

Abstract
Although the nuclear factor-κB (NF-κB)-dependent gene expression is critical to the induction of an efficient immune response to infection or tissue injury, excessive or prolonged NF-κB signalling can contribute to the development of several inflammatory diseases. Therefore, the NF-κB signal transduction pathway is tightly regulated by several intracellular proteins. We have previously identified A20-binding inhibitor of NF-κB activation (ABIN)-3 as an lipopolysaccharide (LPS)-inducible protein in monocytes that negatively regulates NF-B activation in response to tumour necrosis factor (TNF) and LPS. Here we report that ABIN-3 expression is also up-regulated upon TNF treatment of monocytes and other non-myeloid cell types. We also found a significantly enhanced expression of ABIN-3 in monocytes of sepsis patients, which is restored to control levels by corticotherapy. To further understand the transcriptional regulation of ABIN-3 expression, we isolated the human ABIN-3 promoter and investigated its activation in response to TNF and LPS. This revealed that the LPS- and TNF-inducible expression of ABIN-3 is dependent on the binding of NF-κB to a specific B site in the ABIN-3 promoter. Altogether, these data indicate an important role for NF-κB-dependent gene expression of ABIN-3 in the negative feedback regulation of TNF receptor and toll-like receptor 4 induced NF-κB activation.
Keywords: NF-κB, signal transduction, sepsis, lipopolysaccharide, TNF, toll-like receptors
Introduction
The nuclear factor-κB (NF-κB) transcription factor family regulates the expression of multiple genes that are involved in inflammation, immunity, differentiation, proliferation and apoptosis. In mammals, the NF-κB family consists of five members (RelA/p65, c-Rel, p50, p52 and RelB), which function as homo- or heterodimers [1]. Each possesses an N-terminal Rel-homology domain that is responsible for dimerization, nuclear translocation and DNA binding. The most common heterodimer is p50/p65, which is kept inactive in the cytoplasm by binding to the inhibitor of κB (IκB) αprotein. p50/p65 is activated in response to IκB kinase (IKK) β–mediated phosphorylation of IκBα, which triggers its ubiquitination and subsequent proteasome-mediated degradation, thereby releasing NF-κB to translocate to the nucleus and bind to the promoter of NF-κB responsive genes. In the nucleus, NF-κB itself can also be phosphorylated by different kinases, which further regulate its transcriptional activation potential [1, 2].
Given the essential role of NF-κB-dependent gene expression in the regulation of innate and adaptive immune responses, it is not unexpected that unrestrained NF-κB activation is associated with several inflammatory diseases such as Crohn's disease, asthma, rheumatoid arthritis and sepsis [3]. Hence, several proteins act as checkpoints at different levels along the NF-κB signalling pathway and apply various strategies to dampen NF-κB activation, such as intervening with specific protein-protein interactions or specific posttranslational modifications of NF-κB signalling proteins [4, 5]. Many of these negative regulators are themselves regulated by NF-κB and impose a negative feedback mechanism. For example, tumour necrosis factor (TNF) and lipopolysaccharide (LPS) strongly induce the NF-κB-dependent expression of A20, which negatively regulates TNF- and LPS-induced NF-κB activation by changing the ubiquitination status of specific NF-κB signalling proteins [6]. A20-binding inhibitor of NF-κB activation (ABIN)-1, ABIN-2 and ABIN-3 have been suggested to contribute to the NF-κB inhibitory potential of A20 [7–9]. Recently, ABIN-1 was shown to mediate the recruitment of A20 to the IKK adaptor protein IKKγ, leading to the A20-mediated de-ubiquitination of IKK and disruption of the NF-κB activation signal [10]. In contrast to ABIN-1 and ABIN-2, which are constitutively expressed in several cell types, ABIN-3 expression is much more restricted. In this context, ABIN-3 was identified as an LPS-inducible gene in human monocytes, whose expression is regulated at the mRNA level [9]. More recently, ABIN-3 was also identified in a screen for IL-10-induced genes in LPS-stimulated macrophages, and was shown to be superinduced in response to IL-10 and different pro-inflammatory stimuli including TNF, LPS, IL-1, peptidoglycan, poly(I:C) and unmethylated CpG DNA [11]. Here, we have further investigated the LPS- and TNF-inducible expression of ABIN-3 in some other cell types. Moreover, we have compared ABIN-3 expres sion in monocytes from healthy patients and sepsis patients. To further understand the transcriptional regulation of ABIN-3 expression, we have also isolated the ABIN-3 promoter and characterized its activation in response to TNF and LPS.
Materials and methods
Cell lines, plasmids and reagents
Human embryonic kidney HEK293T cells (gift from Dr. M. Hall, University of Birmingham, Birmingham, UK) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum, 2 mM L-Glutamine, 0.4 mM sodium pyruvate and antibiotics. Human cervix carcinoma HeLa cells (gift from Dr. P. Agostinis, University of Leuven, Leuven, Belgium) and human hepatocellular carcinoma HepG2 cells (gift from Dr. H. Schaller, University of Heidelberg, Heidelberg, Germany) were both grown in DMEM supplemented with 10% foetal bovine serum, 2 mM L-Glutamine, 0.4 mM sodium pyruvate, non-essential amino acids and antibiotics. The human myelomonocytic THP-1 cell line (obtained from ATCC, Manassas, VA) was grown in RPMI1640 supplemented with 10% foetal bovine serum, 0.4 mM sodium pyruvate, 2 mM L-Glutamine, 4 μMβ-mercaptoethanol and antibiotics. The human leukaemic monocyte lymphoma cell line U937s (obtained from Biogeneve) and the human T cell leukaemia cell line Jurkat (gift from Dr. J Philippe, Ghent University, Belgium) were grown in the same medium as THP-1 cells. The murine RAW264.7 macrophage cell line was obtained from the ATCC (Manassas, VA) and was cultured in DMEM supplemented with 10% foetal calf serum (FCS), 2mM L-Glutamine, 0.4 mM sodium pyruvate and antibiotics. The plasmid pFlag-CMV1-hTLR-4 was a kind gift from Dr. M. Muzio (Department of Immunology and Cell Biology, Mario Negri Institute, Milano, Italy) [12]. The plasmid pconaLUC (LMBP 3248) [13], encoding the luciferase gene driven by a minimal NF-κB-responsive chicken coalbumine promoter, was provided by Dr. A. Israël (Institute Pasteur, Paris, France) and pActβgal, containing the β-galactosidase gene after the β-actin promoter, was obtained from Dr. Inoue (Institute of Medical Sciences, Tokyo, Japan). The plasmid encoding the NF-κB p65 subunit was a kind gift from Dr. G. Haegeman (University of Ghent, Ghent, Belgium). pCAGGS-E-ABIN-3 [9], pCAGGSEhA20 (LMBP 3778) [14] and the plasmid encoding IKK β-K44A [15] were described elsewhere. The plasmids that have been assigned a LMBP accession number are available from the BCCM/LMBP Plasmid collection, Department of Molecular Biology, Ghent University, Belgium (http: //bccm.belspo.be/about/lmbp.htm). Recombinant human TNF was produced in Escherichia coli in our laboratory and was purified to at least 99% homogeneity. TNF had a specific biological activity of 3 × 107 IU/mg purified protein, as determined with the international standard code 87/650 (National Institute for Biological Standards and Control, Potters Bar, UK). LPS from Salmonella abortus was from Alexis (San Diego, CA) and hydrocortisone hemisuccinate (HC) was from Sigma (Saint Louis, Missouri). MG-132 and sc-514 were obtained from Calbiochem (San Diego, CA).
