

Abstract
A highly sensitive, specific and robust method for the analysis of oxidized metabolites of linoleic, acid (LA), arachidonic acid (AA) and docosahexaenoic acid (DHA) was developed using charge-switch derivatization, LC-ESI MS/MS with selected reaction monitoring (SRM) and quantitation by high mass accuracy analysis of product ions thereby minimizing interferences from contaminating ions. Charge-switch derivatization of LA, AA and DHA metabolites with N-(4-aminomethylphenyl)-pyridinium resulted in a 10-30 fold increase in ionization efficiency. Improved quantitation was accompanied by decreased false positive interferences through accurate mass measurements of diagnostic product ions during SRM transitions by ratiometric comparisons with stable isotope internal standards. The limits of quantification (LOQ) were between 0.05 to 6.0 pg with a dynamic range of 3-4 orders of magnitude (correlation coefficient r2 > 0.99). This approach was utilized to quantitate the levels of representative fatty acid metabolites from WT and iPLA2γ−/− mouse liver identifying the role of iPLA2γ in hepatic lipid 2nd messenger production. Collectively, these results demonstrate the utility of high mass accuracy product ion analysis in conjunction with charge-switch derivatization for the highly specific quantitation of diminutive amounts of LA, AA and DHA metabolites in biologic systems.
Keywords: Eicosanoids, Docosanoids, Metabolite Determination, LC-MS/MS, Phospholipase, iPLA2γ
Introduction
Polyunsaturated fatty acids (PUFAs) such as arachidonic acid (AA, 20:4n-6), docosahexaenoic acid (DHA, 22:6n-3) and linoleic acid (LA, 18:2n-6) are essential components of mammalian membranes that serve multiple roles in cellular function [1-5]. Typically, these polyunsaturated fatty acids are stored in an inactive form esterified to membrane phospholipids and are released during cellular activation by a wide variety of phospholipases to facilitate cellular responses to external perturbations [6-10]. The precise complement of lipid 2nd messengers in each cell type is determined, in large part, by the integrated array of cell type-specific phospholipases and the kinetic flux through specific downstream oxidative pathways tailored to the function of each cell type to generate a highly specific array of polyunsaturated oxidized signaling metabolites. Prominent examples of oxidative enzymes leading to the generation of signaling fatty acids include the cyclooxygenases [11, 12], lipoxygenases [13-15] and P450 monooxygenases [16, 17]. Many hundreds to thousands of oxidized fatty acid metabolites are generated in mammalian cells that coordinate physiologic signaling processes and cellular adaptation in health, but are subject to maladaptive alterations in disease processes promoting disease progression [18-20]. Accordingly, a major goal of lipid 2nd messenger research is the identification of the diversity of oxidized fatty acids in biologic systems, quantitation of alterations in their amounts during physiologic and pathologic perturbations, and mechanistic identification of their roles in disease processes [e.g., 21-29].
Recent approaches for the identification and quantification of the diversity of oxidized fatty acids in biologic systems have largely relied on liquid chromatographic separation followed by electrospray ionization in the negative ion mode and selected reaction monitoring (SRM) [30-38]. However, ionization in the negative ion mode is inefficient in comparison to ionization in the positive ion mode. Although oxidized fatty acids in inflammatory cells, abscesses, exudates or inflammatory fluids can be measured using negative mode ionization [39-51], the much lower abundance of oxidized fatty acid signaling metabolites that regulate cellular function in muscle, liver, brain and other solid organs are typically 20 - 2000 fold lower than that present in inflammatory cells or fluids. Thus, the substantially lower ionization efficiency in the negative ion mode has placed severe limitations on the accurate identification and quantitation of oxidized fatty acids in non-inflammatory cells and tissues.
The inherent limitations in sensitivity in the analysis of oxidized fatty acids in the negative ion mode have previously been appreciated. Accordingly, multiple charge-switch chemical derivatization procedures to improve the ionization efficiency for detection of oxidized fatty acid signaling metabolites have been developed using derivatization and positive ion mode ionization [e.g., 52-59]. Examples of charge switch derivatization of the carboxylic acid group include derivatization with tris(2,4,6-trimethoxyphenyl)phosphonium propylamine (TMPP) bromide [52], cholamine [53], 2-bromo-1-methylpyridinium iodide (BMP) and 3-carbinol-1-methylpyridinium iodide (CMP) [54], p-dimethylaminophenacyl bromide (DmPA-Br) [55], 2-Hydrazinopyridine (HP), 2-picolylamine (PA) and others [56-58]. However, these approaches are suboptimal for analysis of oxidized fatty acid moieties using ESI due to the undesirable fragmentation of the charge switch tag in many cases. This weakness was recognized by Gelb et al. who utilized molecular orbital calculations to develop a novel charge-switch reagent, N-(4-aminomethylphenyl)-pyridinium (AMPP) [59], which possessed the desired charge switch mediated increase in S/N and markedly improved the generation of informative fragment ions. Through using high mass accuracy product ion analysis in conjunction with AMPP charge–switch derivatization we demonstrate the sensitivity and specificity of this method for the detection and quantitation of oxidized linoleic, arachidonic, and docosahexaenoic acid metabolites in the high attomole and low fmol range. Furthermore, high mass accuracy analysis of diagnostic product ions collected during SRM dramatically decreases false positive interferences which compromise identification and quantitation of the targeted analytes that would otherwise go undetected or over estimated using traditional isobaric SRM analyses. The utility of this approach in biological tissues was examined in hepatic tissue from wild-type and iPLA2γ−/− mice we previously generated and characterized [60-62]. Collectively, this approach provides a facile method to dramatically increase the sensitivity and accuracy of analysis of diminutive amounts of oxidized fatty acids in biologic samples while simultaneously decreasing false positive interferences through the use of high mass accuracy product ion analysis during monitored SRM transitions.
