

Abstract
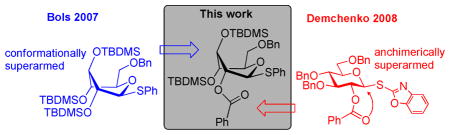
A novel glycosyl donor that combines the concepts of both conformational and electronic superarming has been synthesized. The reactivity and selectivity of the donor has been tested in competition experiments.
Traditionally oligosaccharide synthesis relies on protective group manipulations and the use of orthogonal glycosylation methods, such as different types of glycosyl donors. Due to the increased interest in biologically relevant oligosaccharides the development of new methodologies have been impressive during the last couple of decades and paved the way for more efficient synthesis.1,2 These developments include, but are not limited to, one-pot protection3 and glycosylation strategies,4,5 polymer-supported6 and automated synthesis,7,8 ionic liquid supported,9,10 fluorous tag assisted,11,12 surface-tethered (STICS),13 and HPLC-assisted syntheses.14
The control of the glycosyl donor’s reactivity belongs to the tools available for improving oligosaccharide synthesis. The armed-disarmed concept was introduced by the group of Fraser-Reid,15 and utilizes selective activation of one donor over another with the same anomeric leaving group. The reactivity of the donor relies on the protective groups used; more electron-withdrawing groups reduce (disarm) the donor reactivity and vice versa. Glycosyl donor A1 is armed (benzylated) whereas A2 is disarmed (more electron-withdrawing protective groups) and acts as the glycosyl acceptor (Scheme 1A).15,16
Scheme 1.

Chemoselective strategies for oligosaccharide synthesis: A. Armed-disarmed; B. Conformational superarming; C. Electronic superarming.
With the insight into manipulation of reactivity by protective groups, new methodologies for oligosaccharide have been developed, one example is “one-pot” oligosaccharide strategies, introduced by the groups of Fraser-Reid, Ley, and Wong.17,18,19,20,21,22,23 For instance, a very detailed work by Wong and collaborators22 showed that the reactivity of an armed or a disarmed donor varied considerably depending on stereochemistry and whether deoxy groups were present.24
The stereochemical effect of hydroxyl groups on the development of positive charge in heterocycles have been further demonstrated using piperidines as the model compounds. By comparing the pKa values for the conjugate acid it was found that equatorial substituents are significantly more deactivating (EWD) than their axial counterparts.25,26,27,28 The same effects were found for glycosyl donors and used by Bols and co-workers to conformationally arm glycosyl donors by changing the equatorial rich 4C1 conformation to an axial rich conformation.29,30,31,32,33 The conformational changes were induced by creating steric congestion at the equatorial C-2, C-3 and C-4 positions of D-glucosides,34 resulting in a skew-boat conformation (donor B1, Scheme 1B). The new type of donors showed a 20-fold increase in reactivity as compared to its per-O-benzylated counterpart.32 The superarmed glycosyl donor B1 could be effectively coupled with “armed” acceptor B2 promoted by NIS/TfOH at −78 °C to afford the corresponding disaccharide in 85% yield (Scheme 1B).30
Derived from the discovery of the O2/O5 cooperative effect in glycosylation35 Demchenko and co-workers reported that a glycosyl donor containing a 2-O-benzoyl group, instead of a 2-O-benzyl, increased the reactivity compared with a fully benzylated analogue and hence to be considered “super armed”. The arming was found to be due to anchimeric assistance,41 which overrules the EWD properties of the benzoyl group.36,37 Thus, glycosylation with 2-O-benzoyl-3,4,6-tri-O-benzyl protected S-benzoxazolyl (SBox) glucoside C1 with per-benzylated “armed” glycosyl acceptor C2 in the presence of dimethyl(methylthio)sulfonium triflate (DMTST) provided disaccharide in 70% yield (Scheme 1C). This concept for superarming was found to be universally applicable to common leaving groups including O-pentenyl, S-ethyl, S-phenyl, S-tolyl and S-thiazolinyl.38
With two different approaches to superarm glycosyl donors, we wondered which superarmed donor is more reactive. To investigate this, a direct competition experiment was performed wherein the conformationally superarmed S-phenyl glycosyl donor 1a was set to compete with electronically superarmed glycosyl donor 1b for cyclohexanol (Scheme 2). The most reliable comparison was achieved in the NIS/TfOH-promoted competition experiment starting at −78 °C and slowly warming up to 0 °C, essentially the same reaction conditions as reported by Bols and co-workers.30 Formation of disaccharide 2a derived from the conformationally superarmed glycosyl donor 1a was predominant (2a isolated in 91% yield), whereas 1b was recovered in 94% yield. This result clearly indicated that donor 1a has superior reactivity in comparison to donor 1b under these reaction conditions.