Isolation of monocytes
Peripheral blood mononuclear cells (PBMC) were prepared by diluting fresh blood samples 1: 2 in RPMI1640 Glutamax medium (Biowhittaker; Verviers, Belgium) and centrifuging them over Ficoll (MSL; Eurobio, Les Ulis, France) for 20 min.at 15°C and 600 ×g. Human monocytes were selected from PBMC by adherence to plastic. PBMC were plated at 6 × 106 cells/ml and allowed to adhere for 1 hr at 37°C in a 5% CO2 air incubator in a humidified atmosphere. Non-adherent cells were removed and adherent cells were washed with RPMI1640. The purity of the adherent cells was checked by flow cytometry using an anti-CD14 antibody, and was routinely higher than 95%. To test the effect of corticosteroids on ABIN-3 expression, healthy donors' monocytes were cultured in RPMI1640 supplemented with antibiotics (100 IU/ml penicillin, 100 g/ml streptomycin) and 0.2% normal human serum (Biowhittaker), and stimulated with 100 ng/ml LPS in the presence or absence of 100 μM HC. Monocytes from sepsis patients were also isolated by adherence and were immediately used for RNA extraction.
Cloning of the ABIN-3 promoter
The sequence of the ABIN-3 promoter was deduced from the human genomic sequence in the NCBI genome database (http://www.ncbi.nlm.nih.gov) and the ABIN-3 cDNA (GenBank™ accession number NM024873). The transcription initiation site of the ABIN-3 gene was deduced from multiple EST sequences for ABIN-3 that were present in the NCBI EST database. Although almost all available EST sequences match the initiation site we used, a sequence (BP335455) starting 19 nucleotides more upstream can also be found, indicating that alternative initiation of transcription might occur. Genomic DNA was isolated from HEK293 cells and a 2130 bp ABIN-3 promoter fragment still containing 36 bp of the 5′-end of the ABIN-3 cDNA was isolated by PCR using the 5′-GAAGATCTGAACAGAAGT-GACTGTGGATAG-3′ forward and 5′-ACGCGTCGAC-GATGGAGGTGAAGTGAACAGAG-3′ reverse primers containing respectively a BglII and SalI restriction site at the 5 end for cloning purposes. The PCR mixture contained 100 ng genomic DNA, 0.5 mM dNTP mix, 5 ng/μl of each primer, 2.5 mM MgCl2, 1 μl expand high-fidelity enzyme mix (Roche Applied Science) in a total reaction volume of 100 μl. PCR was performed as follows: 7 min.at 94°C; five cycles of 30 sec.at 94°C, 45 sec.at 70°C, 4 min.at 72°C; five cycles of 30 sec. at 94°C, 45 sec. at 65°C, 4 min. at 72°C; five cycles of 30 sec.at 94°C, 45 sec at 60°C, 4 min. at 72°C; 30 cycles of 30 sec. at 94°C, 45 sec. at 55°C, 4 min.at 72°C; 7 min.at 72°C. The PCR product was cleaved with BglII and SalI (Promega) and cloned in the de-phosphorylated BglII and SalI-linearized pXP2Δ2 luciferase reporter vector (gift from Dr. Jan Tavernier, University of Ghent, Belgium) using T4 DNA ligase (Takara, Cambrex Bio Science) to generate pXP2 Δ2-promABIN-3. The construct pXP2Δ2-promABIN-3mutNF-κB containing the same ABIN-3 promoter fragment but with a mutated NF-κB binding site was made by overlap PCR using two primer pairs. The first PCR round generated two mutant promoter fragments. The 5′-PCR fragment was generated using pXP2Δ2-promABIN-3 as a template and the 5′-GGGGGTACCGAACAGAAGTGACTGTGGATAG-3′ forward (contains KpnI site at 5 end) and reverse 5′-CATC-CTGGGCCATCCCTATTTGG-3′ primers, with the latter containing the mutated NF-κB site (underlined). The 3 -PCR fragment was generated using pXP2 2-promABIN-3 as a template and the 5′-CCAAATAGGGATGGC-CCAGGATG-3′ forward (mutated NF-κB site is underlined) and reverse 5′-GGACGAGCTCAGGGGCGTAGAAC-3′ primers (the latter containing a SacI site at the 5 end). In the second round of PCR, a mixture of these 5 -PCR and 3′-PCR fragments was used as template and amplification was done with the KpnI and Sacl containing forward and reverse primers mentioned above. Conditions for the PCRs were: 7 min.at 94°C, 35 cycles of 30 sec.at 94°C, 45 sec. at 55°C, 2 min.at 72°C, 7 min.at 72°C with the same reaction mixtures as mentioned above but with 5% DMSO added. The resulting product corresponds to the 3 part of the ABIN-3 promoter containing a mutation in the NF-κB binding site. This fragment was cut with KpnI and SacI and cloned in the de-phosphorylated KpnI and SacI- linearized pXP2 2-promABIN-3 vector using T4-DNA ligase to generate pXP2Δ2-promABIN-3mutNF-κB. All constructs were verified by DNA sequencing. No other mutations than those that were introduced in the NF-κB site could be detected.
