

Abstract
Objective
NF-kB is a critical regulator of cell survival genes and the host inflammatory response. The purpose of this study was to investigate the role of enterocyte-specific NF-kB in sepsis through selective ablation of IkB kinase (IKK)-ß.
Design
Prospective, randomized, controlled study.
Setting
Animal laboratories in university medical centers.
Subjects and Interventions
Mice lacking functional NF-kB in their intestinal epithelium (Vil-Cre/Ikkßf/Δ) and wild type (WT) mice were subjected to sham laparotomy or cecal ligation and puncture (CLP). Animals were sacrified at 24 hours or followed seven days for survival.
Measurements and Main Results
Septic WT mice had decreased villus length compared to sham mice while villus atrophy was further exacerbated in septic Vil-Cre/Ikkßf/Δ mice. Sepsis induced an increase in intestinal epithelial apoptosis compared to sham mice which was further exacerbated in Vil-Cre/Ikkßf/Δ mice. Sepsis induced intestinal hyperpermeability in WT mice compared to sham mice, which was further exacerbated in septic Vil-Cre/Ikkßf/Δ mice. This was associated with increased intestinal expression of claudin-2 in septic WT mice, which was further increased in septic Vil-Cre/Ikkßf/Δ mice. Both, pro-inflammatory and anti-inflammatory cytokines were increased in serum following CLP, and IL-10 and MCP-1 levels were higher in septic Vil-Cre/Ikkßf/Δ mice than septic WT mice. All septic mice were bacteremic, but no differences in bacterial load were identified between WT and Vil-Cre/Ikkßf/Δ mice. To determine the functional significance of these results, animals were followed for survival. Septic WT mice had lower mortality than septic Vil-Cre/Ikkßf/Δ mice (47% vs. 80%, p<0.05). Anti-TNF administration decreased intestinal apoptosis, permeability and mortality in WT septic mice and a similar improvement in intestinal integrity and survival were seen when anti-TNF was given to Vil-Cre/Ikkßf/Δ mice.
Conclusions
Enterocyte-specific NF-kB has a beneficial role in sepsis by partially preventing sepsis-induced increases in apoptosis and permeability, which are associated with worsening mortality.
Keywords: Sepsis, gut, NF-kB, Tumor necrosis factor, apoptosis, permeability
INTRODUCTION
Sepsis is the most common cause of mortality in intensive care units, accounting for more than 210,000 deaths in the Unites States each year (1;2). The intestine has long been hypothesized to play a crucial role in the pathophysiology of sepsis and is frequently characterized as the “motor” of the systemic inflammatory response (3-5). Sepsis induces multiple derangements in the intestinal epithelium including increased epithelial apoptosis (6-13), barrier dysfunction (14-17), and production of cytokines (18). Notably, preventing sepsis-induced intestinal apoptosis improves survival 2- to 10-fold in septic peritonitis and pneumonia, although the mechanisms underlying this survival advantage remain unclear (8;11).
The transcription factor NF-kB regulates the transcription of multiple genes that play a central role in regulating both cell survival and the inflammatory response (19;20). Under normal conditions, inactive NF-kB dimers are primarily retained in the cytoplasm via interactions with inhibitors of kB (IkB) proteins (21). When a stimulus is received, IkB kinase (IKK), a complex comprised of two catalytic subunits, IKKα and IKKß, and the regulatory subunit, IKKγ/NEMO, phosphorylates the IkB proteins leading to their degradation. In turn, the NF-kB dimer is free to translocate into the nucleus. In the canonical NF-kB activation pathway, this process is regulated by IKKß (22), which can be activated by a broad range of microbial and host-derived ligands (23). Consequently, NF-kB binds to selective target gene promoters and induces transcriptional activation or suppression of pro-inflammatory and anti-apoptotic mediators (24). Of note, while disruption of the IKKß gene prevents activation of the canonical NF-kB pathway, whole animal knockouts of this gene result in embryonic lethality due to massive apoptosis in the developing liver driven by TNF (25).
NF-kB activation can play either a detrimental role or a beneficial role in the intestinal tract in response to injury and inflammation (26). Small molecule inhibitors of IKKß ameliorate disease in murine models of intestinal inflammation (27;28). Similarly, peptides that bind IKKγ/NEMO reduce the severity of intestinal inflammation in models of colitis (29;30). In contrast, conditional ablation of IKKß in intestinal epithelial cells leads to increased local inflammation and intestinal epithelial apoptosis in an acute colitis model (31). Similarly, enterocyte-specific ablation of IKKß leads to increased intestinal epithelial apoptosis and intestinal hyperpermeability following burn injury (32) and to increased apoptosis and mucosal injury following challenge with Clostridium difficile toxin A (33). Intestine-specific alterations induced by NF-kB inhibition may also be counterbalanced by systemic inflammatory changes as mice with enterocyte-specific ablation of IKKß subjected to ischemia/reperfusion injury have a marked increase in intestinal epithelial apoptosis but a simultaneous decrease in systemic inflammation (34).
Despite the importance of NF-kB activation in regulating epithelial cell pro-inflammatory genes and intestinal epithelial cell survival, little is known about its impact on survival in critical illness. This study examined the impact of enterocyte-specific deletion of IKKß in a murine model of peritonitis on both local gut integrity and systemic inflammation and how these changes affected survival from sepsis.
MATERIALS AND METHODS
Animals
Six to eight week old mice were used for all studies. Mice with selective ablation of IKKß in intestinal epithelial cells were created via Cre/lox recombination by crossing 129/SV mice with a conditional loss-of-function IKKß allele due to flanking exon 3 with two loxP sites (a generous gift from Dr. Michael Karin, UCSD (34)) to C57Bl/6 mice with expression of Cre recombinase under the control of the villin promoter (Jackson Laboratories). Resulting offspring either lacked functional NF-kB in their intestinal epithelium (Vil-Cre/Ikkßf/Δ) or had normal expression of epithelial NF-kB (Ikkßf/Δ, functional wild type referred to hereafter as WT). Of note, mice lacking enterocyte-specific IKKß develop normally without any apparent abnormalities of the gastrointestinal tract as long as they are kept under normal conditions (34). All animal studies were approved by the Animal Studies Committees of Emory University School of Medicine, Washington University School of Medicine, and University of Colorado Anschutz Medical Campus and were conducted in accordance with the National Institutes of Health guidelines for the use of laboratory animals.
