number of studies have shown that the temporal and spatial expression of the cardiac-restricted genes is dependent on single or combinatorial heart-specific transcription factors. In mammals as well as fruit flies, the MADS-box factor myocyte enhancer factor-2 (Mef2), in conjunction with other transcription factors, directly activates the expression of myocardial genes, such as atrial natriuretic peptide (ANP) and alpha-myosin heavy chain [2, 6, 7]. Similarly, srf, a related MADS-box factor, associates with an array of transcription factors including Nkx2-5, Gata4 and myocardin to control the expression of muscle structural genes whose products are incorporated into the contractile apparatus, such as alpha-actin, cardiac-myosin and cardiac-troponins [8, 9]. Moreover, Gata4 and Mef2c each has been confirmed as an activator on the promoter of alphaT-catenin gene mainly restricted to cardiomyocytes [10]. To date, only a handful of such genes regulated by cardiac transcription factors have been identified [11], the mechanisms controlling the expression of the cardiac specific genes are not yet fully understood and the relative contributions of these factors to cardiac events also remain to be established. Investigation of these transcription factors that regulate cardiac-restricted genes is helpful to understand the mechanisms of heart development and the maintenance of cardiac functions.
The cardiac ankyrin repeat kinase (CARK) gene, also named TNNI3K for its interaction with cardiac troponin I, is both a unique expression and heart-enriched gene recently identified in the human heart [12]. In human and rat, CARK is transcribed as a single transcript composed of 25 exons and encodes 835 amino acids. Previous studies indicate that CARK has a high-level expression in foetal and adult heart tissue but not detected in other tissues and belongs to the MAPKKKs superfamily and may play important roles in the cardiac system [12].
In this study, we first cloned the full-length mRNA sequence and characterized the basal promoter region of mouse CARK gene, identified five conserved cis-acting elements in its basal promoter region and verified that Mef2c plays a critical role in regulating basal CARK transcription activity.
Materials and methods
Bioinformatics analysis
Mouse-spliced ESTs homologous to human CARK gene were identified in UCSC Genome Database (http://genome.ucsc.edu) [13]. The CARK promoter sequences were compared and aligned between mouse and other mammals using ClustalW program (http://www.ebi.ac.uk/clustalw/). To characterize the potential sequence-conserved transcription factor binding sites (TFBSs) across these species, we constructed a database ‘CardioSignal’ available at the website http://www.car-diosignal.org, which contained information concerning cardiac specific TFBSs and promoters [14]. Those data were derived from the Eukaryotic Promoter Database (EPD) and the Transcription Factor Database (TRANSFAC).
RT-PCR
Total RNAs were isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) from mouse (C57/6J, Balb/C and KM strains) heart, liver, spleen, lung, brain, kidney, skeletal muscle and testis, and treated with DNase I (Ambion, Austin, TX, USA) according to the manufacturer's instructions. First-strand cDNA was synthesized from 100 ng of the DNase I-treated RNA using oligo(dT)12-18 as a primer and SuperScript II RT-PCR kit (Invitrogen). RT-PCR was performed with gene-specific primers (forward primer NMF: 5′-ACACATCTTTGGCTCTGACGAA; reverse primer NMR: 5′-TCCCGCTTGCTGAACATC) that covered 261 bp length in the mouse CARK coding region using synthesized cDNAs as templates. The PCR cycles were 94°C for 5 min.and 30 cycles at 94°C for 30 sec., 60°C for 30 sec. and 72°C for 30 sec.with a final elongation step of 72°C for 5 min. PCR products were analyzed by 2% agarose gel electrophoresis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control.
Characterization of the CARK transcription start site
5′ RACE analysis was performed to map the transcription start site of the mouse CARK gene using a GeneRacer kit (Invitrogen) according to the manufacturer's instructions. In brief, the total RNA preparation from adult mouse heart tissue was treated with calf intestinal phosphatase to dephosphorylate non-mRNAs or truncated mRNAs, and then treated with tobacco acid pyrophosphatase to remove the 5′ cap structure from the full-length mRNAs. The GeneRacer RNA Oligo was ligated to the 5′ ends of the decapped mRNAs. Subsequently, the cDNAs were synthesized with random primers and SuperScript II reverse transcriptase. Two antisense oligonucleotide primers corresponding to the coding region of the CARK cDNA were synthesized and used for the PCR. The primary PCR was conducted using the synthesized cDNAs as templates, the antisense gene-specific primer NMR (5′-TCCCGCTTGCT-GAACATC) and the GeneRacer 5′-Primer supplied with the kit under the following conditions: 94°C for 5 min. and 30 cycles at 94°C for 30 sec., 60°C for 30 sec. and 72°C for 45 sec. with a final elongation step of 72°C for 5 min. The secondary PCR was conducted using the primary PCR products as templates, the antisense gene-specific primer GSP2 (5′-CGCTTGCTGAACATCTGCTCCGCTA) and the GeneRacer 5′-Nested Primer supplied with the kit under the following conditions: 94°C for 5 min. and 30 cycles at 94°C for 30 sec., 65°C for 30 sec. and 72°C for 45 sec.with a final elongation step of 72°C for 5 min. PCR products were analyzed by 2% agarose gel electrophoresis. The secondary PCR products were purified using QIAquick gel extraction kit (QIAGEN, Hilden, Germany) and cloned into TOPO TA Cloning vector (Invitrogen) and sequenced.