Transfection, luciferase reporter assay and Western blotting
4 × 104 HEK293T cells were grown in 24-well plates and transiently transfected by DNA calcium phosphate co-precipitation with a total of 200 ng DNA. The DNA mixtures comprised 40 ng of specific expression plasmids, 20 ng pAct gal, and either 20 ng pconaLUC, 10 ng pXPΔ2, 10 ng pXP2Δ2-promABIN-3 or 10 ng pXP2Δ2-promABIN-3mutNF-κB. 24 hrs after transfection, cells were either left untreated or stimulated with TNF for 6 hrs, after which they were lysed in 200 μl lysis buffer (25 mM Tris-phosphate pH 7.8, 2 mM dithiothreitol, 2 mM 1, 2-cyclohexaminediaminete-traacetic acid, 10% glycerol and 1% Triton X-100). Luciferase (Luc) activity in cell extracts was measured in a Topcount microplate scintillation reader (Packard Instrument Co., Meriden, CT) upon addition of substrate buffer to a final concentration of 470 μM luciferin, 270 μM co-enzyme A and 530 μM ATP. β-Galactosidase (Gal) activity in cell extracts was assayed with chlorophenol red β-galactopyranoside substrate (Roche Molecular Biochemicals) and measuring of the absorbance with a Benchmark microplate reader (Bio-Rad). Luc values were normalized for Gal values in order to correct for differences in transfection efficiency (plotted as Luc/Gal).
1 × 105 RAW264.7 cells were seeded the day before transfection. Cells were transfected using lipofectamine 2000 and Opti-MEM (Invitrogen) with 250 ng pXp2Δ2 or pXp2Δ2-promABIN-3 together with 250 ng pActβgal. Six hours after transfection, medium was refreshed. Twentyfour hours later, cells were stimulated with 1 μg/ml LPS for 6 hrs, with or without a 2 hrs pre-treatment of the cells with 100 μM HC. Luc activity in cell extracts was measured as described above. Gal activity was assayed with the Galacto-Star™ System (Applied Biosystems, Massachusetts, USA) and measured in a Topcount microplate scintillation reader.
For protein expression studies, THP-1, U937s and Jurkat cells were grown at 8 × 105 cells in 2 ml medium and HeLa and HepG2 cells were grown at 2 × 105 cells/well in a 6-well plate. 24 hrs later, cells were left untreated or pre-treated for 1 hr with MG-132 or sc-514 or for 2 hrs with HC. Cells were then stimulated for 6 or 18 hrs with LPS or TNF. Cell lysates were made in E1A-buffer (50 mM HEPES pH 7.6, 250 mM NaCl, 0.5% NP-40, 5 mM ethylenedi-aminetetraacetic acid [EDTA]) supplemented with protease and phosphatase inhibitors. Cells were left on ice for 15 min. and then centrifugated at 20, 000 g for 15 min. Supernatants was used for Western blotting. Equal amounts of protein were mixed with Laemmli sample buffer and separated by 10% SDS-PAGE, followed by Western blotting. Immunodetection of ABIN-3 was done with a rabbit polyclonal ABIN-3 antibody raised against an ABIN-3 specific peptide (NH2-HFVQGTSRMIAAESSTEHKE-COOH) coupled to keyhole limpet haemocyanin.
RT-PCR
THP-1, U937s and Jurkat cells were grown at 8 × 105 cells in 2 ml RPMI1640 medium and HeLa and HepG2 cells were seeded at 1.5 × 105 cells/well in a 6-well plate. After 24 hrs, cells were either left untreated or pretreated with MG-132 or sc-514 for 1 hr. Cells were then stimulated with LPS or TNF for 3 hrs. Total cellular RNA was isolated by the TRIZOL™-method (Invitrogen) and first strand cDNA was synthesized using ‘Superscript™ First-Strand Synthesis System for RT-PCR’ (Invitrogen). Reverse transcribed cDNA samples were amplified by PCR with gene specific primers (5′-ACTGGACGCCGCGGAAAGAT-3′ and 5′-TGGCGGAAGCTGGTCAAGAG-3′) that amplify a fragment of the open reading frame of ABIN-3. As a control for cDNA integrity, a β-actin fragment was amplified with 5′-GAACTTTGGGGGATGCTCGC-3′ and 5′-TGGTGGGCAT-GGGTCAGAAG-3′ primers.
Total RNA was prepared from primary monocytes selected by adherence, using the RNeasy Mini Kit (Qiagen, Valencia, CA). Purified RNA was reverse-transcribed with Superscript II RNase H (Invitrogen) according to the manufacturer's protocol. The expression levels of ABIN-3 and GAPDH were determined by real-time quantitative PCR, using a FastStart DNA masterplus SYBR Green I and a LightCycler (Roche, Meylan, France). The forward and reverse primers for human ABIN-3 were respectively 5′-CAAAGGAAAAGGAACATTAC-3′ and 5′-TGCTGTAGCTC-CTCTTTCTC-3′ . Primers for glyceraldehyde-3-phos-phatase dehydrogenase (GAPDH) were the RT2 PCR primer set from SuperArray (Frederick, MD). For ABIN-3, each run consisted of an initial denaturation time of 5 min. at 95°C and 40 cycles at 95°C for 8 sec., 56°C for 8 sec., and 72°C for 15 sec. For GAPDH, the run consisted of 40 cycles at 95°C for 15 sec., 58°C for 15 sec.and 72°C for 25 sec. The cDNA copy number of each gene was determined using a six-point standard curve. Standard curves were run with each set of samples, the correlation coefficients (r2) for the standard curves being >0.98. All results were normalized with respect to the expression of GAPDH. To confirm the specificity of the PCR products, the melting profile of each sample was determined using the LightCycler, and by heating the samples from 60°C to 95°C at a linear rate of 0.10°C/sec. while measuring the fluorescence emitted. Analysis of the melting curve demonstrated that each pair of primers amplified a single product. In all cases, the PCR products were checked for size by agarose gel separation and ethidium bromide staining to confirm that a single product of the predicted size was amplified.
Nuclear extract preparations
THP-1 cells (10 × 106) were stimulated with LPS or TNF for various times. After washing in phosphate-buffered saline, cells were resuspended in a buffer containing 10 mM Hepes pH 7.5, 10 mM KCl, 1 mM MgCl2, 5% glycerol, 0.5 mM EGTA, 0.1 mM EDTA, 0.5 mM dithiothreitol, 2 mM Pefabloc and 0.3 mM aprotinin and incubated for 15 min.at 4°C.50 μl of 10% Nonidet P-40 was added and the whole mixture was vortexed and centrifuged at 20,000 g for 15 min. The pellet was re-suspended in a buffer containing 20mM Hepes pH7.5, 1% Nonidet P-40, 1 mM MgCl2, 400 mM NaCl, 10 mM KCl, 20% glycerol, 0.5 mM EGTA, 0.1 mM EDTA, 0.5 mM dithiothreitol, 2 mM Pefabloc and 0.3 mM aprotinin. After an additional incubation for 20 min. on ice, the suspension was centrifuged again at 20,000 g for 5 min. and the supernatant was stored at −70°C.