Sepsis model
Peritonitis was induced in mice via cecal ligation and puncture (CLP) (35). Under isoflurane anesthesia, a small midline abdominal incision was made and the cecum was ligated immediately distal to the ileocecal valve. The cecum was then punctured twice with a 21-gauge needle, gently squeezed to extrude a small amount of stool, and replaced in the abdomen which was closed in layers. Sham mice were treated identically with the exception that the cecum was neither ligated nor punctured. All mice were injected subcutaneously with 1 ml of normal saline to account for insensible fluid losses that occurred during surgery. Animals were sacrificed at 24, 48 or 72 hours (acute studies) or followed 7 days for survival.
NF-kB p65 Activation
Intestinal epithelial cells were collected by scraping the mucosa with a glass slide followed by obtaining nuclear extracts according to manufacturer’s protocol (Nuclear Extract Kit, Active Motif, Carlsbad, CA). NF-kB p65 activation was determined by using the TransAM NF-kB p65 Transcription Factor Assay Kit (Active Motif) according to the manufacturer’s instructions.
Western blots were also performed to determine protein levels of phosphorylated NF-kB p65 in intestinal epithelial cells. Fresh mucosal scrapings were homogenized in a 5x volume of ice-cold homogenization buffer and total protein was extracted (12). Forty μg of protein were added to an equal volume of 2x Laemmli sample buffer and heated at 95°C for 5 min. Samples were run on polyacrylamide gels (Bio-Rad, Hercules, CA) at 180 V for 45 min, and protein was transferred to Immuno-Blot polyvinylidene difluoride membranes (Bio-Rad) at 80 V for 2 hr. Membranes were blocked with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (Sigma) for 1 hr at room temperature and then incubated with either rabbit polyclonal anti-NF-kB p-p65 or beta-actin (1:1000; Cell Signaling Technology) overnight at 4°C. After washing, membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1000; Cell Signaling Technology). Proteins were visualized via a chemiluminescent system (Pierce, Rockford, IL) and exposed to X-ray film.
Intestinal epithelial apoptosis
Apoptosis was detected using two complimentary techniques: active caspase-3 staining and morphologic analysis of hematoxylin and eosin-stained sections (13;36). For active caspase-3, sections were incubated in 3% hydrogen peroxide for 10 min, heated in Antigen Decloaker (Biocare Medical) for 45 min, blocked with 20% normal goat serum (Vector Laboratories) and then incubated with rabbit polyclonal anti-active caspase-3 (1:100; Cell Signaling Technology, Beverly, MA) overnight at 4° C. Sections were then incubated with goat anti-rabbit biotinylated secondary antibody (1:200; Vector Laboratories) for 30 min at room temperature followed by Vectastain Elite ABC reagent (Vector Laboratories) for 30 min. Sections were then developed with diaminobenzidine, and counterstained with hematoxylin. For morphological analysis, apoptotic cells were identified by characteristic abnormalities of cell shrinkage with nuclear condensation and fragmentation. Apoptotic intestinal epithelial cells were quantified in 100 contiguous crypts and 50 well-oriented villi per animal by an examiner blinded to specimen identity.
Morphological analysis
Villus length was determined on hematoxylin and eosin-stained jejunal sections by measuring the distance in μm from the crypt neck to the villus tip using Nikon Elements imaging software (Nikon Instruments, Melville, NY). A minimum of 12 well-oriented villi from each section were measured.
Intestinal inflammation
Intestinal inflammation was assayed by a pathologist blinded to section identity (ABF). Inflammation was scored using two independent scoring systems for intestinal inflammation (37;38). Sections were assessed for inflammatory infiltrate into both the lamina propria and submucosa.
Intestinal permeability
Permeability was measured in vivo to fluorescein isothiocyanate conjugated-dextran (FD-4, 22 mg/ml, molecular mass 4.4 kDa, Sigma, St. Louis, MO) (39). Animals were gavaged with 0.5 ml of FD-4 19 hours after laparotomy. At time of sacrifice (5 hours after gavage), blood was collected and centrifuged at 3000 rpm at 4°C for 20 min. Plasma (50 μl) was mixed with an equal volume of sterile phosphate-buffered saline (pH 7.4) and the concentration of FD-4 was determined by fluorospectrometry (NanoDrop 3300, Thermo Scientific, Wilmington, DE) with an excitation wavelength of 470 nm and an emission wavelength of 515 nm using serially diluted samples as standards.
Tight Junction Expression
Quantitative real-time PCR was used to evaluate gene expression of claudin-2, occludin, and ZO-1. Total RNA was isolated from whole jejunal tissue using the Illustra RNAspin Mini RNA Isolation Kit according to manufacturer’s protocol (GE Healthcare, Piscataway, NJ) and cDNA was synthesized from 0.5 μg of total RNA. Gene expression was detected using pre-developed TaqMan primers and probes (Applied Biosystems, Foster, CA) and run on an ABI 7900HT Sequence Detection System (Applied Biosystems). All samples were run in duplicate and normalized to expression of the endogenous control, glyceraldehyde-3-phosphate (GAPDH) (Applied Biosystems). Relative quantification of PCR products were based upon the value differences between the target gene and GAPDH using the comparative CT method.
Cellular localization of claudin-2 was also evaluated by fluorescent immunohistochemistry. Intestinal sections were incubated in 3% hydrogen peroxide for 10 min, heated in Antigen Decloaker (Biocare Medical, Concord, CA) for 45 min for antigen retrieval, blocked with 20% normal goat serum (Vector Laboratories, Burlingame, CA) and incubated for 3 hours at room temperature with rabbit polyclonal anti-claudin-2 (1:200; Abcam, Cambridge, MA). Slides were then incubated in goat anti-rabbit Alexa 594-conjugated secondary antibody (1:200, Invitrogen) for 30 min at room temperature, and diamidino-2-phenylindole was used to fluorescently label nuclei.
Cytokine levels
Blood was harvested at time of sacrifice, and serum was obtained by centrifugation at 5,000 rpm for 5 min in serum separator tubes and stored at −80°C until further use. Serum cytokine levels of IL-1β, IL-6, IL-10, IL-12, IL-13, IFN-γ, TNF, G-CSF, and MCP-1 were measured using a multiplex cytokine assay (Bio-Rad, Hercules, CA) according to manufacturer’s instructions. All samples were run in duplicate.
Cultures
Blood was serially diluted in sterile normal saline and cultured on sheep blood agar plates. Additionally, A piece of liver and two mesenteric lymph nodes were harvested and weighed and then ground with disposable tissue grinders and suspended in in 100ul of sterile normal saline. Each sample was incubated overnight at 37°C, and colony counts were enumerated. Colony counts were expressed as CFU/ml of fluid and then converted to a logarithmic scale for statistical analysis (40;41).