Materials and Methods
Materials
The following standards from Cayman Chemical (Ann Arbor, MI) were used: thromboxane B2 (TXB2), TXB2-d4, 6-keto prostaglandin F1α (6keto-PGF1α), prostaglandin F2α (PGF2α), prostaglandin F1α (PGF1α), prostaglandin E2 (PGE2), PGE2-d4, prostaglandin E1 (PGE1), prostaglandin D2 (PGD2), leukotriene B4 (LTB4), LTB4-d4, 5-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-HETE), 8-hydroxy-5Z,9E,11Z,14Z-eicosatetraenoic acid (8-HETE), 11-hydroxy-5Z,8Z,12E,14Z-eicosatetraenoic acid (11-HETE), 12-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid (12-HETE), 12-HETE-d8, 15-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HETE), 20-hydroxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid (20-HETE), 5(6)-epoxy-8Z, 11Z,14Z-eicosatrienoic acid (5,6-EET), 8(9)-epoxy-5Z,11Z,14Z-eicosatrienoic acid (8,9-EET), 11(12)-epoxy-5Z,8Z,14Z-eicosatrienoic acid (11,12-EET), 14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid (14,15-EET), arachidonic acid (AA), 9-hydroxy-10E,12Z-octadecadienoic acid (9-HODE), 13-hydroxy-9Z,11E-octadecadienoic acid (13-HODE), 13-HODE-d4, 9-oxo-10E,12Z-octadecadienoic acid (9-OxoODE), 13-oxo-9Z,11E-octadecadienoic acid (13-OxoODE), 9(10)-dihydroxy-12Z-octadecenoic acid (9,10-DiHOME), 9,10-DiHOME-d4, 12,13-dihydroxy-9Z-octadecenoic acid (12,13-DiHOME), 12,13-DiHOME-d4, 9(10)-epoxy-12Z-octadecenoic acid (9(10)-EpOME), 12(13)epoxy-9Z-octadecenoic acid (12(13)-EpOME), linoleic acid (LA), 4-hydroxy-5E,7Z,10Z,13Z,16Z,19Z-docosahexaenoic acid (4-HDoHE), 7-hydroxy-4Z,8E,10Z,13Z,16Z,19Z-docosahexaenoic acid (7-HDoHE), 8-hydroxy-4Z,6E,10Z, 13Z,16Z,19Z-docosahexaenoic acid (8-HDoHE), 10-hydroxy-4Z,7Z,11E,13Z,16Z,19Z-docosahexaenoic acid (10-HDoHE), 11-hydroxy-4Z,7Z,9E,13Z,16Z,19Z-docosahexaenoic acid (11-HDoHE), 13-hydroxy-4Z,7Z,10Z,14E,16Z,19Z-docosahexaenoic acid (13-HDoHE), 14-hydroxy-4Z,7Z,10Z,12E,16Z,19Z-docosahexaenoic acid (14-HDoHE), 16-hydroxy-4Z,7Z,10Z, 13Z,17E,19Z-docosahexaenoic acid (16-HDoHE), 19,20-dihydroxy-4Z,7Z,10Z,13Z,16Z-docosapentaenoic acid (19,20-DiHDPA), 10,17-dihydroxy-4Z,7Z,11E,13Z,15E,19Z-docosahexaenoic acid (10,17-DiHDoHE), 7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid (RvD1), 7S,16R,17S-trihydroxy-4Z,8E,10Z,12E,14E,19Z-docosahexaenoic acid (RvD2), and docosahexaenoic acid (DHA). AMPP was obtained from Alchem Laboratories Corporation (Alachua, FL). HPLC-grade acetonitrile, methanol, chloroform, and water were purchased from Burdick & Jackson (Muskegon, MI). Glacial acetic acid and N, N-dimethylformamide were obtained from Sigma-Aldrich (St Louis, MO) while N-hydroxybenzotriazole and 3-(dimethylamino)propyl)-ethyl carbodiimide hydrochloride were purchased from Advanced Chem Tech (Louisville, KY) and TCI America (Portland, OR), respectively. Ascentis Express C18 reverse phase high performance liquid chromatography (HPLC) columns (15cm × 2.1mm) were obtained from Supelco (Bellefonte, PA) while solid phase extraction Strata-X columns were purchased from Phenomenex (Torrance, CA).
Preparation of Standard Solutions and Quantitation
Stock solutions of reference standards were prepared at a concentration of 10-100 pg/μl in ethanol and stored in a brown glass vial under nitrogen at −80°C. The stock solutions were further diluted by ethanol to prepare standard working solutions. Appropriate amounts of internal standards of oxidized fatty acids in ethanol were injected into samples (e.g., biologic tissues and fluids) for quantitative analysis by ratiometric comparisons of the high mass accuracy diagnostic product ion areas to authentic standards. All samples were flash frozen and stored at −180°C prior to polytron homogenization at 0-4°C.
Hepatic Tissue Samples Preparation
Oxidized fatty acids were analyzed in hepatic tissue obtained from either control C57BL/6 mice or in C57BL/6 mice in which iPLA2γ was globally deleted by removal of the active site in exon 5 by homologous recombination as described previously [60]. All animal breeding, care and sacrifice were carried out by protocols approved by the Animal Studies Committee at Washington University School of Medicine. Following sacrifice, livers were immediately removed, quickly rinsed twice in ice-cold PBS (pH7.4) solution, blotted, snap-frozen in liquid nitrogen and stored at −180°C until homogenization and extraction. Samples were polytron homogenized at 0-4°C prior to extraction using a modified Bligh and Dyer procedure. Briefly, ~20 mg of hepatic tissue was extracted by addition of 2 ml of ice-cold MeOH/CHCl3 (1:1 v/v containing 2% HAc) followed directly by addition of 2 μl of an antioxidant mixture (0.2 mg/ml BHT, 0.2 mg/ml EDTA, 2 mg/ triphenylphosphine, and 2 mg/ml indomethacin in a solution of 2:1:1 methanol/ ethanol/H2O) and stable isotope internal standards (e.g., TXB2-d4, LTB4-d4,13-HODE-d4, 9,10-DiHOME-d4, 12,13-DiHOME-d4, PGE2-d4, and 12-HETE-d8). Next, the samples were polytron homogenized (at 0-4°C as described above), 1 ml of ice-cold H2O was added and the resultant mixture was centrifuged at 15,000g for 15 min to separate the organic and aqueous phases. The CHCl3 layer was transferred to a new tube while the aqueous layer was re-extracted by subsequent addition of 1 ml CHCl3, vortexed and layers were separated by centrifugation at 15,000g for 15 min. The CHCl3 extracts were combined, evaporated under a nitrogen stream and resuspended in 1 ml of 10% methanol/water for solid phase extraction.
Solid Phase Extraction
The sample extracts were immediately applied to a Strata-X solid phase extraction cartridge (30 mg/1ml) that had been previously preconditioned with 1 ml of methanol followed by sequential washing with 1 ml of 10% methanol/90% H2O and 2×1 ml of 5% methanol/95% H2O. The oxidized fatty acid metabolites were eluted with 2×0.5 ml washes with methanol containing 1% glacial acetic acid. The combined methanol washes were evaporated to dryness using a SpeedVac concentrator prior to charge-switch derivatization with N-(4-aminomethylphenyl)-pyridinium (AMPP).
Derivatization Reactions
Derivatization with AMPP was performed as previously described in detail by Gelb et al. [59]. In brief, 15 μl of ice-cold acetonitrile/N,N-dimethylformamide (4:1, v:v) and 15 μl of ice-cold 640 mM (3-(dimethylamino)propyl)ethyl carbodiimide hydrochloride in HPLC grade water were added to the residue in the sample vial. The vial was briefly vortexed and 30 μl of 5 mM N-hydroxybenzotriazole /15 mM AMPP in acetonitrile were added, vortexed and placed in a 60°C water bath for 30 min. Samples were then cooled down and kept in the autosampler rack at 4 °C while queued for injection.
LC-ESI MS/MS Analysis
LC-ESI MS/MS analysis was performed using an LTQ-Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) equipped with a Surveyor HPLC system (Thermo Scientific, San Jose, CA). Briefly, the derivatized metabolites were separated on a C18 reversed phase column (Ascentis Express, 2.7 μm particles, 150 × 2.1 mm) at 23°C using a linear gradient of solvent A (0.1% glacial acetic acid in water) and solvent B (0.1% glacial acetic acid in acetonitrile) at a flow rate of 0.2 ml/min. The following solvent gradient program was used: 0.0-5.0 min, 25% B; 5.0-7.0 min, 25-35% B; 7.0-20.0 min, 35-60% B; 20.0-20.1 min, 60-100% B; 20.1-24.0 min, 100% B; 24.0-25.0 min, 100% B to 25% B and 10 min isocratic hold at 25% B. The sample injection volume was 10 μl and the autosampler tray temperature was kept at 4°C throughout the analysis. Diagnostic product ion transitions were determined by full mass scans of targeted metabolites to identify informative transitions to monitor during chromatography. Quantitative analysis of the derivatized fatty acids metabolites was performed using LC-MS/MS by ratiometric comparisons of the high mass accuracy (< 5ppm) diagnostic product ions in samples to that of stable isotope internal standards.