Electrophoretic mobility shift assay
DNA binding was analysed by incubating 8 μg nuclear proteins for 30 min. at room temperature with a specific 32P-labelled oligonucleotide probe. Binding buffer consisted of 20 mM Hepes pH7.5, 60 mM KCl, 4% Ficoll 400, 2 mM dithiothreitol, 100 mg/ml poly[d(I: C)], and 1 mg/ml bovine serum albumin. Samples were analysed by electrophoresis on a 5% native polyacrylamide gel that was run for 2 hrs at 100 V in 0.5 × Tris-boric acid-EDTA buffer pH 8.0. Gels were dried and bands were visualized by autoradiography. In supershift experiments, 1 μg anti-p65 polyclonal antibody (Santa Cruz Biotechnology) was added to 8 μg nuclear extract and incubated for 1 hr at 4°C before adding the 32P-labelled oligonucleotide probe. The oligonucleotide probe was based on the putative NF-κB binding site in the ABIN-3 promoter, as predicted by the MatInspector (Genomatix Software GmbH, Munich; http://www.genomatix.de) transcription factor search program. NF-κB-specific wild-type and mutant (underlined) oligonucleotide probes were:
5′-AGCTAAATAGGGATTTCCCAGGAT-3′ (NF-κBWT-reverse), 5′-AGCTATCCTGGGAAATCCCTATTT.-3′ (NF-BWT-forward) and 5′-AGCTAAATAGGGATGGC-CCAGGAT-3′ (NF-κBmut-reverse), 5′-AGCTATCCTGGGC-CATCCCTATTT-3′ (NF-κBmut-forward). Double-stranded oligonucleotides were obtained by mixing the single-stranded oligos with their complementary strand in a molar ratio of 1: 1, incubating them for 10 min.at 95°C, and cooling down slowly to 4°C.
Patients characteristics
The study protocol was approved by the institutional review board for human experimentation. Patients with a severe sepsis, as defined by a panel of experts from the American College of Chest Physician/Society of Critical Care Medicine, were included into the study [16]. Patients were excluded if they were under 18 years, had neutropenia, had received chemotherapy during the last 6 months, were presently receiving corticosteroid therapy or any other immunosuppressive therapies or were human immunodeficiency virus positive. The simplified acute physiology score (SAPSII) [17] and theSequential organ failure assessment (SOFA) score [18] were calculated during the first 24 hrs. All patients were included just before and 24 hrs after initiating replacement therapy with low doses of corticosteroids. We also included healthy volunteers recruited amongst health care workers or blood donors (Etablissement Français du Sang, Paris, France).
Data are given as mean ±S. E. M. The significance of the differences between controls, septic shock patients before and after corticosteroid treatment was determined by analysis of variance (ANOVA) and Fischer's protected least significant difference (PLSD). The effect of hydrocortisone treatment on ABIN-3 expression was also analysed using the Wilcoxon signed-rank test. A value of P<0.05 was the criterion for statistical significance. Statistical analysis was performed with Statview software (Abacus concept Inc., Berkeley, CA).
Results
ABIN-3 expression is increased by LPS and TNF, as well as during sepsis
ABIN-3 expression has previously been described as an LPS-inducible gene in the monocytic cell line THP-1 and in bone marrow-derived macrophages [9, 11]. To further analyse the inducible expression of ABIN-3 in other cell lines and cell types, we analysed ABIN-3 mRNA and protein expression in the mono-cytic cell lines THP-1 and U937s, the T cell line Jurkat, the hepatoma cell line HepG2 and the cervix carcinoma cell line HeLa. Cells were treated with LPS or TNF for 3 hrs, after which mRNA was isolated and analysed for ABIN-3 expression by semi-quantitative RT-PCR (Fig. 1A). Basal ABIN-3 mRNA expression could already be observed in THP-1 and HeLa cells, and to a lesser extent in Jurkat and HepG2 cells. LPS treatment up-regulated ABIN-3 mRNA expression in THP-1 and U937s cells. Similarly, also TNF increased ABIN-3 expression in these cells, as well as in HepG2 and HeLa cells. No up-regulation of ABIN-3 mRNA could be observed in Jurkat cells. Interestingly, ABIN-3 protein expression could only be detected in LPS stimulated THP-1 and U937s cells, and in TNF stimulated HeLa cells (Fig. 1B). These data demonstrate that ABIN-3 expression is not restricted to LPS stimulated myeloid cells, but also occurs in other cell types in response to TNF. Moreover, they show that ABIN-3 expression is regulated at the transcriptional and translational level.
1.
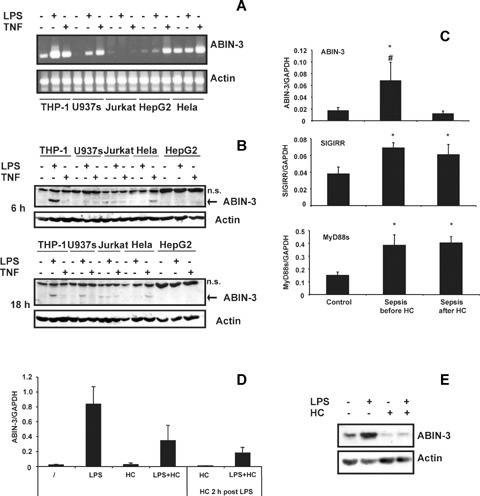
ABIN-3 expression is up-regulated in response to LPS and TNF.(A) Semi-quantitative RT-PCR of ABIN-3 mRNA expression in different cell lines. RNA was isolated from the indicated cell lines that were either left untreated or stimulated for 3 hrs with 100 ng/ml LPS or 1000 IU/ml TNF. Semi-quantitative RT-PCR was performed using ABIN-3 specific primers to amplify a 700 bp fragment of the ABIN-3 open-reading frame. β-actin was used as a control.(B) Western blotting for ABIN-3 protein expression in different cell lines. Cells were either left untreated or stimulated for 6 or 18 hrs with LPS or TNF as indicated. Cell extracts were separated by SDS-PAGE and analysed by Western blotting and immunode-tection with anti-ABIN-3 polyclonal antibodies. β-actin expression was used as a control for equal loading.(C) Real-time quantitative PCR of ABIN-3 expression in monocytes from sepsis patients. Monocytes were selected by adherence from PBMC of healthy donors (n= 12) and septic shock patients (n= 6) before and 24 hrs after low-dose HC therapy. Total RNA was isolated and ABIN-3, SIGIRR and MyD88s expression were analysed by real time quantitative PCR using specific primers. All results were normalized with respect to the expression of GAPDH.*P<0.01 patients versus healthy controls (Anova), P<0.05 patients before HC versus patients after HC (Wilcoxon signed-rank test).(D) Effect of HC on LPS-induced ABIN-3 mRNA expression in primary human monocytes. Monocytes were selected by adherence from PBMC of healthy donors. Cells were stimulated with 100 ng/ml LPS for 20 hrs in the presence or the absence of 100 μM HC, which was given 2 hrs after LPS. Total RNA was isolated and ABIN-3 mRNA expression was analysed by real-time PCR. Results were normalized with respect to the expression of GAPDH and represent the mean ± SD of three experiments performed on cells from different donors.(E) Protein expression of ABIN-3 in monocytes isolated from healthy donors. Cells were stimulated with 100 ng/ml LPS for 20 hrs in the presence or the absence of 100 M HC, which was given at the same time as LPS. Cell extracts were separated by SDS-PAGE and analysed by Western blotting and immunodetection with anti-ABIN-3 polyclonal antibodies. β-actin expression was used as a control for equal loading.