Monoclonal Anti-TNF Antibody
Mice were given a single intraperitoneal injection of 300 μg of monoclonal anti-TNF antibody TN3 19.12 (a generous gift from Dr. Robert Schreiber, Washington University) or an equivalent volume of isotype control 3 hr prior to CLP. Of note, experiments with anti-TNF were performed at the same time as other experiments but are presented separately (figure 6) for ease of presentation.
Figure 6.
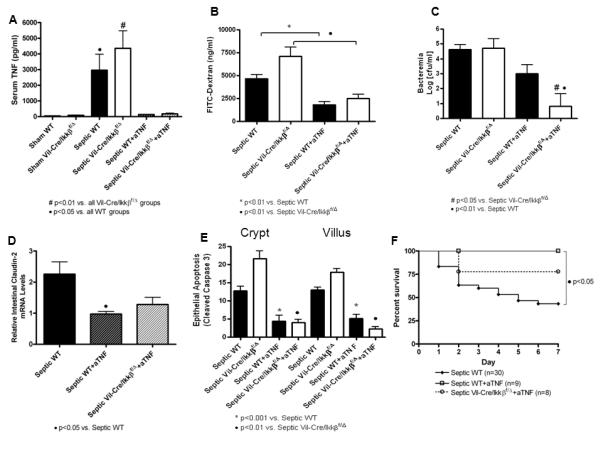
Sepsis-induced alterations in intestinal integrity, permeability and mortality are dependent on TNF. Sepsis-induced increases in serum TNF were completely blocked following TNF neutralization. n=8-18/group (A). Intestinal permeability was lower in septic WT mice following anti-TNF antibody and in septic Vil-Cre/Ikkßf/Δ mice following anti-TNF antibody; however, intestinal permeability was not different between Vil-Cre/Ikkßf/Δ and WT mice following TNF neutralization. n=8-18/group (B). Bacteremia was decreased in septic WT mice following anti-TNF antibody and was further decreased in Vil-Cre/Ikkßf/Δ mice following TNF neutralization. n=8-18/group (C). Claudin-2 expression was lower in septic WT mice following anti-TNF antibody; however, claudin-2 expression was not different between Vil-Cre/Ikkßf/Δ and WT mice following TNF neutralization. n=8-14/group (D). Intestinal epithelial apoptosis was lower in septic WT mice treated with anti-TNF antibody and in septic Vil-Cre/Ikkßf/Δ mice following anti-TNF antibody; however, apoptosis was not different between Vil-Cre/Ikkßf/Δ and WT mice following TNF neutralization. n=5-12/group (E). Anti-TNF treatment significantly improved mortality in both Vil-Cre/Ikkßf/Δ and WT mice compared to untreated groups; however there were no significant differences in the mortality between Vil-Cre/Ikkßf/Δ and WT mice following TNF neutralization (F).
Statistics
Continuous data sets were tested for Gaussian distribution using the D’Agostino-Pearson omnibus normality test. If data were found to have Gaussian distributions, multiple group comparisons were performed using one-way analysis of variance followed by the Newman-Keuls post-test. If data were found to have non-Gaussian distributions, multiple group comparisons were performed using the Kruskal-Wallis non-parametric one-way analysis of variance by ranks followed by the Dunns post-test. Survival studies were analyzed using the Log-Rank test. Data were analyzed using the statistical software program Prism 4.0 (GraphPad Software, San Diego, CA) and are reported as mean ± SEM. A p value <0.05 was considered statistically significant.
RESULTS
In all experiments, comparisons were made between 1) WT sham and septic mice, 2) Vil-Cre/Ikkßf/Δ sham and septic mice, and 3) WT and Vil-Cre/Ikkßf/Δ septic mice. Unless otherwise specified, animals were sacrificed 24 hours after the onset of sepsis.
Sepsis-induced NF-kB activation in the intestinal epithelium is abrogated in Vil-Cre/Ikkßf/Δ mice
To verify that NF-kB activation in the intestinal epithelium was diminished by selective enterocyte ablation of IKKß, NF-kB p65 activation was compared between septic WT and Vil-Cre/Ikkßf/Δ mice (Figure 1A). Activation of NF-kB p65 in the intestinal epithelium was abrogated by selective ablation of IKKß in the intestinal epithelium 24 hours after CLP . A similar pattern was noted for intestinal epithelial expression of phosphorylated NF-kB p65 which was significantly decreased in septic Vil-Cre/Ikkßf/Δ mice in both sham and septic mice (Figure 1B, C).
Figure 1.
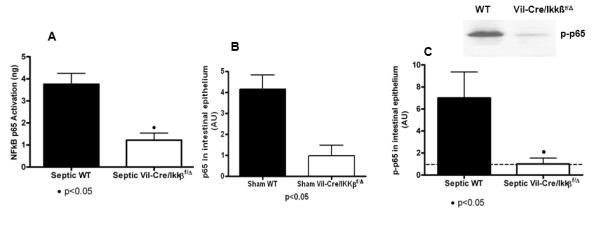
Activation of NF-kB p65 in the intestinal epithelium. Sepsis induced activation of NF-kB p65 in the intestinal epithelium as measured by a transcription factor assay (A). Similar results were seen when levels were assayed by Western blot in both sham (B) and septic mice (C). Ablation of Ikkß in the intestinal epithelium (Vil-Cre/Ikkßf/Δ) prevented activation of NF-kB p65 following induction of septic peritonitis. n=4-7/group.
Enterocyte NF-kB protects the intestinal epithelium from sepsis-induced gut damage
To determine whether enterocyte NF-kB altered sepsis-induced changes in gut integrity, villus length and intestinal epithelial apoptosis were measured in sham and septic WT and Vil-Cre/Ikkßf/Δ mice. Septic WT mice exhibited a 24% decrease in villus length compared to sham mice, while septic Vil-Cre/Ikkßf/Δ mice had worsened villus atrophy with a 22% further decrease in villus length compared to septic WT mice (Figure 2).
Figure 2.