Mass spectrometric analyses were carried out on an LTQ-Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). The LTQ ion source was operated in positive ion mode at sheath, auxiliary, and sweep gas flows (arbitrary units) of 40, 5, and 1, respectively. The capillary temperature was set to 275°C and the electrospray voltage was 4.0 kV. Capillary voltage and the tube lens voltage were set to 2 and 100 V, respectively. Resolving powers (at m/z 400 Th) of 30,000 in full scan mode and 7500 in MS/MS mode were used. The instrument was calibrated externally following the manufacturer’s instructions and mass accuracy was within 3 ppm at mass values from m/z 130 to 2000. For MS/MS analyses, a normalized collision energy of 30% was applied, the activation time was set at 30 ms with an activation parameter q = 0.25. Collision induced dissociation of derivatized oxidized fatty acid metabolites was initially performed by direct infusion of standards and analysis of full mass scan product ion spectra to identify diagnostic transitions. The identified informative precursor and product ion transitions were monitored by isolating the precursor and product ions within a range of ±1 Th. Product ions were collected in the C trap prior to injection into the Orbitrap for accurate mass analysis and ions within 5ppm of the exact mass of the targeted compound were quantified. AGC target settings for FT full mass scan, FT MS2 scan and ion trap MS2 scan were 5 × 105, 2 × 105 and 1 × 104, respectively. Data acquisition was performed using the Xcalibur operating system, Version 2.1 (Thermo Scientific).
Calibration Standards and Quantification
Data analysis was performed using Xcalibur Version 2.1 software. The linearity of the developed method was determined using calibration curves constructed from each of the authentic standards of the targeted metabolites. Metabolite quantitation was performed from ratiometric comparisons of accurate mass measurements corresponding to the accurate mass peak area of the metabolite to the accurate mass peak areas of stable isotope internal standards. Linear regression analysis determined r2 values of 0.99 or greater for each metabolite.
Statistical Analysis
Statistical analysis was performed using the two-tailed Student’s t test. P values of less than 0.05 were considered to be statistically significant. All data are reported as the mean ±SEM unless otherwise noted.
Results and Discussion
Development of LC-ESI MS/MS Methods for the Analysis of Oxidized Metabolites of Linoleic Acid, Arachidonic Acid and Docosahexaenoic Acid
First, authentic standards of LA, AA and DHA metabolites were extracted, derivatized with AMPP and prepared for mass spectrometric analysis by solid phase extraction as described in “Materials and Methods.” The RPHPLC separation of the AMPP-derivatized metabolites of LA, AA and DHA demonstrated excellent chromatographic characteristics during separation on a C18 reversed phase column at 23°C (Fig. 1). Next, each of the targeted oxidized fatty acid metabolites were analyzed by SRM in the LTQ and product ions resulting from diagnostic transitions were collected in the C trap. Product ions from each monitored transition were injected into the Orbitrap for accurate mass determination (<5ppm) to minimize false positive interferences. Examination of each of the representative oxidized derivatized fatty acid metabolites demonstrated full mass product ion fragmentation patterns in the LTQ Orbitrap that were similar to those using QqQ instruments [59], but differed substantially in the relative abundance of each of the product ions produced. This result is consistent with the differing modes of fragmentation produced in ion traps which induce bond cleavage by RF excitation vs. fragmentation in QqQ instruments which induce fragmentation by collisional activation with inert gases.
Figure 1. Selected reaction monitoring (SRM) total ion current chromatograms of the targeted oxidized metabolites of linoleic, arachidonic and docosahexaenoic acids.

The indicated fatty acid metabolite standards were derivatized with N-(4-aminomethylphenyl)-pyridinium (AMPP) and measured by LC/MS/MS via SRM in the positive ion mode following chromatographic separation using a C18 reverse phase HPLC column as described in “Materials and Methods”. Representative SRM chromatograms for linoleic acid (LA) and the indicated oxidized LA metabolites (A); arachidonic acid (AA) and the indicated oxidized AA metabolites (B and C); and docosahexaenoic acid (DHA) and the indicated oxidized DHA metabolites (D) are shown with their retention times and the informative SRM transitions.
The separation of AMPP charge-switch derivatized oxidized fatty acid products was optimized using a customized mobile phase gradient (described in “Materials and Methods”) which resulted in baseline resolution of the majority of targeted metabolites (Fig. 1). Diagnostic fragmentation products of the authentic standards of the targeted analytes were first identified using direct infusion and subsequently substantiated by fragmentation of column chromatographic eluents. Each of the oxidized fatty acids examined were either baseline resolved by RPHPLC or analytes that possessed overlapping chromatographic retention times were discriminated based on diagnostic SRM transitions. Ions corresponding to the monitored transitions were collected in the C trap and injected into the Orbitrap for accurate mass determination to minimize false positive interferences that are not related to the analyte of interest.
As previously discussed by Gelb et al. [59] the utility of the AMPP derivatization lies not only in its salutary effects of increased ionization efficiency from charge-switch derivatization that result in increased S/N, but moreover facilitates the accrual of multiple informative fragment ions that emanate from dual mechanisms of fragmentation of the AMPP tag. Specifically, the AMPP derivatization tag yielded markers for metabolites of derivatized fatty acids through monitoring the high mass accuracy fragmentation of the diagnostic ions present at m/z 183.0917 (benzenemethanimine pyridinium ion) and m/z 169.0886 (tolylpyridinium distonic radical ion) that can physically map the elution profile of candidate derivatized fatty acid metabolites as well as other metabolites containing a carboxylic group present during extraction (Scheme 1).
Scheme 1.
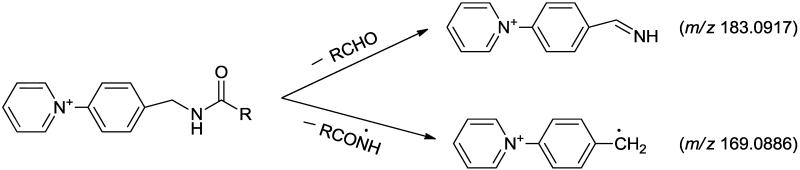
The salutary design of AMPP tag not only delivers markedly enhanced sensitivity but also results in concomitant fragmentations in both the analyte and tag portions as was envisioned using molecular orbital calculations performed by Gelb et al. [59]. As anticipated, this strategy works extremely well with multiple other oxidized fatty acid metabolites of great biologic significance. In some cases, chromatographic elution results in the incomplete resolution of metabolites with similar structures. To circumvent this difficulty, we identified diagnostic transitions that can be utilized to discriminate metabolites that are incompletely resolved. For example, 16- and 17-HDoHE as well as 10- and 11-HDoHE possessed overlapping chromatographic elution profiles which were discriminated by high mass accuracy determination of diagnostic product ions. Specifically, the unique transition in 16-HDoHE (m/z 511→ m/z 401, Fig. 2A) was not present in the coeluting 17-HDoHE (m/z 511→ m/z 441, Fig. 2B) thus allowing discrimination between each coeluting metabolite. As proposed in Scheme 2, formation of the ion at m/z 401.2587 likely results from the loss of 2, 4-heptadienal (C7H10O) from 16-HDoHE-AMPP).