Scheme 2.
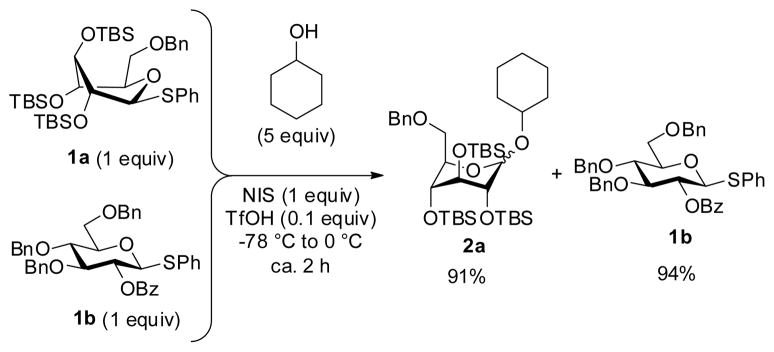
Competition experiment between conformationally superarmed donor 1a and electronically superarmed donor 1b.
This result put the question whether further enhancement in reactivity could be achieved by using a combination of both conformational and electronic effects. To address this question and with the goal of incorporating all key structural features from both approaches into a single hybrid donor 1c, equipped with both the 2-O-benzoyl substituent and remote O-TBS substituents at 3,4-positions in combination with 6-O-benzyl (glycosyl donor 1c, see the SI for its synthesis) was obtained. The conformation of donor 1c was investigated by 1H-NMR, where the coupling constants suggested a skew boat conformation similar to the one found for conformationally armed donors.30
The conformation was further proved by obtaining the first reported crystal structure of the superarmed donor 1a, which has similar (to 1c) 3J coupling constants in its 1H-NMR. The crystal structure confirms the suggested skew-boat conformation and confirms that 1c is in a similar conformation.39,40
Having established that 1c indeed has changed the conformation to an axial-rich skew-boat, the properties of the hybrid donor 1c could be investigated in more detail. For this purpose, glycosyl donors 1a–c were compared under standard reaction conditions (NIS/TfOH, −78 °C to 0 °C). Since 1c has incorporated a 2-O-benzoyl substituent, the stereoselectivity was expected to be excellent due to neighboring group participation. The stereoselectivity obtained with donor 1a having a non-participating TBS-group at O-2 is normally excellent with poor acceptors, such as other carbohydrates, but can be reduced when using simple alcohols as acceptors.30 The results of this study are summarized in Table 1. All glycosylations were giving good-to-excellent yields and for entries 2 and 3 complete β-selectivity was observed, whereas donor 1a (Entry 1) was less selective. Glycosylations of benzylated “armed” glycosyl acceptors equipped with the S-phenyl anomeric group with donor 1c gave moderate-to-good yields and complete stereoselectivity (entries 5 and 6), with an acceptor site. The lower yield observed with the primary acceptor (entry 5) is mainly due to migration of the TBS protective group from donor 1c to the acceptor. The new donor can therefore be considered super armed as it is more reactive than a conventionally armed donor.
Table 1.
Comparative study of glycosyl donors 1a–c.