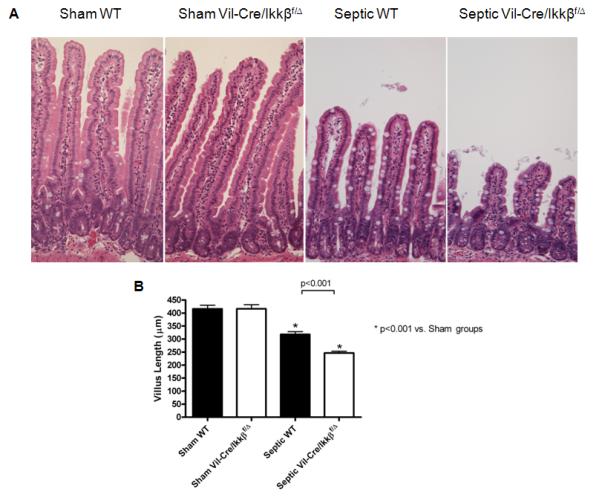
Sepsis-induced villus atrophy is exacerbated in mice lacking functional enterocyte NF-kB. Intestinal morphology (A) was evaluated in hematoxylin and eosin (H&E)-stained intestinal sections. Septic WT mice had markedly shorter villi than sham mice. Villi were even shorter in septic Vil-Cre/Ikkßf/Δ mice. Magnification x20. Villus length was quantified in sections of jejunum (B). Septic WT mice had significantly increased villus atrophy, which was exacerbated in the absence of functional enterocyte NF-kB (septic Vil-Cre/Ikkßf/Δ mice). n=12-13/group.
Sepsis also induced a significant increase in intestinal epithelial apoptosis in both the crypts and villi of WT mice as measured by the number of apoptotic cells per 100 crypts (Figure 3A,B) and per 50 villi (Figure 3C,D) by either active caspase-3 staining (Figure 3E) or H&E staining. Similar to villus length, sepsis-induced apoptosis was further exacerbated in the crypt and villus epithelium in septic Vil-Cre/Ikkßf/Δ mice compared to septic WT mice. In contrast, there was minimal inflammatory infiltration in either the lamina propria or submucosa in either septic WT or Vil-Cre/Ikkßf/Δ mice.
Figure 3.
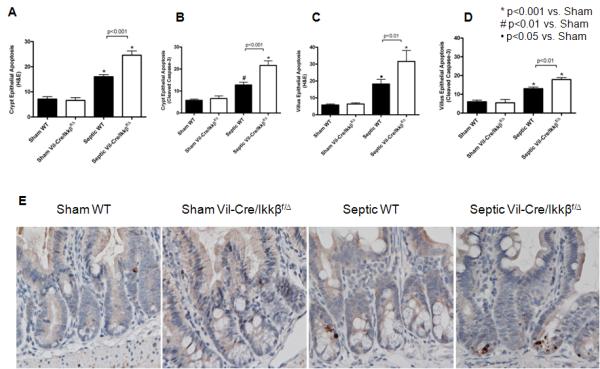
Sepsis-induced intestinal epithelial apoptosis is exacerbated in mice lacking functional enterocyte NF-kB. Intestinal apoptosis was quantified in crypt (A and B) and villus (C and D) epithelium via H&E staining (A and C) and active caspase-3 staining (B and D). Septic WT mice exhibited increased epithelial apoptosis compared to shams by both methods, while septic Vil-Cre/Ikkßf/Δ mice exhibited exacerbated epithelial apoptosis compared to septic WT mice. n=9-12/group. Representative histomicrographs (E) are shown for sham and septic WT and Vil-Cre/Ikkßf/Δ mice stained for active caspase 3. Active caspase 3 staining in the crypt epithelium appears brown.
Enterocyte NF-kB protects the intestinal epithelium from sepsis-induced intestinal barrier dysfunction
Since sepsis-induced intestinal injury was increased in mice lacking functional enterocyte NF-kB, intestinal barrier function was assayed in these mice. Sepsis induced a 1.7-fold increase in intestinal permeability in WT mice compared to sham mice as measured by the appearance of FD-4 in the bloodstream while septic Vil-Cre/Ikkßf/Δ mice had worsened intestinal hyperpermeability with a 34% increase in FD-4 appearance compared to septic WT mice (Figure 4A).
Figure 4.
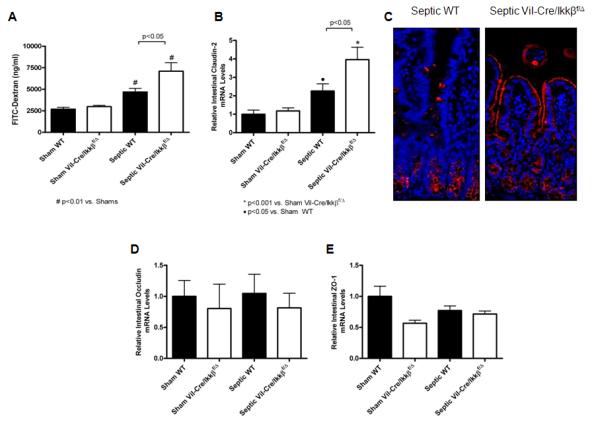
Sepsis-induced intestinal hyperpermeability is exacerbated in mice lacking functional enterocyte NF-kB. Intestinal permeability was evaluated in vivo by measuring the amount of FITC-conjugated dextran (FD-4) in the plasma (A). Septic WT mice exhibited increased permeability compared to shams, while septic Vil-Cre/Ikkßf/Δ mice had a further increase in intestinal permeability compared to septic WT mice. n=9-12/group. Gene expression of claudin-2 (B) was then evaluated by qRT-PCR. Claudin-2 levels were increased in septic WT mice compared to shams, while septic Vil-Cre/Ikkßf/Δ mice had a further increase in claudin-2 expression. n=8-14/group. Localization of claudin-2 was evaluated by fluorescent immunohistochemistry (C). Compared to sham WT and septic Vil-Cre/Ikkßf/Δ mice which had minimal staining (not shown), claudin-2 expression was increased in septic WT mice and localized predominantly in the crypts. In contrast, septic Vil-Cre/Ikkßf/Δ mice exhibited increased claudin-2 that was localized along the apical membrane of the villi as well as in the crypts. Representative images for each group are shown Magnification x20. Gene expression of occludin (D) and ZO-1 (E) was also evaluated by qRT-PCR. No differences were detected in either occludin or ZO-1, independent of whether an animal was subjected to CLP or had enterocyte NF-kB n=8-14/group.
To determine whether alterations in tight junction proteins contributed to intestinal barrier dysfunction, claudin-2, occludin, and ZO-1 expression in the intestinal epithelium were evaluated by real-time PCR. Intestinal gene expression of claudin-2 was increased in septic WT mice compared to shams, and further increased by 43% in septic Vil-Cre/Ikkßf/Δ mice (Figure 4B). Claudin-2 localization was then analyzed via fluorescent immunohistochemistry (Figure 4C). Septic WT mice exhibited increased expression of claudin-2 in the crypts with minimal expression in the villus epithelium compared to sham mice. In contrast, septic Vil-Cre/Ikkßf/Δ mice exhibited increased expression in the crypts as well as a change in subcellular localization with strong localization along the apical membrane of the villi. In contrast, occludin and ZO-1 expression were similar between all groups 24 hr post-CLP (Figure 4D,E), and their subcellular localization was not different between groups (data not shown).