Figure 2. Full mass product ion scans used to discriminate 16- and 17-HDoHE derivatized with N-(4-aminomethylphenyl)-pyridinium (AMPP).

(A) Mass spectrum of the CID of 16-DHoHE-AMPP exhibiting a prominent diagnostic fragment ion at m/z 401 which was utilized for the SRM transition based identification and quantitation of 16-DHoHE. (B) Mass spectrum of the CID of 17-DHoHE-AMPP displaying an informative fragment ion at m/z 441 which was utilized for the SRM transition based identification and quantitation of 17-DHoHE.
Scheme 2.

Similarly, the product ion at m/z 441.2537 was likely generated from the loss of 2-pentene (C5H10) from 17-HDoHE-AMPP. The unique transition of m/z 511→ m/z 321 for 10-HDoHE (Fig.3A) was selected to discriminate the coeluting metabolite 11-HDoHE for which the identified diagnostic transition of m/z 511→ m/z 361 was employed (Fig. 3B). The proposed fragmentation pathways for formation of the m/z 321.1961 and m/z 361.1911 ions from 10- and 11-HDoHE AMPP amides, due to loss of C13H18O and C11H18, respectively, are shown in Scheme 3.
Figure 3. Full mass product ion scans used to discriminate 10- and 11-HDoHE derivatized with N-(4-aminomethylphenyl)-pyridinium (AMPP).

(A) Mass spectrum of the CID of 10-HDoHE-AMPP exhibiting a diagnostic fragment ion at m/z 349 which was selected for the SRM transition based identification and quantitation of 10-HDoHE. (B) Mass spectrum of the CID of 11-HDoHE-AMPP displaying an informative fragment ion at m/z 361 which was selected for SRM transition based identification and quantitation of 11-HDoHE.
Scheme 3.

Accurate mass determination of product ions derived from SRM can substantively reduce false positive identifications. We and others [37] have identified difficulties in metabolite identification and quantitation using precursor and product ions with 1Th selection windows which have the potential to contain multiple different isobaric analytes/interferences in addition to the analyte of interest. In many cases, utilization of SRM results in relatively pure product ions as assessed by accurate mass determination. For example over 99% of murine 12-HETE in serum was authenticated by product ion accurate mass analysis thereby substantiating the integrity of the measured analyte using conventional procedures (Fig. 4A and B). In contrast, when a sample of mouse serum was analyzed for PGE1 using the identified SRM transition of m/z 521 → m/z 493 eluting with retention time 9.3 min (Fig. 4C and D), the resulting product ions from SRM demonstrated that although the majority of the product ions at the monitored transition were likely authentic PGE1 (exact mass=493.3425), two contaminating ions were also present at m/z 492.8650 and 492.5438 (each markedly outside of the 5 ppm accurate mass selection window). Calculations of the combined areas of the two contaminating peaks to that of authentic PGE1 demonstrated that an error of 27% was introduced by the presence of the contaminating ions. Accurate mass from SRM product ion determination eliminates the interfering ions at m/z 492.8650 and 492.5438, thereby providing more accurate quantitative results. Another example of the utility of accurate mass product ion analysis is demonstrated in the false positive identification of LTB4 from mesenteric arteries (Fig. 4E and 4F). Using accurate mass analysis of the product ions generated during the monitored SRM transition (m/z 503→ m/z 391) clearly eliminated the interference of the contaminating ion present at m/z 391.3361. This erroneous assignment of this ion contributes 44% of the total peak area that would be obtained using unit mass resolution during conventional isobaric SRM analysis. These results were not due to alterations resulting from the changes in the mode of ionization (negative to positive) or the derivatization reaction since false positive identification of 13,14-dihydro15keto PGF2α were previously documented in underivatized samples using RPHPLC and LC-MS/MS with SRM in the negative ion mode. Specifically, Volmer et al. reported that the ion peak at m/z 113 which was identified as 13,14-dihydro15keto PGF2α in chronic lymphocytic leukemia cells using SRM (m/z 353 → m/z 113) with unit mass resolution represented a false positive identification (Fig. 2;[37];). We have observed multiple similar errors in identification and quantitation using conventional SRM analysis with isobaric product ion determination which can be deconvoluted using product ion accurate mass analysis. In many cases this problem is more significant, affecting not only quantitation, but leading to false identification of the observed ion peak. For example (Fig. 5), when a sample of a murine mesenteric artery was analyzed for LTB4 (m/z 503 → m/z 391, retention time 10.45min), the results demonstrated abundant product ions resulting from this transition leading to the conclusion that LTB4 was present in mesenteric arteries. However, the accurate mass of the monitored product ion does not correspond to that of the product ion of LTB4 standard (i.e. m/z 391.2016) while the spiked authentic deuterated internal standard LTB4 was present in the anticipated amount as assessed by accurate mass (product ion accurate mass 393.2142 resulting from the transition of tetra deuterated LTB4 m/z 507 → m/z 393) (spectrum not shown). Thus, high mass accuracy determination eliminates the false positive identification of LTB4 that cannot be revealed using SRM with isobaric mass resolution. Thus, one approach to minimize false positive interferences is the use of high mass accuracy analysis of product ions collected during the monitored SRM transitions. Product ions which do not match the accurate mass of their isobarically assigned analytes do not represent the moiety of interest and instead may be generated in a variety of ways from different known and unknown metabolites, their fragmentation products or products from sequential fragmentations to give rise to unanticipated product ions during SRM analysis that do not result from the analyte of interest.
Figure 4. Comparison of quantitative methods for measuring selected eicosanoids in mouse serum using mass unit resolution of product ion SRM and accurate mass product ion SRM.

(A) Peak retention time profile and SRM transition (m/z 487 → m/z 347) of 12-HETE-AMPP with mass unit resolution; (B) Peak retention time profile and high mass accuracy extracted ion chromatogram of the product ion at m/z 347.2188 (within 5 ppm); (C): Peak retention time profile and SRM transition (m/z 521 → m/z 493) of PGE1-AMPP with mass unit resolution; (D) Peak retention time profile and high mass accuracy extracted ion chromatogram of the product ion at m/z 493.3425 (within 5 ppm); (E) Peak retention time profile and SRM transition (m/z 503 → m/z 391) of LTB4-AMPP with mass unit resolution; (F) Peak retention time profile and high mass accuracy extracted ion chromatogram of the product ion at m/z 391.2016 (within 5 ppm).
Figure 5. Determination of the false positive identification of LTB4 in a biologic sample utilizing SRM with accurate product ion mass analyses.