Construction of luciferase reporter plasmids
Five CARK promoter truncation constructs shown in Figures 3A and 1C were constructed by cloning various segment lengths of the CARK promoter into pGL3 basic vector with a luciferase reporting gene (Promega, Madison, WI, USA), and their names are based on the 5′ most oligonucleotide. Briefly, a 1440 bp DNA fragment containing of the CARK promoter was amplified using F1 primer (5′-GCATGGTACCAGTTTCCTGGGCTGGTGAAT) and R1 primer (5′-CGATAGATCTGTCTGCTTTCCTCCTGTGCT) with restriction enzyme digestion sites KpnI and BglI I from mouse genome DNA. The PCR products were then excised with KpnI and BglII and cloned into KpnI and BglII sites of pGL3 basic vector to generate pF1–1361-luc vector. Further 5′ deletions were carried out by PCR with F2 primer (5′-GCATGGTACCTCAGGATCAACTTTGGGCTC) and R1 primer, or F3 primer (5′-GCATGGTACCTGCCAAA-GATGCCAAACACT) and R1 primer, or F4 primer (5′-CGATGGTACCATGCTTGCTGTTTACCTTTC) and R1 primer or F5 primer (5′-GCATGGTACCAACATATT-TTCAACTGGTCT) and R1 primer. pGL3 basic vector containing no promoter element was used as negative control and pGL3 promoter vector fused with SV40 promoter element was used as positive control. A 2.7-kb long promoter region of ANP gene, a gift from Dr. Issei Komuro [15], was also subcloned into pGL3 basic vector (named as pANP-luc) as positive control.
3.
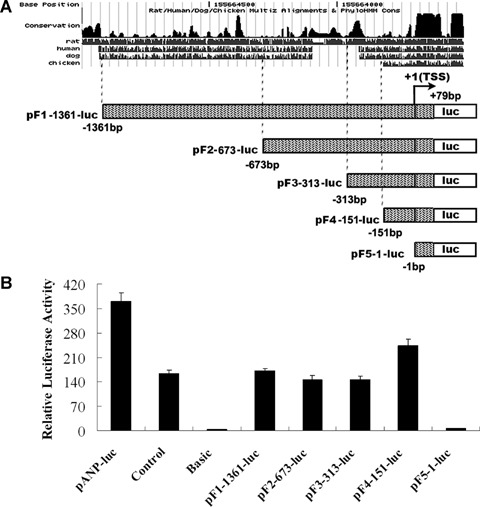
Truncation analysis of promoter regions in the 5′ flanking region of mouse CARK gene. (A) A series of constructs containing various lengths of the 5′ region of the CARK promoter regions based on the conservation between mouse and other mammals using ClustalW program were fused to the luciferase reporter. (B) These constructs were introduced into primary rat cardiomyocytes, HEK293S cells, Hela cells, rat aorta smooth muscle cells and rat cardiac fibroblasts for transient expression assays. After 48 hrs, cells were lysed and luciferase activities were assayed. Firefly luciferase activities expressed had been normalized on the basis of Renilla luciferase activity encoded by the co-transfected control plasmid, pRL-CMV. Data shown are the means ± S. D., n= 3. The results showed that the luciferase activity was detectable only in rat cardiomyocytes. pGL3 basic vector containing no promoter element was used as negative control and pGL3 promoter vector fused with SV40 promoter element was used as positive control. A 2.7-kb long promoter region of ANP gene was sub-cloned into pGL3 basic vector (named as pANP-luc) as positive control.
1.

Characterization of mouse CARK gene.(A) Using the nucleotide sequences of human CARK full-length mRNA as a query for BLAT search of the UCSC mouse ESTs database, the largest three spliced mouse ESTs homologous to human CARK gene were identified and underlined. The arrows indicated the direction of ESTs.(B) Graphic representation of mouse CARK gene in genome. The exons were figured with black boxes. Exons 21 and 23 were skipped in the alternatively spliced isoform AY526096. The 5′ and 3′ UTR are showed in white boxes. (C) Nucleotide and deduced amino acid sequences of mouse CARK gene. Skipped nucleotides in the alternatively spliced isoform are shown in grey background. Black and white arrows showed gene-specific primers and their directions.
Site-directed mutagenesis in vitro
Cis-acting element response sites in CARK minimal promoter region were identified by sequence analysis using the CardioSignalScan program (http://www.cardiosignal.org/tools/scan.html) [14]. Putative sequence-conserved TFBSs were Tbx5 (−95/−80), SRE (−86/−73), M-CAT (−47/−33), MEF2 (−22/−12) and GATA4 (−9/+1; Fig. 4A). Site-directed mutagenesis was performed to modify each binding site based on the pF4–151-luc reporter using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions. Wild-type and modified sequences of each binding site in CARK promoter region were indicated in Fig. 4B. Five mutant luciferase reporter constructs (mut-Tbx5-luc, mut-SRE-luc, mut-MCAT-luc, mut-MEF2-luc and mut-GATA4-luc) were verified by DNA sequencing to confirm the integrity and the presence of the desired mutations in the constructs (Fig. 4C).