The impact of enterocyte NF-kB on cytokines and bacteremia
To determine whether the lack of functional enterocyte NF-kB altered the systemic inflammatory response to sepsis, serum cytokine levels were assayed in sham and septic WT and Vil-Cre/Ikkßf/Δ mice at 24 hours (Table 1). The pro-inflammatory cytokines IL-6 and MCP-1, as well as the anti-inflammatory cytokine IL-10, were significantly increased in serum 24 hr after the onset of sepsis in WT mice. Additionally, serum MCP-1 and IL-10 were higher in septic Vil-Cre/Ikkßf/Δ mice compared to septic WT mice. No differences were seen in cytokine levels between these animals are 48 or 72 hours (supplementary tables 1 and 2). No significant differences in serum levels of IL-1β, IL-12, IL-13, or IFN-γ were detected between septic and sham mice regardless of whether they had functional enterocyte NF-kB.
TABLE 1.
| Serum (pg/ml) |
Sham WT | Sham Vil-Cre/ Ikkßf/Δ | Septic WT | Septic Vil-Cre/ Ikkßf/Δ |
|---|---|---|---|---|
| IL-1ß | 161.1±45.8 | 115.8±34.3 | 202.3±60.1 | 237.8±61.4 |
| IL-6 | 47.5±6.8 | 57.65±10.8 | 3444.0±667.9 a | 5422.0±1103.0 b |
| IL-10 | 33.7±11.4 | 44.5±24.5 | 388.1±87.9 a | 679.1±128.8 b,c |
| IL-12 | 20.34±3.9 | 20.0±5.0 | 16.7±1.9 | 14.5±1.8 |
| IL-13 | 371.7±97.0 | 278.3±49.4 | 297.5±79.5 | 268.9±75.6 |
| IFN-γ | 18.4±3.4 | 23.0±4.7 | 33.2±5.5 | 36.1±4.8 |
| G-CSF | 1472.0±159.4 | 1346.0±349.5 | 21584.0±1067.0a | 19485.0±2494.0 d |
| MCP-1 | 102.4±22.2 | 124.4±31.5 | 1763.0±330.9 | 3204.0±852.7 c,e |
| IL-1ß | 161.1±45.8 | 115.8±34.3 | 202.3±60.1 | 237.8±61.4 |
| IL-6 | 47.5±6.8 | 57.65±10.8 | 3444.0±667.9 a | 5422.0±1103.0 b |
| IL-10 | 33.7±11.4 | 44.5±24.5 | 388.1±87.9 a | 679.1±128.8 b,c |
| IL-12 | 20.34±3.9 | 20.0±5.0 | 16.7±1.9 | 14.5±1.8 |
| IL-13 | 371.7±97.0 | 278.3±49.4 | 297.5±79.5 | 268.9±75.6 |
| IFN-γ | 18.4±3.4 | 23.0±4.7 | 33.2±5.5 | 36.1±4.8 |
| G-CSF | 1472.0±159.4 | 1346.0±349.5 | 21584.0±1067.0a | 19485.0±2494.0 d |
| MCP-1 | 102.4±22.2 | 124.4±31.5 | 1763.0±330.9 | 3204.0±852.7 c,e |
To determine if differences in sepsis-induced intestinal hyperpermeability between WT and Vil-Cre/Ikkßf/Δ mice impacted systemic bacterial load, blood cultures were obtained 24 hours after CLP. Sepsis induced bacteremia in both WT and Vil-Cre/Ikkßf/Δ mice; however, there were no statistically significant differences in bacterial burden measured (data not shown) despite their differences in gut barrier function. Of note, while bacteremia was detectable at 24, 48 and 72 hours following CLP, the concentrations were 3 log lower at 48 and 72 hours than 24 hours. Bacteria were also detectable in cultures of mesenteric lymph nodes and liver 48 and 72 hours following CLP, but there were no differences between WT and Vil-Cre/Ikkßf/Δ mice (data not shown).
Mortality from sepsis is worsened in mice lacking functional enterocyte NF-kB
To determine the functional significance of enterocyte NF-kB, WT and Vil-Cre/Ikkßf/Δ mice were subjected to CLP and followed 7 days for survival (Figure 5). Septic WT mice had a 47% seven-day mortality, while septic Vil-Cre/Ikkßf/Δ mice had an 80% mortality (p<0.05). All sham mice survived their operation.
Figure 5.
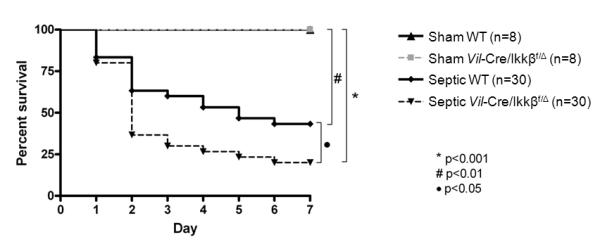
Sepsis-induced mortality is higher in mice lacking functional enterocyte NF-kB. Mice were subjected to CLP and followed 7 days for survival. Septic mice lacking functional enterocyte NF-kB (Vil-Cre/Ikkßf/Δ mice) had worsened mortality compared to septic WT mice (p<0.05). All sham animals survived.
Sepsis-induced hyperpermeability and intestinal apoptosis are mediated by TNF
The pro-inflammatory cytokine TNF is a key mediator of both intestinal barrier dysfunction and sepsis-induced inflammation. CLP increased levels of serum TNF in both WT and Vil-Cre/Ikkßf/Δ mice, but enterocyte NF-kB did not affect TNF levels as TNF levels were similar in septic WT and Vil-Cre/Ikkßf/Δ mice (Figure 6A) as were TNF levels in the jejunual epithelium by western blot (data not shown). To determine whether sepsis-induced intestinal hyperpermeability is mediated by TNF, WT and Vil-Cre/Ikkßf/Δ mice animals were treated with anti-TNF antibody 3 hours prior to CLP which completely blocks the increase in serum TNF induced by CLP (Figure 6A). Neutralizing TNF prevented sepsis-induced intestinal hyperpermeability (Figure 6B), suggesting TNF is a key mediator of barrier dysfunction in sepsis. A similar decrease in intestinal hypermeability was seen in septic Vil-Cre/Ikkßf/Δ mice following administration of anti-TNF antibody (Figure 6B). To determine whether intestinal hyperpermeability correlated to severity of bacteremia, blood cultures were also obtained in a different set of animals subjected to the same insults. Anti-TNF administration led to a non-significant decrease in bacteremia in WT mice, consistent with the decrease in permeability (Figure 6C). A further decrease in bacteremia was observed in septic Vil-Cre/Ikkßf/Δ mice given anti-TNF antibody (Figure 6C). Of note, changes in claudin-2 gene expression mirrored those of intestinal permeability (Figure 6D), and subcellular localization of claudin 2 was similar in WT and Vil-Cre/Ikkßf/Δ septic mice treated with anti-TNF antibody (data not shown). Anti-TNF antibody had no effect on intestinal expression of either occludin or ZO-1 (data not shown).