(A) Peak retention time profile of LTB4-AMPP standard utilizing SRM (m/z 503 → m/z 391); (B) high mass accuracy extracted ion chromatogram of the product ion, m/z 391.2016 (within an error of 2 ppm) of the standard LTB4-AMPP; (C) corresponding full MS/MS spectrum of the LTB4-AMPP standard; (D) Peak retention time profile indicative of LTB4-AMPP obtained from murine mesenteric arterial tissue utilizing the SRM transition (m/z 503 → m/z 391); (E) high mass accuracy extracted ion chromatogram of the product ion, m/z 391.3355, present in the biologic sample which does not correspond to the accurate mass of the product ion of LTB4-AMPP thereby revealing a false positive identification if only mass unit resolution is utilized (F). The MS/MS spectra of D and E confirm substantial differences between the LTB4 standard and compounds in the biological sample.
Recovery of Metabolites during Solid Phase Extraction, Linearity of Quantitation and the Limits of Quantitation Using High Mass Accuracy Product Ion Analysis
Recoveries of metabolites from Strata-X cartridges were measured following the extraction of samples of each metabolite by ratiometric comparison to high mass accuracy measurements of stable isotope internal standard. Extraction yields were obtained by comparing the LC-ESI MS/MS peak integrals to those obtained from AMPP derivatized samples that were directly injected onto the LC column. The extraction recoveries for each metabolite were excellent (Table 1). Using the optimized conditions in “Materials and Methods” as described above, the sensitivity, linearity and accuracy of the developed method were examined. Table 1 summarizes the linearity of quantitation, lower limit of quantitation, dynamic range and accuracy of quantitation. The limits of quantitation for LA and AA oxidized metabolites were between 0.05 to 1.0 pg and linear calibration ranges up 300 to 1000 pg were observed. The limits of quantitation for DHA oxidized metabolites were between 0.5 to 6.0 pg with linear calibration ranges up 500 to 1000 pg. Linear regression analysis determined correlation coefficient r2 values of 0.99 or greater for each analyte. Analytes were quantified using standard curves created through high mass accuracy (<5 ppm) determinations of peak areas of standards by ratiometric comparisons to high mass accuracy product ion determinations of metabolites. The accuracy was calculated as the percentage of measured value to the expected values in quality control samples. Table 1 lists the figures of merit for the developed high mass accuracy charge switch approach for oxidized fatty acid metabolites.
Table 1. Relative response factor (R/R0)a, linearity of measurement, correlation coefficient (r2), limits of quantitation (LOQs)b, extraction yield, and method accuracyc analyses.
| Analytes | Extraction Yield (%) |
Accuracy (% ± SD) |
R/R0 | LoQ (pg) | Calibrated range (pg) |
Correlation coefficient (r2 ) |
Internal standard |
|---|---|---|---|---|---|---|---|
| 9-HODE | 98 | 95.0 ± 8.5 | 1.10 ± 0.11 | 0.05 | 0.05–300 | 0.9977 | 13-HODE-d4 |
| 13-HODE | 90 | 105.0 ± 12.5 | 1.08 ± 0.08 | 0.07 | 0.07–300 | 0.9956 | 13-HODE-d4 |
| 9-OxoODE | 84 | 86.2 ± 5.0 | 0.72 ± 0.05 | 0.5 | 0.5-1000 | 0.9990 | 13-HODE-d4 |
| 13-OxoODE | 88 | 97.8.0 ± 7.3 | 0.88 ± 0.12 | 1.0 | 1.0–1000 | 0.9987 | 13-HODE-d4 |
| 9,10-DiHOME | 80 | 103.0 ± 15.0 | 1.05 ± 0.15 | 0.08 | 0.08-300 | 0.9991 | 9,10-DiHOME-d4 |
| 12,13-DiHOME | 77 | 94.2.0 ± 3.4 | 0.99 ± 0.10 | 0.04 | 0.04-300 | 0.9989 | 12,13-DiHOME-d4 |
| 9(10)-EpOME | 76 | 91.5 ± 9.5 | 0.78 ± 0.10 | 0.5 | 0.5-1000 | 0.9999 | 9,10-DiHOME-d4 |
| 12(13)-EpOME | 80 | 95.0 ± 8.5 | 0.75 ± 0.11 | 0.5 | 0.5-1000 | 0.9945 | 9,10-DiHOME-d4 |
| Resolvin D1 | 75 | 94.1 ± 10.4 | 0.40 ± 0.10 | 5.0 | 5.0-1000 | 0.9998 | PGE2-d4 |
| Resolvin D2 | 79 | 110.0 ± 14.8 | 0.55 ± 0.12 | 6.0 | 6.0-1000 | 0.9937 | PGE2-d4 |
| 10,17- DiHDoHEA |
85 | 98.0 ± 10.3 | 0.40 ± 0.08 | 0.5 | 0.5-500 | 0.9960 | PGE2-d4 |
| 19,20-DiHDPA | 103 | 97.2 ± 7.8 | 5.80 ± 0.75 | 0.5 | 0.5-500 | 0.9948 | PGE2-d4 |
| 17-HDoHE | 82 | 95.2 ± 10.1 | 1.20 ± 0.20 | 2.0 | 2.0–1000 | 0.9950 | 12-HETE-d8 |
| 16-HDoHE | 78 | 99.0 ± 3.5 | 1.25 ± 0.16 | 2.0 | 2.0–1000 | 0.9984 | 12-HETE-d8 |
| 14-HDoHE | 78 | 93.8 ± 8.8 | 1.18 ± 0.10 | 1.0 | 1.0–1000 | 0.9989 | 12-HETE-d8 |
| 13-HDoHE | 83 | 105.0 ± 11.2 | 1.32 ± 0.20 | 2.0 | 2.0–1000 | 0.9995 | 12-HETE-d8 |
| 11-HDoHE | 77 | 101.0 ± 8.5 | 1.15 ± 0.22 | 2.0 | 2.0–1000 | 0.9966 | 12-HETE-d8 |
| 10-HDoHE | 81 | 88.0 ± 7.8 | 1.40 ± 0.08 | 2.0 | 2.0–1000 | 0.9984 | 12-HETE-d8 |
| 8-HDoHE | 82 | 90.2 ± 9.9 | 1.05 ± 0.15 | 1.0 | 1.0–1000 | 0.9919 | 12-HETE-d8 |
| 7-HDoHE | 79 | 102.0 ± 8.5 | 1.20 ± 0.11 | 1.0 | 1.0–1000 | 0.9963 | 12-HETE-d8 |
| 4-HDoHE | 83 | 108.0 ± 4.5 | 1.00 ± 0.08 | 1.0 | 1.0–1000 | 0.9994 | 12-HETE-d8 |