4.
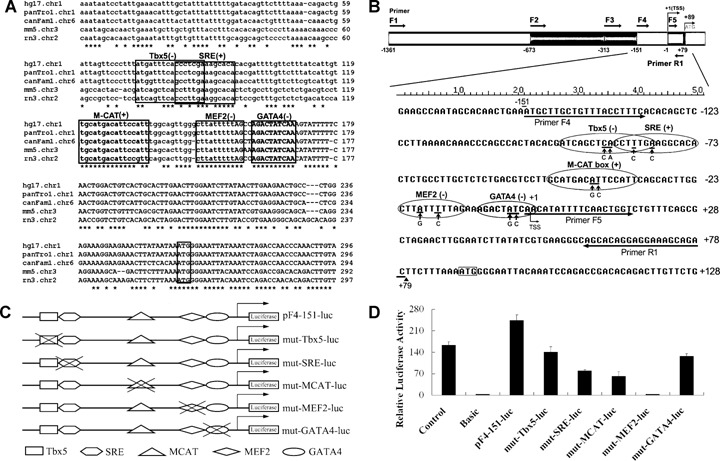
Mutational analysis of putative transcription factor binding sites of mouse CARK promoter.(A) Homology comparison of minimal promoter region of mouse CARK gene with human, chimpanzee, dog and rat revealed the presence of several conserved putative transcription factor binding sites, including Tbx5, SRE, M-CAT box, MEF2 and GATA4.(B) The putative transcription factor binding sites are shown in grey background and the modified nucleotides are underlined, the substitutes are illustrated below.(C) Five site-directed mutants were prepared in the –151/+79 promoter fragment (pF4-151-luc), as described under ‘Materials and methods’.(D) These mutated constructs were introduced into primary rat cardiomyocytes for transient expression assays. Firefly luciferase activities expressed had been normalized on the basis of Renilla luciferase activity. Data shown are the means ± S. D., n= 3.
Cell culture
Rat aorta smooth muscle cells were derived as described before [16]. To obtain rat primary cardiomyocytes and fibroblasts, the rat hearts were dissected from 1–2-day-old Sprague-Dawley neonates under sterile conditions. Atrial and other tissues were removed, the ventricles were minced and the cells were dissociated using eight cycles of 0.08% trypsin (HyClone, Logan, UT, USA) degradation at 37°C with gentle agitation. Trypsin digestion was halted after 8 min. by transferring dissociated cells to Dulbecco's modified Eagle's medium (DMEM, HyClone) containing 10% foetal bovine serum (FBS, HyClone). After filtering (300 μm), the cells were centrifuged for 5 min.at 1000 rpm and then resuspended in DMEM with 10% FBS, 100 μg/ml streptomycin (Sigma, St. Louis, MO, USA) and 100U/ml penicillin (Sigma). These cells were then placed in a large petri dish in a humidified incubator (5% CO2, 95% air at 37°C) for 60 min. to allow early adherence of fibroblasts. The nonadherent cells were then plated on 12-well cell culture plates in DMEM, with 10% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin in a humidified incubator (5% CO2, 95% air at 37°C). HEK293S cells, Hela cells, rat aorta smooth muscle cells and rat cardiac fibroblasts were cultured under the same condition.
Transfection and luciferase assay
Primary rat cardiomyocytes, HEK293S cells, Hela cells, rat aorta smooth muscle cells and rat cardiac fibroblasts were cultured in 12-well plates. These cells were transfected transiently with the CARK promoter vectors by using Lipofectamine 2000 (Invitrogen). For each transfection, 2.5 μl Lipofectamine 2000 and 125 μl Opti-MEM I (Invitrogen) were incubated for 5 min. and then mixed with 2 μg of the luciferase reporter construct, 80 ng of the Renilla luciferase reporter vector (pRL-CMV, Promega) and 125 μl Opti-MEM I. After 20 min., 750 μl of serum-free DMEM was added and poured over the cells. Cells were incubated at 37°C for 6 hrs. The medium was then replaced with 1 ml of DMEM supplemented with 10% FBS, and cells were incubated at 37°C for an additional 42 hrs. The cell lysates were prepared with the Dual-Luciferase reporter assay system (Promega), and both firefly and Renilla luciferase activities were measured in Veritas Microplate Luminometer (Turner, Sunnyvale, CA, USA). The transfection efficiency was normalized according to the Renilla luciferase activity. Each transfection was carried out in triplicate and repeated three times.