The impact of TNF neutralization on sepsis-induced gut epithelial apoptosis was also assayed. Similar to permeability and claudin-2 gene expression, anti-TNF antibody significantly decreased sepsis-induced intestinal epithelial apoptosis in the crypts and villi (Figure 6E), A similar decrease in intestinal apoptosis was seen in septic Vil-Cre/Ikkßf/Δ mice following administration of anti-TNF antibody (Figure 6E).
To determine the impact of TNF on mortality, anti-TNF antibodies were given to WT and Vil-Cre/Ikkßf/Δ mice subjected to CLP, which were followed seven days for survival. Anti-TNF treatment significantly improved mortality in WT septic mice (Figure 6F). A similar improvement in mortality was seen in septic Vil-Cre/Ikkßf/Δ mice following administration of anti-TNF antibody since mortality in septic Vil-Cre/Ikkßf/Δ mice was 80% (Figure 5) while mortality in septic Vil-Cre/Ikkßf/Δ mice given anti-TNF was 25% (Figure 6E).
DISCUSSION
This study demonstrates that inhibition of NF-kB in enterocytes exacerbates sepsis-induced intestinal injury and worsens mortality in mice subjected to CLP. Although NF-kB inhibition has been proposed as a therapeutic approach for sepsis (42), findings in gene-targeted mice and studies with pharmacological inhibitors indicate that NF-kB regulates both protective and destructive responses (43-45). As such, the net effects of NF-kB inhibition need to be evaluated carefully.
By isolating the enterocyte-specific effects of NF-kB, this study expands our understanding of the role the intestine plays as the “motor” of the systemic inflammatory response syndrome. One possible way enterocyte-specific NF-kB apears to be protective in sepsis is by abrogating intestinal epithelial apoptosis. It is known that NF-kB is a critical regulator of the transcriptional activation of a multitude of genes involved in apoptosis (32), and this could profoundly influence mucosal repair in sepsis. For example, NF-kB can upregulate anti-apoptotic molecules such as c-FLIP, c-IAPs, and anti-apoptotic Bcl-2 family members (24). Consistent with this, failure to activate enterocyte NF-kB resulted in exacerbated intestinal epithelial apoptosis following CLP. Our results are consistent with previous studies of mice that lack enterocyte IKKß exhibit showing marked increases in intestinal epithelial apoptosis following radiation (46), C. difficile toxin A (33), and thermal injury (32).
Intestinal barrier function was dependent, at least in part, on enterocyte NF-kB in this study, since intestinal hypermpermeability was worsened following CLP in Vil-Cre/Ikkßf/Δ mice compared to WT mice. It is possible that this was secondary to the upregulation in sepsis-induced apoptosis seen in Vil-Cre/Ikkßf/Δ mice, as there is data suggesting that intestinal permeability is dependent upon apoptosis in models of critical illness (47;48). The increase in sepsis-induced hyperpermeability was associated with an increase in claudin-2. This pore-forming protein is associated with increased paracellular permeability and has previously been shown to be upregulated in the inflamed mucosa of patients with inflammatory bowel disease (49;50). Similarly, previous data from our laboratory demonstrated that claudin-2 is significantly increased in the intestine following CLP (14). The observation that claudin-2 is increased and changes subcellular localization after the onset of sepsis but is exacerbated following CLP in Vil-Cre/Ikkßf/Δ mice suggests that its regulation in the intestinal tight junction is at least partially NF-kB-dependent. In contrast, neither occludin nor ZO-1 were affected by CLP or enterocyte NF-kB in this study.
While enterocyte NF-kB plays a protective role in the intestine following sepsis, this does not mean that its effects are globally protective. For example, in mice subjected to ischemia/reperfusion, enterocyte NF-kB inhibition induces a marked increase in intestinal epithelial apoptosis (as is seen following CLP) but simultaneously leads to a marked decrease in systemic inflammation (34). The functional significance of these findings in ischemia/reperfusion is unclear since animals were not followed for survival. However, altering the host inflammatory response might be expected to have a profound impact on mortality, although whether this would be beneficial or detrimental is likely dependent upon the severity of the pro- and anti- inflammatory response as well as the timing of the response (51-54). As a surrogate for the systemic inflammatory response, serum cytokine levels were therefore examined following CLP. As might be expected, sepsis induced a significant increase in both pro-inflammatory IL-6 and MCP-1 as well as anti-inflammatory IL-10 (55). However, enterocyte NF-kB generally did not alter systemic cytokines 24, 48 or 72 hours following CLP since there were no differences in cytokine levels between Vil-Cre/Ikkßf/Δ and WT mice except for higher IL-10 and MCP-1 levels in septic Vil-Cre/Ikkßf/Δ mice at 24 hours. While this differs from results seen in ischemia/reperfusion, this may be explained by differences in when cytokines were measured (4 hours after ischemia/reperfusion, 24-72 hours after CLP) (34). Additionally, no differences in bacteremia were noted between Vil-Cre/Ikkßf/Δ and WT septic mice. These results are consistent with findings in mice lacking the cRel subunit of NF-kB, in which there are no differences in bacterial counts in the liver, spleen, or blood compared to WT mice after CLP (56). Taken together, these data suggest that the increased mortality observed in mice that lack functional enterocyte NF-kB is not a result of increased systemic inflammation or impaired pathogen clearance. Instead, sepsis-induced alterations in intestinal integrity that are dependent upon enterocyte NF-kB may have played a role in mediating the worsened mortality in Vil-Cre/Ikkßf/Δ septic mice.
The pro-inflammatory cytokine TNF is involved in both NF-kB signaling and the pathophysiology of sepsis. TNF binds to TNFR1, which induces NF-kB signaling and induces both MAPK pro-inflammatory signaling and apoptosis. However, NF-kB activation protects cells from TNFR1-mediated apoptosis by inducing anti-apoptotic proteins. As such, NF-kB activation determines cellular fate following TNF binding, with induction of pro-inflammatory signals in NF-kB-competent cells but induction of death of NF-kB-deficient cells (26;57). In addition to its pro-apoptotic role in the intestinal epithelium, TNF is a key mediator of inflammation-induced barrier dysfunction, and causes a time- and dose-dependent increase in the expression of claudin-2 in human intestinal epithelial cell monolayers, leading to increased paracellular permeability (58).