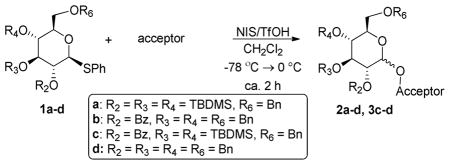
| |||
|---|---|---|---|
| entry | donor | acceptor | product, yield (α/β ratio) |
| 1 | 1a | CHexanol | 2a, 82% (1:2.8) |
| 2 | 1b | CHexanol | 2b, 91% (β only) |
| 3 | 1c | CHexanol | 2c, 97% (β only) |
| 4 | 1d | CHexanol | 2d, 84% (1:2) |
| 5 | 1c |
|
3c, 53% (β only) |
| 6 | 1c |
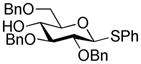
|
4c, 70% (β only) |
With the glycosylation properties of donor 1c established, its reactivity was studied by competition experiments with donors 1a and 1b. using essentially the same reaction conditions as those described in Scheme 2. From the first experiment between the hybrid donor 1c and the electronically superarmed donor 1b it was obvious that compound 1c was significantly more reactive, i.e. complete conversion of 1c to glycoside 2c and almost full recovery of unreacted 1b was observed (Scheme 3). Competition between donor 1c and the conformationally superarmed donor 1a revealed that donor 1a was much more reactive. High conversion of donor 1a to glycoside 2a was observed, whereas most of donor 1c was recovered.
Scheme 3.
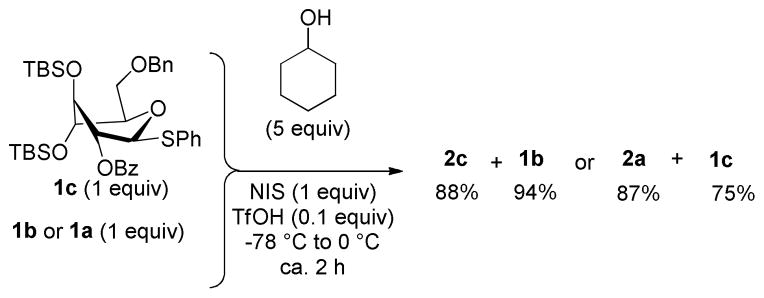
Competition experiment between the hybrid donor 1c and previously developed donors 1a and 1b.
Puzzled by the lower reactivity of the hybrid donor 1c the question arose whether the trans-vicinal 2-O-benzoyl group in 1c is able to increase the reactivity by the anchimeric effect or if it is overall disarming due to its electron-withdrawing nature. To investigate this α-counterpart 1e of β-donor 1c was synthesized (see the SI for its synthesis). Due to the 1,2-cis orientation in donor 1e, the 2-O-benzoyl group is unable to provide the anchimeric assistance. Therefore, if this effect prevails α-linked donor 1e would be less reactive than its β-linked counterpart 1c.
A competition experiment between donors 1c and 1e carried out under the standard conditions clearly showed that β-linked donor 1c (80% conversion) was more reactive than its α-linked conterpart 1e (10% conversion). The higher reactivity of 1c compared to 1e suggests that the 2-O-benzoyl group is providing an arming effect by means of the anchimeric assistance.41 On the other hand, reactivity difference between 1c and 1e could also be partly due to the anomeric effect lowering the ground state energy of the α-anomer.42,43 In the absense of the anchimeric assistance, axial thioglycosides have been found to be more reactive than their equatorial counterparts. This has been explained with the importance of an anti-periplanar arranggement between the leaving group and one of the ring-oxygens lone-pairs.44,43
To gain a better understanding of the effects caused by the 2-O-protective group, 2-O-benzyl superarmed donor 1f was synthesized.30 A competition experiment between donors 1c and 1f was performed and in this case donor 1f (80% conversion) was found to be more reactive than donor 1c (10% conversion). This result suggests that the 2-O-benzoyl in 1c is having an overall disarming effect in comparison to that of the 2-O-benzyl group in donor 1f. Since donor 1c is more reactive than 1e, but less reactive than 1f, the arming anchimeric assistance of the 2-O-benzoyl group is overruled by the electron-withdrawing properties. The higher reactivity of 1c compared to 1b is arguably due to the altered axial-rich conformation, which results in a smaller electron-withdrawing effect from substituents on the sugar ring. The altered, and not so flexible, conformation could, however, also diminish the effect of the anchimeric assistance since the 2-O-benzoyl is not perfectly antiperiplanar to the anomeric leaving group in the skew-boat conformation.
In conclusion, we have successfully synthesized a new type of donor 1c that combines conformational arming and anchimeric assistance effects. Glycosylations with this donor are high yielding and stereoselective. From this work it is clear that conformational arming is the more powerful tool when it comes to increasing the reactivity of the glycosyl donor. Anchimeric assistance does not increase the reactivity further in this particular case, but does lead to stereocontrol. Thus the combined donor obtained is highly reactive (superarmed), useful in one-pot glycosylations and stereoselective. Alternative promoter systems for thioglycosides were investigated, but without providing additional insight – the NIS/TfOH promotor system remains the most succesful in terms of yields and simplicity.