Electrophoretic mobility shift assay (EMS assay)
Nuclear extracts were prepared from primary neonatal rat cardiomyocytes with NE-PER nuclear and cytoplasmic extraction reagents (Pierce, Rockford, IL, USA) containing multiple protease inhibitors, such as aprotinin, leupeptin and phenylmethylsulfonyl fluoride, according to the manufacturer's instructions. Synthesized oligonucleotides were 3′-end-labelled with biotin-N4-CTP and terminal deoxynucleotidyl transferase according to the instructions for the Biotin 3′-end DNA labelling kit (Pierce), and then the biotinylated complementary oligonucleotides were annealed to generate the double-stranded oligonucleotides as probes. The sequences of MEF2 oligonucleotides were as follows: wild-type sense oligo, 5′-GCACTTGGCT-TATTTTTAGAAAGACT and wild-type antisense oligo, 5′-AGTCTTTCTAAAAATAAGCCAAGTGC; mutant sense oligo, 5′-GCACTTGGCTTGTTCTTAGAAAGACT and mutant antisense oligo, 5′-AGTCTTTCTAAGAACAAGC-CAAGTGC. EMS assays were performed according to the instructions for the LightShift Chemiluminescent EMS assay kit (Pierce). To detect DNA protein complexes, the membrane was incubated with streptavidin-conjugated horseradish peroxidase (HRP) and then visualized with LightShift luminol/enhancer solution and LightShift stable peroxide solution (Pierce) according to the manufacturer's instructions. For competition experiments, unlabelled wild-type and mutated MEF2 oligonucleotides were added in a 200-fold molar excess prior to the addition of the biotinylated probes. To identify the transcription factor comprising the DNA protein complexes by the supershift assay, the nuclear extracts were incubated in the binding buffer for 60 min. at 4°C with anti-rat Mef2c antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA or anti-rat srf antibody (Santa Cruz) prior to the addition of the biotinylated probes.
MEF2C morpholino antisense oligonucleotide
The morpholino antisense oligonucleotide used was 5′-CTG-AATCTTTTTTCTCCCCATAGTC. The control morpholino oligonucleotidewas 5′-CCTCTTACCTCAGTTACAATTTATA. They were transiently transfected into primary rat cardiomyocytes following the manufacturer's procedures (Gene Tools, LLC, Philomath, OR, USA. Briefly, morpholino DNA duplex and endoporter (every 1 ml medium containing 6 μl endoporter) were mixed and added to the cells in DMEM with 10% FBS, the samples were incubated for 24 hrs at 37°C in a humidified atmosphere of 5% CO2. Then the cells were transfected transiently with the pF4–151-luc CARK promoter plasmids using Lipofectamine 2000 as above described and after 48 hrs, cells were harvested for luciferase assay and western blot analysis.
Western blot analysis
Cells were washed in cold PBS and prepared with NE-PER nuclear and cytoplasmic extraction reagents (Pierce) containing multiple protease inhibitors, according to the manufacturer's instructions. Cell lysates were analyzed by SDS/PAGE and transferred electrophoretically to nylon membrane (Pierce). The bands were probed with primary antibodies specific for Mef2c or Gapdh (Santa Cruz) and HRP-labelled secondary antibodies. Detection was carried out by the chemiluminescent system (ECL, Amersham Biosciences, Piscataway, NJ, USA.
Quantitative real-time RT-PCR analysis
SYBR green-based quantitative real-time RT-PCR was used to examine changes in levels of CARK mRNAs in RNAs prepared from primary rat cardiomyocytes transfected with MEF2C morpholino antisense or sense oligonucleotide as described above. Total RNAs were isolated using Trizol reagent (Invitrogen) and treated with DNase I (Ambion) according to the manufacturer's instructions. cDNAs were generated using oligo(dT)12-18 as a primer and SuperScript II RT-PCR kit (Invitrogen).cDNA was added to 25 μl of reaction volume containing 12.5 μl of 2× SYBR Green Premix EX Taq (TaKaRa, Otsu, Japan), and 400 nmol/L CARK gene-specific primers (forward: 5′-GACAAAGCAGCCAGGGAA; reverse: 5′-AGCAGGCAATCGGAAGAA). To confirm amplification specificity, the PCR products were subjected to a melting curve analysis. Assays were performed in triplicate with a DNA Engine Opticon 2 Real Time PCR Detector (Bio-Rad, Richmond, CA, USA). GAPDH gene was used as internal control.
Statistical analysis
All data were expressed as mean ± S. D. Differences between groups were examined for statistical significance using Student's t-test. A P value less than 0.05 was considered statistically significant.
Results
Identification of alternative splicing variants of mouse CARK isoform
To characterize and clone the mouse CARK cDNA sequences, we used the human full-length CARK mRNA sequence (GenBank accession no. NM_015978) as a query for BLAST search in the UCSC mouse ESTs database (http://genome.ucsc.edu). Ten mouse-spliced ESTs orthologous to human CARK were found (Fig. 1A). Based on the three largest EST sequences (GenBank accession nos. BB658710, AU051569 and BB474379), the full-length cDNA of mouse CARK was identified by RT-PCR. Two fragments, 1921bp and 1641bp, were amplified between AU051569 and BB474379 with primers AK1 and AK2 (Fig. 1C). After aligning the fragments with known ESTs, two transcripts were identified and sequenced, isoform 1 was of the length 2505 bp and isoform 2 was 2028-bp long. Sequences of the two transcripts were submitted to GenBank and assigned with accession numbers AY526095 and AY526096. The isoform 1 encoded a putative polypeptide of 834 amino acids and isoform 2 encoded a shorter 675 amino acids polypeptide because exons 21 and 23 were spliced out (Fig. 1B).