| 6keto-PGF1α | 85 | 106.3 ± 8.5 | 0.95 ± 0.05 | 0.1 | 0.1–5000 | 0.9984 | TXB2-d4 |
| TXB2 | 88 | 100.2 ± 3.5 | 1.03 ± 0.03 | 0.1 | 0.1–500 | 0.9990 | TXB2-d4 |
| PGF2α | 78 | 98.1 ± 7.1 | 0.91 ± 0.03 | 0.4 | 0.4–500 | 0.9988 | PGE2-d4 |
| PGF1α | 80 | 108.2 ± 5.2 | 2.10 ± 0.03 | 0.1 | 0.1–500 | 0.9917 | PGE2-d4 |
| PGE2 | 77 | 99.1 ± 4.2 | 1.02 ± 0.03 | 0.3 | 0.3–500 | 0.9981 | PGE2-d4 |
| PGE1 | 73 | 103.1 ± 7.1 | 1.42 ± 0.11 | 0.2 | 0.2–500 | 0.9972 | PGE2-d4 |
| PGD2 | 73 | 92.1 ± 10.2 | 0.66 ± 0.07 | 0.5 | 0.5–1000 | 0.9981 | PGE2-d4 |
| LTB4 | 65 | 98.8 ± 9.1 | 1.02 ± 0.02 | 0.7 | 0.7–1000 | 0.9992 | LTB4-d4 |
| 20-HETE | 76 | 105.1 ± 3.1 | 2.20 ± 0.10 | 0.3 | 0.3–500 | 0.9977 | 12-HETE-d8 |
| 15-HETE | 68 | 97.2 ± 9.2 | 1.10 ± 0.18 | 0.6 | 0.6–1000 | 0.9953 | 12-HETE-d8 |
| 12-HETE | 66 | 99.9 ± 8.1 | 1.00 ± 0.05 | 0.7 | 0.7–1000 | 0.9969 | 12-HETE-d8 |
| 11-HETE | 73 | 97.9 ± 4.2 | 1.32 ± 0.13 | 0.5 | 0.5–1000 | 0.9973 | 12-HETE-d8 |
| 8-HETE | 80 | 103.1 ± 5.5 | 1.50 ± 0.12 | 0.5 | 0.5–1000 | 0.9972 | 12-HETE-d8 |
| 5-HETE | 70 | 108.4 ± 7.2 | 0.82 ± 0.08 | 0.8 | 0.8–1000 | 0.9999 | 12-HETE-d8 |
| 14,15-EET | 75 | 94.3.1 ± 4.4 | 0.91 ± 0.11 | 0.8 | 0.8–1000 | 0.9989 | 12-HETE-d8 |
| 11,12-EET | 80 | 98.9 ± 6.7 | 0.80 ± 0.08 | 0.8 | 0.8–1000 | 0.9982 | 12-HETE-d8 |
| 8,9-EET | 79 | 101.0 ± 9.9 | 0.75 ± 0.14 | 0.8 | 0.8–1000 | 0.9991 | 12-HETE-d8 |
| 5,6-EET | 85 | 106.2 ± 7.7 | 0.95 ± 0.08 | 0.5 | 0.5–1000 | 0.9990 | 12-HETE-d8 |
Relative response factor (R/R0) was calculated as the ratio of an equivalent amount of analyte (R) to its internal standard (R0) and expressed as the mean ± standard deviation (N=3).
LOQ was defined as 3 × the limit of detection which is expressed as pg injected on column resulting an signal to noise ratio of 3:1.
Method accuracies were calculated as the percent deviation from the expected concentrations and expressed as the mean ± standard deviation (n= 3 experiments).
Analysis of the Oxidized Metabolites of LA, AA and DHA in Hepatic Tissues from Wide-Type and iPLA2γ Genetic Knockout Mice
To demonstrate the utility of this approach in biologic samples, oxidized metabolites of LA, AA and DHA in hepatic tissues from wild-type mice and iPLA2γ−/− mice were examined. Using the developed method, we were able to identify and quantitate 37 targeted metabolites derived from LA, AA and DHA in murine liver (Fig. 6). The metabolites from linoleic acid (Fig. 6A) revealed significant decreases for 9(10)- and 12(13)-EpOME as well as their epoxide hydrolase products 9,10-DiHOME and 12,13-DiHOME (p < 0.05). In contrast, the hepatic content of 9- and 13-HODE and their allylic hydroxyl oxidization products (9- and 13-oxoODE) were unchanged in WT vs. iPLA2γ−/− mice. Analysis DHA metabolites (Fig. 6B) demonstrated specific alterations in hepatic docosanoids after genetic ablation of iPLA2γ. These included a 43% decrease of 19,20-DiHDPA which was the major DHA metabolite in liver, a 68% decrease in RvD2 and 17-HDoHE, a 52% decrease in 14-HDoHE, and a 55% decrease in 4-HDoHE. The other measured DHA oxidized molecular species were statistically unchanged in comparisons between iPLA2γ−/− mice and their WT littermates. Additionally, oxidized AA metabolites revealed significant decreases in the content of 5-, 8-, 11-, 12- and 15-HETEs in the iPLA2γ−/− mouse. In contrast, the content of EETs was unchanged in hepatic tissue of the iPLA2γ−/− mouse (Fig. 6C). Significant changes were also found in prostaglandins PGD2, TXB2, PGF2α, and 6keto-PGF1α. We also measured the content of non-esterified AA, LA and DHA in WT murine liver (AA, 14.1 ± 2.5 ng/mg; LA, 21.7 ± 4.2 ng/mg; and DHA, 34.8 ± 2.1 ng/mg) as well as liver from iPLA2γ−/− mice (AA, 10.9 ± 1.3 ng/mg; LA, 12.9 ± 1.1 ng/mg; and DHA, 24.8 ± 1.8 ng/mg). The results demonstrated the decreased polyunsaturated fatty acids in iPLA2γ−/− liver and indicate the important role of iPLA2γ in murine liver lipid signaling and metabolism.
Figure 6. Oxidized metabolites of linoleic acid, arachidonic acid and docosahexaenoic acid in hepatic tissues from wild-type (WT) and iPLA2γ Knockout (KO) mice.

Oxidized fatty acid metabolites from murine hepatic tissue were extracted into solvent, isolated by solid phase extraction, derivatized with AMPP and quantitated by LC/MS/MS via selected reaction monitoring (SRM) in the positive ion mode following separation of molecular species using a reverse phase column as described in “Materials and Methods”. Significant decreases in the production of multiple identified oxidized metabolites of LA, DHA and AA as a result of genetic ablation of iPLA2γ are displayed in panels A, B and C, respectively. Values presented are the mean ±S.E. and comparisons were made using Student’s t-test (n=4). *: P< 0.05 level, **: P< 0.01 level.