Supplementary Material
Figure 1.
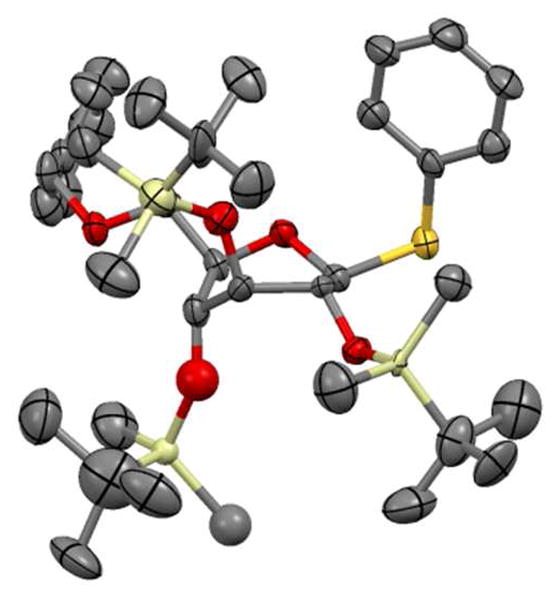
Crystal structure of 1a.
Scheme 4.

Competition experiment between donor 1c and donor 1e and donor 1f.
Acknowledgments
A.V.D. thanks NIGMS (award GM077170) for supporting this work. H.D.P. is grateful to UMSL Graduate School for proving her with the Dissertation Fellowship. Dr. Winter and Mr. Kramer (UM – St. Louis) are thanked for HRMS determinations. M.H. thanks FTP for supporting this work.
Footnotes
Supporting Information Available
Experimental details for preparation of new compounds, NMR spectra, and X-ray crystal data.
Contributor Information
Christian M. Pedersen, Email: cmp@chem.ku.dk.
Mikael Bols, Email: bols@chem.ku.dk.
Alexei V. Demchenko, Email: demchenkoa@umsl.edu.
References
- 1.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 2.Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 3.Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee CC, Chang KL, Hung SC. Nature. 2007;446:896–899. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wang Y, Ye XS, Zhang LH. Org Biomol Chem. 2007;5:2189. doi: 10.1039/b704586g. [DOI] [PubMed] [Google Scholar]; (b) Francais A, Urban D, Beau JM. Angew Chem Int Ed. 2007;46:8662–8665. doi: 10.1002/anie.200703437. [DOI] [PubMed] [Google Scholar]
- 5.Kaeothip S, Demchenko AV. Carbohydr Res. 2011;346:1371–88. doi: 10.1016/j.carres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osborn HMI, Khan TH. Tetrahedron. 1999;55:1807–1850. [Google Scholar]
- 7.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 8.Krock L, Esposito D, Castagner B, Wang CC, Bindschadler P, Seeberger PH. Chem Sci. 2012;3:1617–1622. [Google Scholar]
- 9.Pathak AK, Yerneni CK, Young Z, Pathak V. Org Lett. 2008;10:145–148. doi: 10.1021/ol702743x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran AT, Burden R, Racys DT, Galan MC. Chem Commun. 2011;47:4526–4528. doi: 10.1039/c0cc05580h. [DOI] [PubMed] [Google Scholar]
- 11.Hogendorf WFJ, Lameijer LN, Beenakker TJM, Overkleeft HS, Filippov DV, Codée JDC, der Marel GA. Org Lett. 2012;14:848–851. doi: 10.1021/ol2033652. [DOI] [PubMed] [Google Scholar]
- 12.Jaipuri FA, Pohl NL. Org Biomol Chem. 2008;6:2686–2691. doi: 10.1039/b803451f. [DOI] [PubMed] [Google Scholar]
- 13.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem Commun. 2009:1834–1836. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 14.Tran AT, Burden R, Racys DT, Galan MC. Chem Commun. 2011;47:4526–4528. doi: 10.1039/c0cc05580h. [DOI] [PubMed] [Google Scholar]
- 15.Mootoo DR, Konradson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]
- 16.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068–6070. [Google Scholar]
- 17.Fraser-Reid B, Wu Z, Andrews CW, Skowronski E, Bowen JP. J Am Chem Soc. 1991;113:1434–1435. [Google Scholar]
- 18.Wilson BG, Fraser-Reid B. J Org Chem. 1995;60:317–320. [Google Scholar]
- 19.Douglas NL, Ley SV, Lücking U, Warriner SL. J Chem Soc, Perkin Trans 1. 1998:51–66. [Google Scholar]
- 20.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 21.Ye XS, Wong CH. J Org Chem. 2000;65:2410–2431. doi: 10.1021/jo991558w. [DOI] [PubMed] [Google Scholar]