Heart-specific expression of the CARK gene
The specific expression of the CARK gene in human heart tissue has been reported previously [12]. To detect the expression profile of mouse CARK gene, RT-PCR was performed on a panel of various adult mouse tissues. The CARK-specific sequences were detectable exclusively in heart, no bands were observed in liver, spleen, lung, brain, kidney, skeletal muscle or testis (Fig. 2). These findings were in consistency with previous studies and further demonstrated that the CARK is a heart-restricted gene.
2.
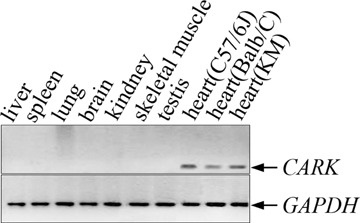
Tissue distribution of mouse CARK gene. Total RNAs were extracted from different mouse tissues using Trizol reagent. Reverse transcribed cDNA including liver, spleen, lung, brain, kidney, skeletal muscle, testis, heart (C57BL/6J), heart (Balb/C) and heart (KM; lanes 1–10) were amplified to give rise to the 261-bp-specific fragment of CARK. The 261 bp band was only detected in mouse heart from three different strains (C57BL/6J, Balb/C and KM). To control for the amount of cDNA in each reaction, a parallel PCR reaction using the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was carried out. RT-PCR products were analyzed on 2% agarose gel.
Mapping of the transcription start site
As the first step towards characterizing the promoter of the mouse CARK gene, the transcription start site was mapped by the 5′ RACE analysis. Two sets of oligonucleotide primers, 5′ primer and NMR, and 5′ nested primer and GSP2 were used, respectively, in the 5′ RACE analysis (Fig. 1C). The nested PCR products were extracted from the agarose gel and cloned into TOPO-TA cloning vector for sequencing. The results showed that RNA oligo was linked to an adenine residue at nucleotide position 88 bp upstream from the translation start codon, indicating that the transcription of the CARK gene starts at this position. Prolonging the extension time in the primary and secondary PCRs did not amplify any large PCR products. Therefore, the transcription start site of the CARK gene was located 88 bp upstream from the translation start codon (Fig. 4B).
Functional analysis of the CARK promoter using sequential deletions
To determine whether the 5′ flanking sequence of the mouse CARK gene possessed functional transcription activity, a sequence-conserved region of 1440 bp (from −1361 to +79 relative to the transcription start site) was amplified as potential promoter and cloned into the pGL3 basic luciferase plasmid to generate pF1-1361-luc plasmid (Fig. 3A), which was transiently co-transfected with Renilla luciferase plasmid pRL-CMV into HEK293S cells, Hela cells, rat aorta smooth muscle cells, cardiomyocytes and cardiac fibroblasts. A significant high level of luciferase activity was only detected in transfected cardiomyocytes (Fig. 3B), not in other cell types (data not shown), and the CARK gene promoter region (−151/+79) showed higher level of transcription activity than other four promoter regions, indicating that this region (−151/+79) contains the cis-acting elements sufficient to initialize the cardiac-specific transcription activity (Fig. 3B).
To locate the region essential for transcription activation, a series of truncation constructs of the promoter region was prepared and transfected into cardiomyocytes (Fig. 3A). Increasing the 5′ truncations from −1361 to −673 and −313 resulted in a small but significant decrease in luciferase expression (−1361/+79 vs. −673/+79, P < 0.05; −1361/+79 vs. −313/+79, P < 0.05). An increase in expression levels observed upon further truncation to −151 suggests the location of a repressor element between nucleotides −313 and −151. Transcription activity was abrogated after truncation to −1, indicating that sequences in close upstream proximity of 151 are crucial for basal CARK transcription activity. In the region from −151 to −1, no typical TATA box or CAAT box motif was found.
The transcription activity of all deletional promoter regions was only detected in cardiomyocytes, but not in other four cell types (data not shown), which indicated that the silence of CARK in noncardiac cells was due to the lack of cardiac-specific transcription factors.
Functional impact of site-directed mutagenesis of CARK promoter elements
To map cis-acting elements that were required for transcriptional activity of the CARK minimal promoter region (from −151 to −1), a multiple alignment of this region between mouse, human, chimpanzee, dog and rat was performed. By searching all the consensus sequences in this region for cardiac TFBSs in the CardioSignal database (http://www.cardiosignal.org/tools/scan.html), five sequence-conserved cardiac TFBSs including Tbx5, SRE, M-CAT box, MEF2 and GATA4 were identified (Fig. 4A).
To examine whether these binding sites were indeed responsible for the transcriptional activity of CARK, site-directed mutations were introduced into the sequences within the CARK promoter that conform to the consensus (Fig. 4B). Five mutant constructs, mut-Tbx5-luc, mut-SRE-luc, mut-MCAT-luc, mut-MEF2-luc and mut-GATA4-luc, were generated based on the pF4-151-luc plasmid (Fig. 4C). The relative luciferase activities of these mutant constructs in cardiomyocytes were shown in Figure 4D. Mutations in MEF2 binding site caused drastic decrease to only approximate 1% luciferase activity of the original level and mutations in other TFBSs, Tbx5, SRE, M-CAT box and GATA4, resulted in a decrease of the luciferase activity by 42%, 67%, 74% and 48%, respectively. These findings suggested that MEF2 binding site was critical for the cardiac-specific transcriptional activity of the CARK promoter.