To further investigate ABIN-3 expression under more physiological conditions, we compared ABIN-3 mRNA expression by real-time quantitative PCR in monocytes from healthy patients versus sepsis patients (Fig. 1C). As a positive control, we also analysed the expression of single Ig IL-IR-related molecule (SIGIRR) and MyD88s, which are two other negative regulators of NF-κB activation whose expression was previously shown to be specifically up-regulated in monocytes from sepsis patients [19]. In addition, we also analysed ABIN-3, SIGIRR and MyD88s expression in monocytes from sepsis patients that had received low doses of hydrocortisone (HC) treatment, which is currently used for septic shock patients showing adrenal insufficiency [20, 21]. The patients characteristics were the following: six patients with septic shock were studied; they were two males and four females with a mean age of 65 ± 3.5 years, a mean SAPS II score of 63 ± 6, and mean SOFA score 10.6 ± 0.9. The site of sepsis was pneumonia (n = 4), peritonitis (n = 1), and an endocarditis in the last case. Mean serum cortisol was 41 ± 10.5 μg/dl with only one responder defined as serum increase higher than 9 μg/dl after a corticotrophin test (250 μg bolus). This replacement therapy with low doses of corticosteroids was discontinuated later on. The patients were compared with 12 healthy controls (four males) with a mean age of 36±2 years. Similar to SIGIRR and MyD88s, ABIN-3 expression was significantly up-regulated in monocytes from sepsis patients before they received the low-dose corticotherapy. These results demonstrate the up-regulation of multiple NF-κB inhibitory proteins during sepsis, consistent with the previously described re-programming of monocytes to a hyporesponsive state during sepsis [22]. Interestingly, ABIN-3 expression in monocytes from the same patients was profoundly decreased 24 hrs after the onset of the corti-cotherapy, and came back to the levels found for healthy controls. In contrast, corticotherapy had no effect on the up-regulation of SIGIRR and MyD88s. To demonstrate that the inhibitory effect of HC on ABIN-3 expression was due to a direct effect on the monocytes, we also tested its effect on the LPS-induced expression of ABIN-3 mRNA in cultured primary monocytes from healthy patients (Fig. 1D). HC was given at the same time of LPS or 2 hrs later, with the latter more closely mimicing the clinical situation. In both cases, HC treatment significantly inhibited the stimulatory effect of LPS on ABIN-3 mRNA expression, indicating that the reduced monocyte ABIN-3 expression upon treatment of sepsis patients with HC is at least partially due to a direct effect on the patient's monocytes. We were unable to analyse ABIN-3 protein expression in monocytes from sepsis patients because it was ethically not justified to obtain a sufficient number of cells from these patients. However, we analysed ABIN-3 protein expression in LPS and HC-treated monocytes from healthy donors. Consistent with the effects of LPS and HC on ABIN-3 mRNA expression in primary human monocytes (Fig. 1D), we could also demonstrate in these cells the LPS-induced expression of ABIN-3 at the protein level, as well as its inhibition by HC treatment (Fig. 1E).
NF-κB inhibitors prevent the up-regulation of ABIN-3 in response to LPS
Because corticosteroids are known to negatively regulate NF-κB activation [23], and because the patho-physiologic mechanism behind the clinical effects of corticosteroids in septic shock has been directly related to the inhibition of NF-κB in PBMC [24], we hypothesized a role for NF-κB in the regulation of ABIN-3 expression. To further investigate this, we tested the effect of pre-treatment of THP-1 cells with MG-132 and sc-514 on LPS-induced ABIN-3 mRNA and protein expression. MG-132 is a proteasome inhibitor that inhibits NF-κB activation by preventing the proteasome-dependent degradation of IκBα, thereby preventing nuclear translocation of NF-κB [25]. Sc-514 is a selective IKKβ inhibitor that binds specifically to the ATP-binding site of IKKβ[26]. Consistent with our previous observations, LPS clearly induced ABIN-3 mRNA and protein expression in THP-1 cells (Fig. 2A and B). However, pretreatment of the cells with the NF-κB inhibitors MG-132 and sc-514 significantly inhibited the effect of LPS, whereas no effect was observed when the cells were pre-treated with the solvent dimethyl sulfoxide (DMSO). Similar inhibitory effects were obtained when THP-1 cells were pre-treated with HC (data not shown), which is consistent with the results obtained in primary human monocytes (Fig. 1E). Altogether, these results indicate that the LPS-induced expression of ABIN-3 is NF-κB dependent.
2.
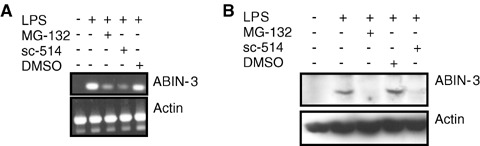
NF-κB inhibitors prevent the LPS-induced expression of ABIN-3. (A) Semi-quantitative RT-PCR of ABIN-3 mRNA. RNA was isolated from THP-1 cells that were either left untreated or stimulated for 3 hrs with 100 ng/ml LPS in the presence or the absence of 50 μM sc-514, 2.5 μM MG-132 or the solvent control DMSO, which were given 1 hr before LPS. Semi-quantitative RT-PCR was performed using ABIN-3 specific primers to amplify a 700 bp fragment of the ABIN-3 open-reading frame. β-actin was used as a control. (B) Western blotting for ABIN-3 protein expression. THP-1 cells that were either left untreated or stimulated for 6 hrs with 100 ng/ml LPS in the presence or the absence of 50 μM sc-514, 2.5 μM MG-132 or the solvent control DMSO, which were given 1 hr before LPS. Cell extracts were separated by SDS-PAGE and analysed by Western Blotting and immunodetection with anti-ABIN-3 polyclonal antibodies. β-actin expression was used as a control for equal loading.