In light of the fact that a) NF-kB signaling can be induced by TNF and b) the critical role TNF plays in sepsis, TNF blockade was therefore examined in septic Vil-Cre/Ikkßf/Δ and WT mice. The results indicated that sepsis-induced changes in intestinal epithelial apoptosis, permeability, bacteremia and mortality were all mediated by TNF as each of these variables was partially or fully normalized by giving mice anti-TNF antibodies as shown in figure 6. The results also demonstrated that mice that lack enterocyte NF-kB had increased gut apoptosis, permeability and mortality compared to WT animals (Figures 2-5). To study a potential relationship between these findings, we examined apoptosis, permeability, bacteremia and mortality in Vil-Cre/Ikkßf/Δ mice treated with or without anti-TNF. The results showed that apoptosis, permeability and mortality were lower in Vil-Cre/Ikkßf/Δ mice that were treated with anti-TNF and that TNF blockade decreased bacteremia in these animals. Together, these data indicate that a) enterocyte NF-kB plays a critical role in mediating intestinal integrity and mortality in the murine model of CLP, b) TNF also plays a critical role in mediating intestinal integrity and mortality in the murine model of CLP, and c) all variables worsened by the absence of NF-kB were improved by administering anti-TNF antibody.
This study has a number of limitations. While serum cytokines were used as a surrogate of the systemic inflammatory response, absolute cytokine levels frequently do not correlate to inflammatory status on a tissue level, and performing ex vivo studies on stimulated leukocytes may have given a more complete picture of systemic inflammation. Additionally, we examined RNA on tight junction proteins from whole jejunal tissue rather than jejunal mucosal scrapings. Vil-Cre/Ikkßf/Δ mice also have a lifelong genetic deletion in IKKß. While these animals have no gross phenotypic abnormalities in their intestinal tract under basal conditions, there may be unknown long-standing compensatory abnormalities induced by deletion of enterocyte NF-kB, which predisposed animals to have a negative outcome when subjected to the acute septic stress induced by CLP.
While we wish to emphasize that the purpose behind performing the anti-TNF experiments was to determine whether the impact of enterocyte NF-kB on gut integrity and mortality was TNF-dependent (rather than examining the effect of anti-TNF in isolation), there were also a number of limitations related to our studies using anti-TNF. First, we found that anti-TNF significantly improved survival in mice subjected to CLP. These findings are somewhat at odds with 12 human trials on over 6000 patients suggesting that anti-TNF has limited impact on survival, (59) although TNF blockade appears to be more efficacioius in sicker patients. Additionally, there have been at least 23 studies examining the impact of TNF blockade on mortality from CLP, of which 41% showed benefit by inhibiting TNF and which globally showed neither benefit nor harm (60). While our findings are thus consistent with some (but not all) prior preclinical studies, their clinical relevance is unclear. Additionally, while our studies were done in a relatively high-mortality model (where anti-TNF has been shown to have the most clinical effiacy in patients in meta-analyses), the impact of the agent was much greater in mice than is seen in patients. It should also be noted that the levels of systemic TNF we identified in septic mice were significantly higher than those found in patients 24 hours after they were diagnosed with peritonitis (61). This may, in part, be related to the amount of cecum ligated as there are widely varying levels of TNF identified in the bloodstream of septic animals based upon this variable (62); however, this may lessen the clinical relevance of our findings.
Despite these limitations, this study demonstrates that activation of enterocyte NF-kB has a protective role in sepsis-induced intestinal injury. While activation of NF-kB induces multiple proteins that can be either adaptive or maladaptive, enterocyte NF-kB appears beneficial, as mortality is increased in its absence. These results have implications for the potential therapeutic use of IKKß/NF-kB inhibitors and raise concerns about prolonged inhibition of this pathway.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sebastian Perez for statistical review of the manuscript.
This work was supported by funding from the National Institutes of Health (GM66202, GM072808, GM082008, P30 DK52574, Shock Society Early Career Investigator Fellowship).
REFERENCES
- (1).Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- (2).Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- (3).Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. 2007;28:384–393. doi: 10.1097/shk.0b013e31805569df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hassoun HT, Kone BC, Mercer DW, et al. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15:1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- (5).Carrico CJ, Meakins JL, Marshall JC, et al. Multiple-organ-failure syndrome. The gastrointestinal tract: the “motor” of MOF. Arch Surg. 1986;121:196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]
- (6).Clark JA, Clark AT, Hotchkiss RS, et al. Epidermal growth factor treatment decreases mortality and is associated with improved gut integrity in sepsis. Shock. 2008;30:36–42. doi: 10.1097/shk.0b013e31815D0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hiramatsu M, Hotchkiss RS, Karl IE, et al. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- (8).Coopersmith CM, Stromberg PE, Dunne WM, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- (9).Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- (10).Stromberg PE, Woolsey CA, Clark AT, et al. CD4+ lymphocytes control gut epithelial apoptosis and mediate survival in sepsis. FASEB J. 2009;23:1817–1825. doi: 10.1096/fj.08-119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Coopersmith CM, Chang KC, Swanson PE, et al. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002;30:195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- (12).Dominguez JA, Vithayathil PJ, Khailova L, et al. Epidermal Growth Factor Improves Survival and Prevents Intestinal Injury in a Murine Model of Pseudomonas Aeruginosa Pneumonia. Shock. 2011;36:381–389. doi: 10.1097/SHK.0b013e31822793c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vyas D, Robertson CM, Stromberg PE, et al. Epithelial apoptosis in mechanistically distinct methods of injury in the murine small intestine. Histol Histopathol. 2007;22:623–630. doi: 10.14670/hh-22.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Clark JA, Gan H, Samocha AJ, et al. Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G471–G479. doi: 10.1152/ajpgi.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).De-Souza DA, Greene LJ. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit Care Med. 2005;33:1125–1135. doi: 10.1097/01.ccm.0000162680.52397.97. [DOI] [PubMed] [Google Scholar]
- (16).Neal MD, Leaphart C, Levy R, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- (17).Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9:143–151. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- (18).Mainous MR, Ertel W, Chaudry IH, et al. The gut: a cytokine-generating organ in systemic inflammation? Shock. 1995;4:193–199. [PubMed] [Google Scholar]