Identification of transcription factors binding to MEF2 element in CARK promoter
In order to determine whether putative MEF2 binding site of the CARK promoter could actually bind protein, nuclear extracts from cardiomyocytes were analyzed by EMSA using a labelled wild-type or mutated MEF2 oligonucleotide probe. As illustrated in Figure 5, DNA protein complexes were detected using wild-type labelled MEF2 probe (lane 1). These DNA protein complexes were specific, and they could be competitively blocked in the presence of excess amounts of unla-belled MEF2 competitors (lane 4), but not in the excess amount of unlabelled mutated MEF2 oligonucleotide (lane 3). To confirm the specificity of MEF2 binding, supershift experiments were performed. The addition of antibody specifically recognizing Mef2c produced a single supershifted complex (lane 5), and anti-srf-specific antibody, as a negative control, did not to affect the mobility of the complex (lane 6). These results indicated that Mef2c is a component of the complex.
5.

Mef2c binding to the CARK promoter sequence by EMSA. Oligonucleotides probe containing bp –30 to –5 of the CARK promoter were 3′-end-biotin-labelled and incubated with primary cardiomyocytes nuclear extracts in the absence or presence of unlabelled wild-type or mutated competitor. Lane 1, MEF2 wild-type probe labelled by biotin; lane 2, MEF2 mutated probe labelled by biotin; lane 3, MEF2 wild-type probe labelled by biotin with 200-fold molar excess of MEF2 mutated probe not labelled by biotin (mt); lane 4, MEF2 wild-type probe labelled by biotin with 200-fold molar excess of MEF2 wild-type probe not labelled by biotin (wt); All mutations were located within the –30/–5 sequence of the CARK promoter as shown in Figure 4B. Supershift EMSA experiments were performed using MEF2 labelled wild-type probe incubated with primary cardiomyocytes nuclear extracts in the presence of anti-Mef2c-specific antibody (lane 5) or anti-srf-specific antibody (lane 6). The arrow indicates the supershifted band with anti-Mef2c-specific antibody.
Loss-of-function analysis of mef2c
To verify the transcription start of the CARK gene was truly regulated by Mef2c, and MEF2C antisense morpholino oligonucleotide-based knockdown strategies was applied. After cardiomyocytes were transfected with the MEF2C antisense morpholino or control oligonucleotides, pF4–151-luc reporter plasmid was introduced and luciferase activity was assayed. The results showed that luciferase activity in cardiomyocytes transfected with the MEF2C antisense oligonucleotides dramatically decreased by 80% compared to the sense oligonucleotides (Fig. 6A). Immunoblot revealed that MEF2C antisense depressed Mef2c protein production (Fig. 6B). Quantitative real-time RT-PCR assay showed that the level of CARK-mRNA expression in the cardiomyocytes transfected with MEF2C antisense oligonucleotides significantly decreased compared to the control oligonucleotides (Fig. 6C). These results confirmed that Mef2c functioned as a transcriptional activator of the CARK promoter.
6.
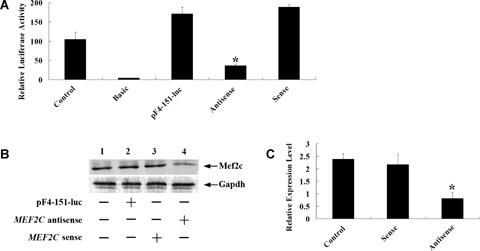
The effect of mef2c low level of expression on transcription activity of CARK in cardiomyocytes. (A) Cardiomyocytes were pretreated with MEF2C-specific antisense or control oligo before being transfected with pF4-151-luc reporter. After 24 hrs of incubation in medium containing antisense or control oligo, cardiomyocytes were trans-fected with pF4-151-luc reporter and cultured for an additional 48 hrs. Then the cells were lysed and luciferase activities were assayed. Firefly luciferase activities expressed had been normalized on the basis of Renilla luciferase activity. Data shown are the means ± S. D., n= 3.(*P < 0.01 compared with the relative luciferase activity of pF4-151-luc).pGL3 basic vector containing no promoter element was used as negative control and pGL3 promoter vector fused with SV40 promoter element was used as positive control. (B) Western blot analysis of Mef2c protein expression in cardiomyocytes after transfection of antisense or control. Gapdh used as an internal control.(C) Cardiomyocytes trans-fected with MEF2C-specific antisense or control oligos were harvested, and total RNA was isolated to perform quantitative real-time RT-PCR. Data shown are the means ± S. D., n= 3 (*P < 0.01 compared to sense oligo transfected cells). All data are the relative expression of CARK after normalization with the internal GAPDH expression. The cells not transfected were used as control.