NF-κB binding to the human ABIN-3 promoter
To further analyse the regulation of ABIN-3 expression by NF-κB, we isolated from the genomic DNA of HEK293 cells a 2130 basepairs(bp) long fragment of the ABIN-3 promoter region, still containing at its 3′ end 36 bp of the ABIN-3 cDNA, and cloned this fragment in the pXP2Δ2 vector in front of the Luc reporter gene to generate pXP2Δ2-promABIN-3. The sequence of the ABIN-3 promoter fragment was deposited to GenBank under accession number EU126609. Analysis of the cloned promoter fragment using MatInspector software (Genomatix Software GmbH, Munich; http://www.genomatix.de) did not result in the prediction of a core promoter region. No TATA-box was found in the proximity of the transcription initiation site. However, we could identify several putative transcription factor binding sites in the ABIN-3 promoter region (Fig. 3; MatInspector, Genomatix Software GmbH, Munich, perfect core similarity [=1] and optimized matrix similarity). Most importantly, also a putative NF-κB binding site (GGGAAATCCC) could be pinpointed between nt −50 and −60 starting from the transcription initiation site (core similarity = 1, matrix similarity = 0.973). To investigate whether the predicted B binding element in the ABIN-3 pro- moter binds NF-κB, we subjected nuclear extracts of LPS- and TNF-treated THP-1 cells to an electrophoretic mobility shift assay with respectively a wild type B (WT-κB) and a mutant B (mut-κB) oligonucleotide probe (B element GGGCCATCCC; mutated residues underlined) that were generated based on the predicted B binding sequence (GGGAAATCCC) of the ABIN-3 promoter. LPS and TNF both induced the rapid and sustained binding of NF-κB to the WT-κB probe, but not to the mut-κB probe (Fig. 4). Pre-incubation of the nuclear extract with anti-p65 specific antibodies before incubation with the 32P-labelled WT-κB probe induced a super-shift of the κB-specific band, confirming the presence of p65 in the κB element binding complex. Because the supershift was only partial, it is likely that other NF-κB family members are also able to bind the κB element in the ABIN-3 promoter. These results clearly illustrate the inducible binding of NF-κB to the ABIN-3 promoter in response to LPS and TNF treatment of human monocytes.
3.

Nucleotide sequence of the ABIN-3 promoter.2130 bp fragment of the ABIN-3 promoter still containing at its 3′ end 36 bp of the ABIN-3 cDNA (shown in italics). Binding sites for transcription factors were predicted using the MatInspector program. Only a selection of putative binding sites is shown. The putative κB element is shown in bold.
4.
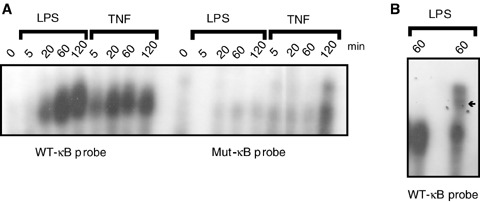
NF-κB binds to the κB site in the ABIN-3 promoter.(A) THP-1 cells were stimulated for the indicated times with 100 ng/ml LPS or 1000 IU/ml TNF. Nuclear extracts were prepared and analysed by electrophoretic mobility shift assay using 32P-labelled WT-κB or mut-κB probes corresponding to the κB site in the ABIN-3 promoter.(B) Binding of p65 NF-κB to the WT-κB probe was demonstrated by a supershift upon pre-incubation of the nuclear extract (obtained from cells treated for 60 min.with LPS) with anti-p65 NF-κB antibodies. The arrow indicates the location of the supershifted NF-κB complex.
The κB element in the ABIN-3 promoter is functionally active
We showed above that ABIN-3 expression can be up-regulated by LPS and TNF in a NF-κB dependent way and that NF-κB can bind to the B site in the ABIN-3 promoter. To confirm that the identified B element of the ABIN-3 promoter is functionally active and involved in the LPS- and TNF-induced expression of ABIN-3, we transiently transfected HEK293T cells with the promoterless pXP2Δ2 vector [27], or with pXP2Δ2-promABIN-3 or pXP2Δ2-promABIN-3mutNF-κB, respectively containing the ABIN-3 WT-B and mut-κB promoter fragment in front of a Luc reporter gene, and analysed the corresponding cellular extracts for Luc activity. Transfected cells were either left untreated or stimulated with 1000 IU/ml TNF for 6 hr. Because HEK293T cells are LPS-unresponsive, we mimicked the LPS signal by overex-pression of toll-like receptor 4 (TLR4), which can already activate NF-κB independent of LPS stimulation. In unstimulated cells, we could already observe a constitutive expression of the Luc reporter gene driven by the ABIN-3 promoter (Fig. 5A). Because we did not detect any constitutive endogenous ABIN-3 expression in these cells (data not shown), constitutive Luc expression driven by the 2130 bp promoter fragment most likely reflects the absence of negative regulatory elements or chromatin-mediated silencing, which can be expected to prevent constitutive expression of the endogenous ABIN-3 gene. However, both TNF as well as TLR4 overexpression resulted in a clear induction of the Luc reporter gene driven by the ABIN-3 promoter, which was not the case when cells were transfected with pXP2Δ2-promABIN-3mutNF-κB containing the mutated B element. Furthermore, overexpression of the p65 NF-κB subunit in HEK293T cells was also able to induce Luc expression from the WT-κB promoter construct, but not from the mut-κB promoter construct (Fig. 5B). Finally, cotransfection of different NF-B inhibitory proteins such as A20, IKKβ-K44A (a dominant-negative kinase dead mutant of IKKβ; [15]), and ABIN-3 itself, inhibited the TNF- and LPS-induced activation of the ABIN-3 promoter (Fig. 5C). In the experiment that is shown, IKKβ-K44A seems to inhibit also the constitutive activity of the ABIN-3 promoter. However, it should be mentioned that this is not a reproducible event. Because HC was found in this study to inhibit ABIN-3 expression in sepsis patients and LPS-treated primary monocytes, we also analysed the effect of HC on LPS-induced ABIN-3 promoter activation in RAW264.7 macrophages. Consistent with the stimulatory effect of TLR4 overexpression on ABIN-3 promoter activity in HEK293T cells, LPS treatment also increased the ABIN-3 promoter dependent expression of a luciferase reporter gene in RAW264.7 cells (Fig. 6). In addition, pre-treatment of these cells with HC significantly diminished the effect of LPS. Altogether, these results demonstrate that the TNF- and LPS-induced expression of ABIN-3 is at least partially mediated by the inducible binding of NF-κB to the κB element in the ABIN-3 promoter.