- (19).Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;32:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- (20).Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- (21).Spehlmann ME, Eckmann L. Nuclear factor-kappa B in intestinal protection and destruction. Curr Opin Gastroenterol. 2009;25:92–99. doi: 10.1097/MOG.0b013e328324f857. [DOI] [PubMed] [Google Scholar]
- (22).Eckmann L, Nebelsiek T, Fingerle AA, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci U S A. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- (24).Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- (25).Li Q, Van AD, Mercurio F, et al. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- (26).Wullaert A, Bonnet MC, Pasparakis M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–158. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lawrance IC, Wu F, Leite AZ, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- (28).MacMaster JF, Dambach DM, Lee DB, et al. An inhibitor of IkappaB kinase, BMS-345541, blocks endothelial cell adhesion molecule expression and reduces the severity of dextran sulfate sodium-induced colitis in mice. Inflamm Res. 2003;52:508–511. doi: 10.1007/s00011-003-1206-4. [DOI] [PubMed] [Google Scholar]
- (29).Shibata W, Maeda S, Hikiba Y, et al. Cutting edge: The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol. 2007;179:2681–2685. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- (30).Dave SH, Tilstra JS, Matsuoka K, et al. Amelioration of chronic murine colitis by peptide-mediated transduction of the IkappaB kinase inhibitor NEMO binding domain peptide. J Immunol. 2007;179:7852–7859. doi: 10.4049/jimmunol.179.11.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- (32).Chen L, Chen P, Chang W, et al. IkB-kinase/NF-kB signaling prevents thermal injury-induced gut damage by inhibiting JNK activation. Crit Care Med. 2007;35:1332–1340. doi: 10.1097/01.CCM.0000261891.30360.F0. [DOI] [PubMed] [Google Scholar]
- (33).Chae S, Eckmann L, Miyamoto Y, et al. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J Immunol. 2006;177:1214–1220. doi: 10.4049/jimmunol.177.2.1214. [DOI] [PubMed] [Google Scholar]
- (34).Chen LW, Egan L, Li ZW, et al. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- (35).Baker CC, Chaudry IH, Gaines HO, et al. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- (36).Fox AC, Robertson CM, Belt B, et al. Cancer causes increased mortality and is associated with altered apoptosis in murine sepsis. Crit Care Med. 2010;38:886–893. doi: 10.1097/CCM.0b013e3181c8fdb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Danielsson A, Hellers G, Lyrenas E, et al. A controlled randomized trial of budesonide versus prednisolone retention enemas in active distal ulcerative colitis. Scand J Gastroenterol. 1987;22:987–992. doi: 10.3109/00365528708991947. [DOI] [PubMed] [Google Scholar]
- (38).D’Haens GR, Geboes K, Peeters M, et al. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- (39).Clark JA, Gan H, Samocha AJ, et al. Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G471–G479. doi: 10.1152/ajpgi.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wells CL, Hess DJ, Erlandsen SL. Impact of the indigenous flora in animal models of shock and sepsis. Shock. 2004;22:562–568. doi: 10.1097/01.shk.0000145935.24344.2d. [DOI] [PubMed] [Google Scholar]
- (41).Robertson CM, Perrone EE, McConnell KW, et al. Neutrophil Depletion Causes a Fatal Defect in Murine Pulmonary Staphylococcus aureus clearance. J Surg Res. 2008;150:278–285. doi: 10.1016/j.jss.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zingarelli B, Sheehan M, Wong HR. Nuclear factor-kappaB as a therapeutic target in critical care medicine. Crit Care Med. 2003;31:S105–S111. doi: 10.1097/00003246-200301001-00015. [DOI] [PubMed] [Google Scholar]
- (43).Li X, Su J, Cui X, et al. Can we predict the effects of NF-kappaB inhibition in sepsis? Studies with parthenolide and ethyl pyruvate. Expert Opin Investig Drugs. 2009;18:1047–1060. doi: 10.1517/13543780903018880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wheeler DS, Zingarelli B, Wheeler WJ, et al. Novel pharmacologic approaches to the management of sepsis: targeting the host inflammatory response. Recent Pat Inflamm Allergy Drug Discov. 2009:396–112. doi: 10.2174/187221309788489779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Uwe S. Anti-inflammatory interventions of NF-kappaB signaling: potential applications and risks. Biochem Pharmacol. 2008;75:1567–1579. doi: 10.1016/j.bcp.2007.10.027. [DOI] [PubMed] [Google Scholar]
- (46).Egan LJ, Eckmann L, Greten FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chin AC, Flynn AN, Fedwick JP, et al. The role of caspase-3 in lipopolysaccharide-mediated disruption of intestinal epithelial tight junctions. Can J Physiol Pharmacol. 2006;84:1043–1050. doi: 10.1139/y06-056. [DOI] [PubMed] [Google Scholar]
- (48).Sun Z, Wang X, Deng X, et al. The influence of intestinal ischemia and reperfusion on bidirectional intestinal barrier permeability, cellular membrane integrity, proteinase inhibitors, and cell death in rats. Shock. 1998;10:203–212. doi: 10.1097/00024382-199809000-00009. [DOI] [PubMed] [Google Scholar]
- (49).Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- (51).Hotchkiss RS, Opal S. Immunotherapy for sepsis--a new approach against an ancient foe. N Engl J Med. 2010;363:87–89. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- (53).McConnell KW, McDunn JE, Clark AT, et al. Streptococcus pneumoniae and Pseudomonas aeruginosa pneumonia induce distinct host responses. Crit Care Med. 2010;38:223–241. doi: 10.1097/CCM.0b013e3181b4a76b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hotchkiss RS, Coopersmith CM, McDunn JE, et al. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Osuchowski MF, Welch K, Siddiqui J, et al. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- (56).Courtine E, Pene F, Cagnard N, et al. Critical role of cRel subunit of NF-kappaB in sepsis survival. Infect Immun. 2011;79:1848–1854. doi: 10.1128/IAI.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Papa S, Zazzeroni F, Bubici C, et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- (58).Mankertz J, Amasheh M, Krug SM, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336:67–77. doi: 10.1007/s00441-009-0751-8. [DOI] [PubMed] [Google Scholar]
- (59).Qiu P, Cui X, Barochia A, et al. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Expert Opin Investig Drugs. 2011;20:1555–1564. doi: 10.1517/13543784.2011.623125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Lorente JA, Marshall JC. Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock. 2005;24(Suppl 1):107–19. doi: 10.1097/01.shk.0000191343.21228.78. [DOI] [PubMed] [Google Scholar]
- (61).Riche FC, Cholley BP, Panis YH, et al. Inflammatory cytokine response in patients with septic shock secondary to generalized peritonitis. Crit Care Med. 2000;28:433–437. doi: 10.1097/00003246-200002000-00024. [DOI] [PubMed] [Google Scholar]
- (62).Singleton KD, Wischmeyer PE. Distance of cecum ligated influences mortality, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Eur Surg Res. 2003;35:486–491. doi: 10.1159/000073387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.