Discussion
CARK is a novel cardiac-specific kinase gene; tissue array analyses showed that CARK is highly expressed in heart, but is undetectable in other tissues [12]. Previous studies indicate that CARK contains similar domain structure with Ilk and may shares some functions similar to those of Ilk and may be implicated in related signalling pathways. CARK can interact directly with cTnI, the disease-causing gene product of hypertrophic cardiomyopathy. As the homolog of Ilk, CARK may transduce the integrin-related signal, thereby connecting sarcomeric protein deficits with integrin pathways. Thus CARK may participate in the pathway linking the primary contractile machinery deficits with secondary cardiac hypertrophy. Investigation of cardiac-restricted genes and their regulating factors is helpful to understand the mechanisms of heart development and the maintenance of cardiac function. To date, only a few cardiac-restricted genes are reported, and the mechanism of their transcriptional control is not very well characterized.
In this study, we first cloned the full-length mRNA sequence and mapped the transcription start site of the mouse CARK gene. By aligning the promoter region sequences of mouse CARK gene with other species, we identified the 1.4-kb long 5′ flanking sequence-conserved region of the CARK gene.
When cloned into a luciferase reporter and transfected into cardiomyocytes and other cell types, this 1.4-kb long sequence showed strong transcriptional activity only in cardiomyocytes, but no signals in other cell types. By sequential deletions mapping, the core promoter region of the CARK gene was mapped between −151 and −1 relative to the transcription start site.
Bioinformatics analysis revealed five potential conserved cardiac-specific TFBSs in the core promoter region of the CARK gene, including Tbx5, SRE, M-CAT box, MEF2 and GATA4. We demonstrated that mutation of the MEF2 sites in the CARK promoter greatly impaired its transcription activity and co-transfection of the CARK promoter-driven luciferase reporter with the MEF2C antisense oligonucleotides dramatically decreased luciferase gene expression. These observations suggest that Mef2c plays a critical role in regulating basal CARK transcription activity.
The Mef2 family of transcription factors comprises a group of transcriptional activators, Mef2A, -B, -C and -D, that show homology in a MADS box and an adjacent motif known as the MEF2 domain [17–19]. MEF2 factors bind to the consensus site, C/TTA(A/T)4TAG/A, which is found in the control regions of numerous muscle-specific genes and has been demonstrated to be important for cardiac and skeletal muscle gene expression [20]. The four vertebrate MEF2 gene products have greater than 85% amino acid identity within the MADS domain and an adjacent 27-amino acid region referred to as the MEF2 domain. During embryogenesis, MEF2 transcripts appear initially as precursors of the cardiac and skeletal muscle lineages and are subsequently expressed at high levels in these differentiated muscle cell types [18, 21]. Previous studies confirm that Mef2 factors can induce dilated cardiomyopathy in transgenic mice and activates a genetic program promoting chamber dilation and mechanical dysfunction in calcineurin-induced heart failure [22, 23]. In light of the potent transcriptional activity of Mef2 proteins and the presence of MEF2 sites in many cardiac and skeletal muscle genes, it is likely that the early activation of MEF2 gene expression in the cardiac and skeletal muscle lineages is important for the establishment of these specific programs of muscle gene expression.
MEF2C is unique in that its transcripts are the most restricted, largely to the cardiac and skeletal muscle lineages and is the earliest expressed gene in embryos among four members of the MEF2 family [19, 24, 25]. The early expression of MEF2C is necessary for proper development of the murine heart and precedes that of cardiac muscle structural genes such as α-cardiac actin and myosin heavy chain [26–28], supporting that it is involved in establishing the transcriptional program associated with cardiac determination or differentiation or both. In MEF2C lacking mouse embryos, heart development arrests at the looping stage (embryonic day 9.0), the future right ventricular chamber fails to form, and cardiomyocyte differentiation is disrupted and a subset of cardiac muscle genes was not expressed [29]. Recent studies indicate that MEF2C is essential for the normal allocation of cells between the ventricular and sino-atrial precursors of the primary heart field [30]. Therefore, it is not surprising that the cardiac-enriched transcription factor Mef2c is the most critical for cardiac-specific CARK gene expression.
The presence of common DNA sequences within the promoter of different genes makes the interpretation of developmental and tissue-specific gene expression quite complex. Because only a limited number of cis-acting factors are defined, diversity in gene expression is likely because of differences in promoter context. Variations in promoter context may be the result of temporal or tissue-specific expression of transcription factors. Our isolation and characterization of a functional CARK promoter will facilitate new investigations into the mechanisms by which transcription factors influence transcriptional events of this cardiac-specific gene.
Acknowledgments
We thank Dr. Issei Komuro (Department of Cardiovascular Medicine, University of Tokyo Graduate School of Medicine, Tokyo) for providing the ANP promoter vector. This study was supported by National 863 High Tech Program of China (granted to Dr. Rutai Hui) and National Natural Science Foundation of China (granted to Dr. Hu Wang).