5.
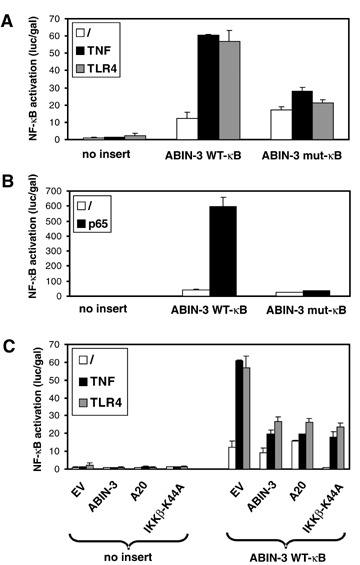
TNF, TLR4 and p65 induce the NF-κB-dependent activation of the ABIN-3 promoter. The Luc reporter vector pXPΔ2 containing either no insert, the WT-κB ABIN-3 promoter fragment or the mut-κB ABIN-3 promoter fragment was transiently transfected in HEK293T cells together with pAct gal. NF-κB was activated by treating the cells for 6 hrs with 1000 IU/ml TNF or by co-expression of TLR4 or p65 as indicated. In (C), cells were also transfected with expression vectors for ABIN-3, A20 or IKKβ-K44A as indicated. Luc activity and Gal activity in cell lysates were assayed 24 hrs after transfection and values are plotted as Luc/Gal to adjust for differences in transfection efficiency. Each error bar represents the mean ±SD of two samples. Results are representative for at least two independent experiments.‘/’: non-stimulated.
6.
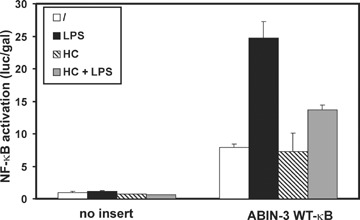
Hydrocortisone inhibits LPS-induced activation of the ABIN-3 promoter in macrophages. The Luc reporter vector pXP2Δ2 containing either no insert or the WT-κB ABIN-3 promoter fragment was transiently transfected in RAW264.7 cells together with pAct βgal. Cells were either left untreated or stimulated for 6 hrs with 1 μg/ml LPS in the presence or absence of 100 μM HC that was given 2 hrs prior to LPS. Luc activity and Gal activity in cell lysates were assayed 24 hrs after transfection and values are plotted as Luc/Gal to adjust for differences in transfection efficiency. Each error bar represents the mean ± SD of two samples. Results are representative for at least two independent experiments.‘/’: non-stimulated.
Discussion
ABIN-3 has previously been proposed as a negative regulator of TNF- and TLR-induced NF-κB activation whose expression is induced in monocytes or macrophages treated with different pro-inflammatory stimuli, and which can be superinduced by additional treatment with IL-10 [9, 11]. Here, we showed that ABIN-3 expression is also up-regulated upon TNF treatment of non-myeloid cell types. We also found a significantly enhanced expression of ABIN-3 in monocytes of sepsis patients, which is in accordance with the reported monocyte re-programming during sepsis [28]. It can be expected that enhanced expression of an NF-κB inhibitory protein, such as ABIN-3 contributes to the depressed immune status that is regularly reported in patients with sepsis or non-infectious systemic inflammatory response syndrome [29]. Many studies have addressed the intracellular signalling associated with the de-regulated production of cytokines in animal models of sepsis, but very few have attempted to decipher the mechanisms responsible of the altered immune status particularly in humans. Also IRAK-M [30], MyD88s [19] and SIGIRR [19], were previously shown to be expressed more rapidly in monocytes from sepsis patients as compared to healthy donors, which has been confirmed in the present study for MyD88s and SIGIRR. IRAK-M negatively regulates TLR signalling by associating with and preventing the dissociation of the IRAK1/IRAK4/TRAF6 complex from the TLR signalling complex [31]. MyD88s is an LPS-inducible splice variant of the TLR adaptor protein MyD88, and behaves as dominant-negative inhibitor of LPS-induced NF-κB activation by preventing the recruitment of IRAK-4 to the TLR signalling complex [32, 33]. SIGIRR represents a unique member of the TLR/IL-1 receptor family that functions as a negative regulator for IL-1 and LPS signalling through its interaction with the TLR4 and IL-1 receptor complex [34]. Although the up-regulation of all of these molecules in monocytes from sepsis patients suggests an important role, the specific contribution of each of these molecules in human sepsis remains to be fully established. In recent years, there is general use of low to moderate doses of corticosteroids in the treatment of septic shock [20, 21]. Interestingly, we noticed that corticosteroid treatment of sepsis patients restored ABIN-3 expression levels back to its level in healthy patients, whereas the levels of MyD88s and SIGIRR remained unaltered. It will therefore be interesting to study whether the beneficial effects of corticotherapy in the case of sepsis are somehow linked with its inhibitory effects on ABIN-3 expression.
The TNF- and LPS-inducible expression of ABIN-3 in monocytes observed in the present study could be prevented by NF-κB inhibitors, indicating an essential role for NF-κB in the regulation of ABIN-3 expression. The latter could be further validated upon isolation of a 2130 bp ABIN-3 promoter fragment and the identification of a functional κB element within the first 100 bp upstream of the transcription initiation site of the ABIN-3 gene. More specifically, site specific mutagenesis combined with gelshift and luciferase reporter experiments revealed the binding of NF-κB to this B element and its essential role in the LPS-and TNF-induced expression of ABIN-3. We could identify several putative consensus binding sequences for other transcription factors as well, including IFN regulatory factor (IRF)-1, IRF-2 and IRF-3, activator protein 1 (AP1), and three half sites for steroid receptors. Consistent with the inhibitory effect of corticotherapy on ABIN-3 expression in monocytes from sepsis patients and the inhibitory effect of HC on LPS-induced ABIN-3 expression in in vitro cultured monocytes, we also observed an inhibitory effect of HC on the LPS-induced activation of the ABIN-3 promoter. Whether this is mediated by the direct binding of glucocorticoid receptors to one of the steroid receptor binding sites in the ABIN-3 promoter or involves the interaction of glucocorticoid receptors with other transcription factors or the inhibition of NF-κB activation is currently unknown [23].