- 22.Koeller KM, Wong CH. Chem Rev. 2000;100:4465–4494. doi: 10.1021/cr990297n. [DOI] [PubMed] [Google Scholar]
- 23.Hsu Y, Lu XA, Zulueta MML, Tsai CM, Lin KI, Hung SC, Wong CH. J Am Chem Soc. 2012;134:4549–4552. doi: 10.1021/ja300284x. [DOI] [PubMed] [Google Scholar]
- 24.Premathilake HD, Demchenko AV. In: Topics in Current Chemistry: Reactivity Tuning in Oligosaccharide Assembly. Fraser-Reid B, Lopez JC, editors. Vol. 301. Springer-Verlag; Berlin-Heidelberg: 2011. pp. 189–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen HH, Lyngbye L, Bols M. Angew Chem Int Ed. 2001;40:3447–3449. doi: 10.1002/1521-3773(20010917)40:18<3447::aid-anie3447>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Jensen HH, Lyngbye L, Jensen A, Bols M. Chem Eur J. 2002;8:1218–26. doi: 10.1002/1521-3765(20020301)8:5<1218::aid-chem1218>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 27.Heuckendorff M, Pedersen CM, Bols M. Chem Eur J. 2010;16:13982–13994. doi: 10.1002/chem.201002313. [DOI] [PubMed] [Google Scholar]
- 28.Jensen HH, Bols M. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 29.McDonnell C, López O, Murphy P, Fernández Bolaños JG, Hazell R, Bols M. J Am Chem Soc. 2004;126:12374–85. doi: 10.1021/ja047476t. [DOI] [PubMed] [Google Scholar]
- 30.Pedersen CM, Nordstrøm LU, Bols M. J Am Chem Soc. 2007;129:9222–35. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- 31.Jensen HH, Pedersen CM, Bols M. Chem Eur J. 2007;13:7576–82. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]
- 32.Pedersen CM, Marinescu LG, Bols M. Chem Commun. 2008:2465–7. doi: 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]
- 33.Heuckendorff M, Pedersen CM, Bols M. J Org Chem. 2012;77:5559–68. doi: 10.1021/jo300591k. [DOI] [PubMed] [Google Scholar]
- 34.Hosoya T, Ohashi Y, Matsumoto T, Suzuki K. Tetrahedron Lett. 1996;37:663–666. [Google Scholar]
- 35.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 36.Mydock LK, Demchenko AV. Org Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mydock LK, Demchenko AV. Org Lett. 2008;10:2107–2110. doi: 10.1021/ol800648d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Premathilake HD, Mydock LK, Demchenko AV. J Org Chem. 2010;75:1095–1100. doi: 10.1021/jo9021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walford C, Heath LS. Chem Commun. 1997:1855–1856. [Google Scholar]
- 40.Yamada H, Nakatani M, Ikeda T, Marumoto Y. Tetrahedron Lett. 1999;40:5573–5576. [Google Scholar]
- 41.Crich D, Li M. Org Lett. 2007;9:4115–4118. doi: 10.1021/ol701466u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heuckendorff M, Pedersen CM, Bols M. Org Lett. 2011;13:5956–5959. doi: 10.1021/ol202308y. [DOI] [PubMed] [Google Scholar]
- 43.The reactivity difference between anomers can be more complicated when having conformational restricted glycosyl donors. See; Heuckendorff M, Pedersen CM, Bols M. J Org Chem. 2013;78:7234–7248. doi: 10.1021/jo4012464. for a discussion of this.
- 44.Mydock LK, Kamat MN, Demchenko AV. Org Lett. 2011;13:2928–2931. doi: 10.1021/ol2009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.