References
- 1.Firulli AB, Thattaliyath BD. Transcription factors in cardiogenesis:the combinations that unlock the mysteries of the heart. Int Rev Cytol. 2002;214:1–62. doi: 10.1016/s0074-7696(02)14002-2. [DOI] [PubMed] [Google Scholar]
- 2.McKinsey TA, Zhang CL, Olson EN. MEF2:a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–7. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 3.Biben C, Harvey RP. Homeodomain factor Nkx2-5 controls left/right asymmetric expression of bHLH gene eHand during murine heart development. Genes Dev. 1997;11:1357–69. doi: 10.1101/gad.11.11.1357. [DOI] [PubMed] [Google Scholar]
- 4.Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat Genet. 1998;18:266–70. doi: 10.1038/ng0398-266. [DOI] [PubMed] [Google Scholar]
- 5.Wang DZ, Valdez MR, McAnally J, Richardson J, Olson EN. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development. 2001;128:4623–33. doi: 10.1242/dev.128.22.4623. [DOI] [PubMed] [Google Scholar]
- 6.Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–55. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zang MX, Li Y, Wang H, Wang JB, Jia HT. Cooperative interaction between the basic helix-loop-helix transcription factor dHAND and myocyte enhancer factor 2C regulates myocardial gene expression. J Biol Chem. 2004;279:54258–63. doi: 10.1074/jbc.M408502200. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–62. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 9.Belaguli NS, Sepulveda JL, Nigam V, Charron F, Nemer M, Schwartz RJ. Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol. 2000;20:7550–8. doi: 10.1128/mcb.20.20.7550-7558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanpoucke G, Goossens S, De Craene B, Gilbert B, Van Roy F, Berx G. GATA-4 and MEF2C transcription factors control the tissue-specific expression of the alphaT-catenin gene CTNNA3. Nucleic Acids Res. 2004;32:4155–65. doi: 10.1093/nar/gkh727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small EM, Krieg PA. Molecular regulation of cardiac chamber-specific gene expression. Trends Cardiovasc Med. 2004;14:13–8. doi: 10.1016/j.tcm.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Meng XM, Wei YJ, Zhao XW, Liu DQ, Cao HQ, Liew CC, Ding JF. Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J Mol Med. 2003;81:297–304. doi: 10.1007/s00109-003-0427-x. [DOI] [PubMed] [Google Scholar]
- 13.Sigrist CJ, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M, Bairoch A, Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–74. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 14.Zhen Y, Wang Y, Zhang W, Zhou C, Hui R. CardioSignal:a database of transcriptional regulation in cardiac development and hypertrophy. Int J Cardiol. 2007;116:338–47. doi: 10.1016/j.ijcard.2006.03.069. [DOI] [PubMed] [Google Scholar]
- 15.Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001;28:276–80. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Han Y, Lin C, Zhen Y, Song X, Teng S, Chen C, Chen Y, Zhang Y, Hui R. PDGF-D contributes to neointimal hyperplasia in rat model of vessel injury. Biochem Biophys Res Commun. 2005;329:976–83. doi: 10.1016/j.bbrc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 17.Leifer D, Krainc D, Yu YT, McDermott J, Breitbart RE, Heng J, Neve RL, Kosofsky B, Nadal-Ginard B, Lipton SA. MEF2C, a MADS/MEF2-family transcription factor expressed in a laminar distribution in cerebral cortex. Proc Natl Acad Sci USA. 1993;90:1546–50. doi: 10.1073/pnas.90.4.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin JF, Miano JM, Hustad CM, Copeland NG, Jenkins NA, Olson EN. A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol Cell Biol. 1994;14:1647–56. doi: 10.1128/mcb.14.3.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shore P, Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 20.Cserjesi P, Lilly B, Hinkley C, Perry M, Olson EN. Homeodomain protein MHox and MADS protein myocyte enhancer-binding factor-2 converge on a common element in the muscle creatine kinase enhancer. J Biol Chem. 1994;269:16740–5. [PubMed] [Google Scholar]
- 21.Yu YT, Breitbart RE, Smoot LB, Lee Y, Mahdavi V, Nadal-Ginard B. Human myocyte-specific enhancer factor 2 comprises a group of tissue-restricted MADS box transcription factors. Genes Dev. 1992;6:1783–98. doi: 10.1101/gad.6.9.1783. [DOI] [PubMed] [Google Scholar]
- 22.Van Oort RJ, Van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, Van Der Nagel R, Doevendans PA, Schneider MD, Van Echteld CJ, De Windt LJ. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. doi: 10.1161/CIRCULATIONAHA.105.608968. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH, Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem. 2006;281:9152–62. doi: 10.1074/jbc.M510217200. [DOI] [PubMed] [Google Scholar]
- 24.Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–63. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- 25.Olson EN, Perry M, Schulz RA. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev Biol. 1995;172:2–14. doi: 10.1006/dbio.1995.0002. [DOI] [PubMed] [Google Scholar]
- 26.Molkentin JD, Markham BE. Myocyte-specific enhancer-binding factor (MEF-2) regulates alpha-cardiac myosin heavy chain gene expression in vitro and in vivo. J Biol Chem. 1993;268:19512–20. [PubMed] [Google Scholar]
- 27.Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–74. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- 28.Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, Olson EN. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci USA. 2006;103:11189–94. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–7. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vong L, Bi W, O'Connor-Halligan KE, Li C, Cserjesi P, Schwarz JJ. MEF2C is required for the normal allocation of cells between the ventricular and sinoatrial precursors of the primary heart field. Dev Dyn. 2006;235:1809–21. doi: 10.1002/dvdy.20828. [DOI] [PubMed] [Google Scholar]