In conclusion, our data demonstrate the essential role of NF-κB in the TNF- and LPS-inducible expression of ABIN-3. Together with previous observations showing that ABIN-3 inhibits NF-κB activation in response to TNF and LPS, these data indicate an important role in the negative feedback regulation of TNF- and LPS-induced NF-κB activation in human cells. Dysregulation of ABIN-3 expression might therefore contribute to the development of sepsis or other inflammatory diseases.
Acknowledgments
This work was supported in part by grants from the ‘Interuniversitaire Attractiepolen’(IAP6/18), the ‘Fonds voor Wetenschappelijk Onderzoek-Vlaanderen’ (FWO; grant 3G010505), and the ‘Geconcerteerde Onderzoeksacties’of the Ghent University (GOA; grant 01G06B6). L. V. did the work with a predoctoral fellowship from the Fund for Scientific Research-Flanders and a fellowship from the Emmanuel Vanderschueren Foundation.
Ann Meeus is acknowledged for the technical assistance with cell culture work.
References
- 1.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann A, Natoli G, Gosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–16. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 3.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–48. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 4.Lang T, Mansell A. The negative regulation of Toll-like receptor and associated pathways. Immunol Cell Biol. 2007;85:425–34. doi: 10.1038/sj.icb.7100094. [DOI] [PubMed] [Google Scholar]
- 5.Wullaert A, Heyninck K, Janssens S, Beyaert R. Ubiquitin: tool and target for intracellular NF-kappaB inhibitors. Trends Immunol. 2006;27:533–40. doi: 10.1016/j.it.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, Haegeman G, Beyaert R. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol. 1999;145:1471–81. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Huffel S, Delaei F, Heyninck K, De Valck D, Beyaert R. Identification of a novel A20-binding inhibitor of nuclear factor-kappa B activation termed ABIN-2. J Biol Chem. 2001;276:30216–23. doi: 10.1074/jbc.M100048200. [DOI] [PubMed] [Google Scholar]
- 9.Wullaert A, Verstrepen L, Van Huffel S, Adib-Conquy M, Cornelis S, Kreike M, Haegman M, El Bakkouri K, Sanders M, Verhelst K, Carpentier I, Cavaillon JM, Heyninck K, Beyaert R. LIND/ABIN-3 is a novel lipopolysaccharide-inducible inhibitor of NF-kappaB activation. J Biol Chem. 2007;282:81–90. doi: 10.1074/jbc.M607481200. [DOI] [PubMed] [Google Scholar]
- 10.Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–8. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 11.Weaver BK, Bohn E, Judd BA, Gil MP, Schreiber RD. ABIN-3: a molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol Cell Biol. 2007;27:4603–16. doi: 10.1128/MCB.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6) J Exp Med. 1998;187:2097–101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura A, Israel A, Le Bail O, Kourilsky P. Detailed analysis of the mouse H-2Kb promoter: enhancer-like sequences and their role in the regulation of class I gene expression. Cell. 1986;44:261–72. doi: 10.1016/0092-8674(86)90760-9. [DOI] [PubMed] [Google Scholar]
- 14.De Valck D, Heyninck K, Van Criekinge W, Vandenabeele P, Fiers W, Beyaert R. A20 inhibits NF-kappaB activation independently of binding to 14-3-3 proteins. Biochem Biophys Res Commun. 1997;238:590–4. doi: 10.1006/bbrc.1997.7343. [DOI] [PubMed] [Google Scholar]
- 15.Zandi E, Rothwarf DM, Delhase M, Hayakawasa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–52. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 16.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 17.Le Gall JR, Lemeshow S, Saulnier F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270:2957–63. doi: 10.1001/jama.270.24.2957. [DOI] [PubMed] [Google Scholar]
- 18.Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, Reinhart CK, Suter PM, Thijs LG. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care. Medicine Intensive Care Med. 1996;22:707–10. doi: 10.1007/BF01709751. [DOI] [PubMed] [Google Scholar]
- 19.Adib-Conquy M, Adric C, Fitting C, Gattolliat O, Beyaert R, Cavaillon JM. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med. 2006;34:2377–85. doi: 10.1097/01.CCM.0000233875.93866.88. [DOI] [PubMed] [Google Scholar]
- 20.Annane D, Sébille V, Troché G, Raphaël JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283:1038–45. doi: 10.1001/jama.283.8.1038. [DOI] [PubMed] [Google Scholar]
- 21.Annane D, Sébille V, Charpentier C, Bollaert PE, François B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troché G, Chaumet-Riffaut P, Bellissant E. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–71. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Morrison DC. Lipopolysaccharide structure-function relationship in activation versus reprogramming of mouse peritoneal macrophages. J Leukoc Biol. 1993;54:444–50. doi: 10.1002/jlb.54.5.444. [DOI] [PubMed] [Google Scholar]
- 23.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 24.Van Leeuwen HJ, Van Der Bruggen T, Van Asbeck BS, Boerboom FT. Effect of corticosteroids on nuclear factor-kappaB activation and hemodynamics in late septic shock. Crit Care Med. 2001;29:1074–7. doi: 10.1097/00003246-200105000-00041. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 26.Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, Bonar S, Mielke C, Albee L, Weier R, Graneto M, Hanua C, Perry T, Tripp CS. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278:32861–71. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- 27.Grimm SL, Nordeen SK. Luciferase reporter gene vectors that lack potential AP-1 sites. Biotechniques. 1999;27:220–2. doi: 10.2144/99272bm01. [DOI] [PubMed] [Google Scholar]
- 28.Muñoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–54. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112:235–43. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 30.Escoll P, Del Fresno C, Garcia L, Valles G, Lendinez M, Arnalich F, Lopez-Collazo E. Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun. 2003;311:465–72. doi: 10.1016/j.bbrc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi K, Hernandez LD, Galan JE, Janeway CAJ, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 32.Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr Biol. 2002;12:467–71. doi: 10.1016/s0960-9822(02)00712-1. [DOI] [PubMed] [Google Scholar]
- 33.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–8. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin J, Qian Y, Yao J, Grace C, Li X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem. 2005;280:25233–41. doi: 10.1074/jbc.M501363200. [DOI] [PubMed] [Google Scholar]