Conclusions
The identification and quantitation of diminutive amounts of oxidized fatty acid signaling metabolites in biologic tissues is essential to understanding the roles of lipid 2nd messengers in biologic systems. However, the identification and quantitation of the diminutive amounts of oxidized non-esterified fatty acids in non-inflammatory cells or exudates is challenging task. In this study, we have utilized a high mass accuracy charge-switch approach to gain deep penetrance into the extremely low abundance regime of oxidized fatty acids that enables their identification, improves quantitation and can be effectively multiplexed as the acquisition rate of tandem mass spectra in next generation instrumentation increases. Through the exquisite sensitivity of charge-switch derivatization in conjunction with the judicious use of informative transitions whose integrity is substantiated by high mass accuracy product ions analyses, new insights into the roles of oxidized fatty acids in cellular biology can be accrued.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants RO1HL41250 and RO1HL118639. R. W. G. has financial relationships with LipoSpectrum and Platomics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Benatti P, Peluso G, Nicolai R, Calvani M. Polyunsaturated Fatty Acids: Biochemical, Nutritional and Epigenetic Properties. J. Am. Coll. Nutr. 2004;23:281–302. doi: 10.1080/07315724.2004.10719371. [DOI] [PubMed] [Google Scholar]
- [2].Gillies PJ, Harris WS, Kris-Etherton PM. Omega-3 fatty acids in food and pharma: the enabling role of biotechnology. Curr. Atheroscler. Rep. 2011;13:467–473. doi: 10.1007/s11883-011-0206-z. [DOI] [PubMed] [Google Scholar]
- [3].Jenkins CM, Cedars A, Gross RW. Eicosanoid signaling pathways in the heart. Cardiovasc. Res. 2009;82:240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sears B, Ricordi C. Role of fatty acids and polyphenols in inflammatory gene transcription and their impact on obesity, metabolic syndrome and diabetes. Eur. Rev. Med. Pharmacol. Sci. 2012;16:1137–54. [PubMed] [Google Scholar]
- [5].Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS - Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metabolism. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gross RW. High plasmalogen and arachidonic acid content of canine myocardial sarcolemma: a fast atom bombardment mass spectroscopic and gas chromatography-mass spectroscopic characterization. Biochemistry. 1984;23:158–165. doi: 10.1021/bi00296a026. [DOI] [PubMed] [Google Scholar]
- [7].Hazen SL, Ford DA, Gross RW. Activation of a membrane-associated phospholipase A2 during rabbit myocardial ischemia which is highly selective for plasmalogen substrate. J. Biol. Chem. 1991;266:629–5633. [PubMed] [Google Scholar]
- [8].Ford DA, Hazen SL, Saffitz JE, Gross RW. The rapid and reversible activation of a calcium-independent plasmalogen-selective phospholipase A2 during myocardial ischemia. J. Clin. Invest. 1991;88:331–335. doi: 10.1172/JCI115296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gross RW, Han X. Lipidomics at the Interface of Structure and Function in Systems Biology. Chem. Biol. 2011;18:284–291. doi: 10.1016/j.chembiol.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ziboh VA, Miller CC, Cho Y. Metabolism of polyunsaturated fatty acids by skin epidermal enzymes: generation of antiinflammatory and antiproliferative metabolites. Am. J. Clin. Nutr. 2000;71:S361S–366. doi: 10.1093/ajcn/71.1.361s. [DOI] [PubMed] [Google Scholar]
- [11].Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J. Lipid Res. 2009;50:S29–34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- [13].Fürstenberger G, Krieg P, Müller-Decker K, Habenicht AJ. What are cyclooxygenases and lipoxygenases doing in the driver’s seat of carcinogenesis? Int. J. Cancer. 2006;119:2247–2254. doi: 10.1002/ijc.22153. [DOI] [PubMed] [Google Scholar]
- [14].Radmark O, Samuelsson B. 5-Lipoxygenase: mechanisms of regulation. J. Lipid Res. 2009;50:S40–45. doi: 10.1194/jlr.R800062-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pidgeon GP, Lysaght J, Krishnamoorthy S, Reynolds JV, O’Byrne K, Nie D, Honn KV. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–524. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- [16].Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- [17].Sarkis A, Roman RJ. Role of cytochrome P450 metabolites of arachidonic acid in hypertension. Curr. Drug Metab. 2004;5:245–256. doi: 10.2174/1389200043335603. [DOI] [PubMed] [Google Scholar]
- [18].Moon SH, Jenkins CM, Liu X, Guan S, Mancuso DJ, Gross RW. Activation of mitochondrial calcium-independent phospholipase A2γ (iPLA2γ) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J. Biol. Chem. 2012;287:14880–14895. doi: 10.1074/jbc.M111.336776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Greene ER, Huang S, Serhan CN, Penigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins and Other Lipid Mediators. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- [21].Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc. Natl. Acad. Sci. USA. 2009;44:18820–18824. doi: 10.1073/pnas.0905415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Patwardhan AM, Akopian AN, Ruparel NB, Diogenes A, Weintraub ST, Uhlson C, Murphy RC, Hargreaves KM. Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J. Clin. Invest. 2010;120:1617–26. doi: 10.1172/JCI41678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells autacoids in anti-inflammation. J. Biol. Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- [25].Bailes JE, Mills JD. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head Injury model. J. Neurotrauma. 2010;27:1617–1624. doi: 10.1089/neu.2009.1239. [DOI] [PubMed] [Google Scholar]
- [26].Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007;28:176–183. doi: 10.1016/j.it.2007.02.007. [DOI] [PubMed] [Google Scholar]
- [27].Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, Toner M, Serhan CN. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J. Immunol. 2008;181:8677–8687. doi: 10.4049/jimmunol.181.12.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, Serhan CN, Ji RR. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010;16:592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Takabatake M, Hishinuma T, Suzuki N, Chiba S, Tsukamoto H, Nakamura H, Saga T, Tomioka Y, Kurose A, Sawai T, Mizugaki M. Simultaneous quantification of prostaglandins in human synovial cell-cultured medium using liquid chromatography/tandem mass spectrometry. Prostaglandins Leukot. Essent. Fatty Acids. 2002;67:51–56. doi: 10.1054/plef.2002.0381. [DOI] [PubMed] [Google Scholar]
- [31].Deems RA, Buczynski MW, Bowers-Gentry RC, Harkewicz R, Dennis EA. Detection and quantitation of eicosanoids via high performance liquid chromatography/electrospray ionization mass spectrometry. Meth. Enzymol. 2007;432:59–82. doi: 10.1016/S0076-6879(07)32003-X. [DOI] [PubMed] [Google Scholar]
- [32].Harkewicz R, Dennis EA. Applications of mass spectrometry to lipids and membranes. Annu. Rev. Biochem. 2011;80:301–325. doi: 10.1146/annurev-biochem-060409-092612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Buczynski MW, Stephens DL, Bowers-Gentry RC, Grkovich A, Deems RA, Dennis EA. TLR-4 and sustained calcium agonists synergistically produce eicosanoids independent of protein synthesis in RAW264.7 cells. J. Biol. Chem. 2007;282:22834–22847. doi: 10.1074/jbc.M701831200. [DOI] [PubMed] [Google Scholar]
- [34].Dickinson JS, Murphy RC. Mass spectrometric analysis of leukotriene A4 and other chemically reactive metabolites of arachidonic acid. J. Am. Soc. Mass Spectrom. 2002;13:1227–1234. doi: 10.1016/S1044-0305(02)00456-7. [DOI] [PubMed] [Google Scholar]
- [35].Zhang J, Pearson T, Matharoo-Ball B, Ortori CA, Warren AY, Khan R, Barrett DA. Quantitative profiling of epoxyeicosatrienoic, hydroxyeicosatetraenoic, and dihydroxyeicosatetraenoic acids in human intrauterine tissues using liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Biochem. 2007;365:40–51. doi: 10.1016/j.ab.2007.03.001. [DOI] [PubMed] [Google Scholar]
- [36].Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass Spectrometry. Anal. Chem. 2009;81:8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Masoodi M, Eiden M, Koulman A, Spaner D, Volmer DA. Comprehensive lipidomics analysis of bioactive lipids in complex regulatory networks. Anal. Chem. 2010;82:8176–8185. doi: 10.1021/ac1015563. [DOI] [PubMed] [Google Scholar]
- [38].Kita Y, Takahashi T, Uozumi N, Nallan L, Gelb MH, Shimizu T. Pathway-oriented profiling of lipid mediators in macrophages. Biochem. Biophys. Res. Commun. 2005;330:898–906. doi: 10.1016/j.bbrc.2005.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gregus AM, Doolen S, Dumlao DS, Buczynski MW, Takasusuki T, Fitzsimmons BL, Hua XY, Taylor BK, Dennis EA, Yaksh TL. Spinal 12-lipoxygenase-derived hepoxilin A3 contributes to inflammatory hyperalgesia via activation of TRPV1 and TRPA1 receptors. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6721–6726. doi: 10.1073/pnas.1110460109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18820–18824. doi: 10.1073/pnas.0905415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gregus AM, Dumlao DS, Wei SC, Norris PC, Catella LC, Meyerstein FG, Buczynski MW, Steinauer JJ, Fitzsimmons BL, Yaksh TL, Dennis EA. Systematic analysis of rat 12/15-lipoxygenase enzymes reveals critical role for spinal eLOX3 hepoxilin synthase activity in inflammatory hyperalgesia. The FASEB J. 2013;27:1939–1949. doi: 10.1096/fj.12-217414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dolęgowska B, Blogowski W, Stępniewska J, Safranow K, Jakubowska K, Olszewska M. Presence of glucose in dialyzing fluid and synthesis of selected lipoxygenase-derived eicosanoids during hemodialysis. Int. Urol. Nephrol. 2012;44:1799–1804. doi: 10.1007/s11255-011-0089-5. [DOI] [PubMed] [Google Scholar]
- [44].Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kühn H, Hazen SL, Goodall AH, Hamali HA, Collins PW, O’Donnell VB. Phospholipid-esterified Eicosanoids Are Generated in Agonist-activated Human Platelets and Enhance Tissue Factor-dependent Thrombin Generation. J. Biol. Chem. 2010;285:6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schuck RN, Theken KN, Edin ML, Caughey M, Bass A, Ellis K, Tran B, Steele S, Simmons BP, Lih FB, Tomer KB, Wu MC, Hinderliter AL, Stouffer GA, Zeldin DC, Lee CR. Cytochrome P450-derived eicosanoids and vascular dysfunction in coronary artery disease patients. Atherosclerosis. 2013;227:442–448. doi: 10.1016/j.atherosclerosis.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Theken KN, Deng Y, Kannon MA, Miller TM, Poloyac SM, Lee CR. Activation of the acute inflammatory response alters cytochrome P450 expression and eicosanoid metabolism. Drug Metab. Dispos. 2011;39:22–29. doi: 10.1124/dmd.110.035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gauthier KM, Goldman DH, Aggarwal NT, Chawengsub Y, Falck JR, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in acetylcholine-induced relaxations of mouse arteries. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H725–H735. doi: 10.1152/ajpheart.00696.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bukhari IA, Shah AJ, Gauthier KM, Walsh KA, Koduru SR, Imig JD, Falck JR, Campbell WB. 11,12,20-Trihydroxy-eicosa-8(Z)-enoic acid: a selective inhibitor of 11,12-EET-induced relaxations of bovine coronary and rat mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1574–583. doi: 10.1152/ajpheart.01122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zarini S, Murphy RC. Biosynthesis of 5-oxo-6,8,11,14-eicosatetraenoic acid from 5-hydroperoxyeicosatetraenoic acid in the murine macrophage. J. Biol. Chem. 2003;278:11190–11196. doi: 10.1074/jbc.M208496200. [DOI] [PubMed] [Google Scholar]
- [50].Maekawa K, Hirayama A, Iwata Y, Tajima Y, Nishimaki-Mogami T, Sugawara S, Ueno N, Abe H, Ishikawa M, Murayama M, Matsuzawa Y, Nakanishi H, Ikeda K, Arita M, Taguchi R, Minamino N, Wakabayashi S, Soga T, Saito Y. Global metabolomic analysis of heart tissue in a hamster model for dilated cardiomyopathy. J. Mol. Cell Cardiol. 2013;59:76–85. doi: 10.1016/j.yjmcc.2013.02.008. [DOI] [PubMed] [Google Scholar]
- [51].Moita E, Gil-Izquierdo A, Sousa C, Ferreres F, Silva LR, Valentao P, Dominguez-Perles R, Baenas N, Andrade PB. Integrated Analysis of COX-2 and iNOS Derived Inflammatory Mediators in LPS-Stimulated RAW Macrophages Pre-Exposed to Echium plantagineum L. Bee Pollen Extract. PLoS ONE. 2013;8:e59131. doi: 10.1371/journal.pone.0059131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cartwright AJ, Jones P, Wolff JC, Evans EH. Derivatization of carboxylic acid groups in pharmaceuticals for enhanced detection using liquid chromatography with electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005;19:1058–1062. doi: 10.1002/rcm.1883. [DOI] [PubMed] [Google Scholar]
- [53].Lamos SM, Shortreed MR, Frey BL, Belshaw PJ, Smith LM. Relative quantification of carboxylic acid metabolites by liquid chromatography-mass spectrometry using isotopic variants of cholamine. Anal. Chem. 2007;79:5143–5149. doi: 10.1021/ac062416m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang WC, Adamec J, Regnier FE. Enhancement of the LC/MS analysis of fatty acids through derivatization and stable isotope coding. Anal. Chem. 2007;79:5150–5157. doi: 10.1021/ac070311t. [DOI] [PubMed] [Google Scholar]
- [55].Guo K, Li L. High-performance isotope labeling for profiling carboxylic acid containing metabolites in biofluids by mass spectrometry. Anal. Chem. 2010;82:8789–8793. doi: 10.1021/ac102146g. [DOI] [PubMed] [Google Scholar]
- [56].Higashi T, Ichikawa T, Inagaki S, Min JZ, Fukushima T, Toyo’oka T. Simple and practical derivatization procedure for enhanced detection of carboxylic acids in liquid chromatography-electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010;52:809–818. doi: 10.1016/j.jpba.2010.03.001. [DOI] [PubMed] [Google Scholar]
- [57].Pettinella C, Lee SH, Cipollone F, Blair IA. Targeted quantitative analysis of fatty acids in atherosclerotic plaques by high sensitivity liquid chromatography/tandem mass spectrometry. J. Chromatogr. B. 2007;850:168–176. doi: 10.1016/j.jchromb.2006.11.023. [DOI] [PubMed] [Google Scholar]
- [58].Johnson DW. Alkyldimethylaminoethyl ester iodides for improved analysis of fatty acids by electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2000;14:2019–2024. doi: 10.1002/1097-0231(20001115)14:21<2019::AID-RCM121>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [59].Bollinger JG, Thompson W, Lai Y, Oslund RC, Hallstrand TS, Sadilek M, Turecek F, Gelb MH. Improved sensitivity mass spectrometric detection of eicosanoids by charge reversal derivatization. Anal. Chem. 2010;82:6790–6796. doi: 10.1021/ac100720p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, Gross RW. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 2007;282:34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW. Genetic Ablation of Calcium-independent Phospholipase A2γ Leads to Alterations in Hippocampal Cardiolipin Content and Molecular Species Distribution, Mitochondrial Degeneration, Autophagy, and Cognitive Dysfunction. J. Biol. Chem. 2009;284:35632–35644. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F, Schappe T, Moore K, Han X, Abumrad NA, Gross RW. Genetic Ablation of Calcium-independent Phospholipase A2γ Prevents Obesity and Insulin Resistance during High Fat Feeding by Mitochondrial Uncoupling and Increased Adipocyte Fatty Acid Oxidation. J. Biol. Chem. 2010;285:36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]