

Abstract
Virulent Clostridium difficile strains produce toxin A and/or toxin B that are the etiological agents of diarrhea and pseudomembranous colitis. Treatment of C. difficile infections (CDI) has been hampered by resistance to multiple antibiotics, sporulation, emergence of strains with increased virulence, recurrence of the infection, and the lack of drugs that preserve or restore the colonic bacterial flora. As a result, there is new interest in non-antibiotic CDI treatments. The human conjugated bile salt taurocholate was previously shown in our laboratory to inhibit C. difficile toxin A and B activities in an in vitro assay. Here we demonstrate for the first time in an ex vivo assay that taurocholate can protect Caco-2 colonic epithelial cells from the damaging effects of the C. difficile toxins. Using caspase-3 and lactate dehydrogenase assays, we have demonstrated that taurocholate reduced the extent of toxin B-induced apoptosis and cell membrane damage. Confluent Caco-2 cells cultured with toxin B induced elevated caspase-3 activity. Remarkably, addition of 5 mM taurocholate reduced caspase-3 activity in cells treated with 2, 4, 6, and 12 µg/ml of toxin B by 99%, 78%, 64%, and 60%, respectively. Furthermore, spent culture medium from Caco-2 cells incubated with both toxin B and taurocholate exhibited significantly decreased lactate dehydrogenase activity compared to spent culture medium from cells incubated with toxin B only. Our results suggest that the mechanism of taurocholate-mediated inhibition functions at the level of toxin activity since taurocholate did not affect C. difficile growth and toxin production. These findings open up a new avenue for the development of non-antibiotic therapeutics for CDI treatment.
Introduction
Clostridium difficile infection (CDI) is the most common definable cause of hospital-acquired and antibiotic-associated diarrhea in the United States [1]. Pathogenic strains of C. difficile possess a 19.6 kb pathogenicity locus responsible for the production of toxin A (308 kDa) and toxin B (269 kDa). These toxins are crucial to C. difficile pathogenesis [2], [3], [4], [5], [6], such that strains that do not produce either of these toxins are not associated with disease. Both toxins have similar enzymatic cleavage activities [7], [8], [9] and are cytotoxic to cultured cells; however, toxin B is 100-1,000-fold more potent than toxin A [5], [10], [11]. During infection, these toxins are released into the intestinal lumen where they bind to surface receptors on colonic epithelial cells via their receptor-binding domain and are then internalized by host cells via receptor-mediated endocytosis [12], [13]. The acidic environment within the endosomes activates the cysteine protease activity of the toxins, which cleaves and releases the glucosyltransferase domain located at the N-terminus into the cytosol of the mammalian host [14], [15], [16], [17], [18]. The glucosyltransferase domain monoglucosylates low molecular weight GTPases of the Rho family (RhoA, B, C, Rac, and Cdc42) in the cytosol using cellular uridine diphosphoglucose (UDP-glucose) as the glucose donor [8], [11]. This monoglucosylation interrupts the normal function of the Rho GTPases leading to various deleterious effects including apoptosis, inflammation, cell rounding, actin cytoskeleton dysregulation, and altered cellular signaling [8], [11], [14], [19], [20].
Primary bile salts (cholate and chenodeoxycholate) are biosynthesized from cholesterol in the liver and are conjugated with either glycine or taurine prior to their release into the gall bladder [21], [22], [23]. Conjugation makes bile salts less hydrophobic, more soluble, and prevents passive re-absorption as they traverse the gastrointestinal tract [24]. In addition to their role in fat digestion and absorption, bile salts also inhibit bacterial overgrowth in the small intestine [25], [26], a major site of absorption of nutrients and other metabolites. Consequently, various bacteria synthesize hydrolases that modify conjugated bile salts by deconjugation. Further alterations can occur through dehydroxylation, dehydrogenation, and sulfation, resulting in the generation of secondary and tertiary bile salts [22], [27], [28], [29]. These bacterial modifications render bile salts essentially insoluble, resulting in decreased aqueous concentrations and bacteriostaticity. Moreover, the release of amino acids as a result of these modifications may also act as alternative electron acceptors in this anaerobic environment [30], [31], [32], improving bacterial growth. Bile-salt hydrolases are produced by several genera of enteric bacteria including Clostridium, Bifidobacterium, Bacteroides, Lactobacillus, and Enterococcus [29]. We suggest that modifications to bile salts have evolved to enable the intestinal microbiota to gain a survival advantage by counteracting this host defense mechanism.
The foundation of CDI is the ability of this multiple antibiotic-resistant bacterium to overpopulate the human gastrointestinal tract following reduction of the normal gut microbiota by antibiotic therapy [33], [34]. It is well documented that antibiotic treatment is the greatest risk factor for CDI [35], [36], [37], [38], [39]. A large number of C. difficile isolates have shown an alarming pattern of resistance to the majority of antibiotics currently used in hospitals and outpatient settings [40], [41], [42], [43]. The dwindling number of antibiotics available to effectively clear CDI and prevent recurrence has sparked new interest in identifying and developing alternative non-antibiotic treatments, either as stand-alone therapies or as adjunctive therapies designed to augment the efficacy of the current antibiotic regimens. The proposed non-antibiotic treatments include: infusion of stool from healthy donors [44], [45], [46], [47], adjunctive use of monoclonal antibodies specific to the toxins [48], probiotics [49], and use of non-toxigenic C. difficile strains to out-compete toxigenic strains [50], [51], [52]. As the toxins play an essential role in C. difficile pathogenesis, inhibition of either toxin production or toxin activity is another promising approach.
Taurocholate, a major human conjugated bile salt, was previously reported by our laboratory to inhibit the in vitro substrate cleavage activity of the C. difficile toxins A and B in a dose-dependent manner [53]. In the present study, we demonstrate that physiologic concentrations of taurocholate protect Caco-2 colonic epithelial cells from the damaging effects of the toxins. These findings open up a new avenue that could be harnessed for the development of non-antibiotic therapeutic treatments for CDI.
Materials and Methods
Bacterial Strains and Growth Conditions
Active toxin producing C. difficile strains VPI 10463 [ATCC (American Type Culture Collection) 43255 (tcdA+ tcdB+)], 630 [ATCC BAA-1382 (tcdA+ tcdB+)], 5325 [ATCC BAA-1875 (tcdA+ tcdB+)], VPI 11186 [ATCC 700057 (tcdA− tcdB+)], and the hypervirulent strain ATCC BAA-1805 (tcdA+ tcdB+) were purchased from the ATCC (Manassas, VA). Brain heart infusion (BHI) medium was purchased from Becton Dickinson (Cockeysville, MD). The bacteria were grown in either liquid culture in BHI medium or on Cdifftox Agar plates [54] and incubated anaerobically in an atmosphere of 85% N2, 10% H2, and 5% CO2 at 37°C in a Controlled Atmosphere Anaerobic Chamber (PLAS LABS, Lansing, MI). Sodium taurocholate was purchased from Sigma-Aldrich (St. Louis, MO).
Growth of C. difficile Strains in BHI Medium Containing Taurocholate
Overnight cultures (OD600nm = 1.4) of each strain of C. difficile were diluted 1∶100 in 30 ml of fresh BHI broth medium in the presence or absence of 5 mM taurocholate and incubated anaerobically at 37°C. Portions of the cultures were removed every 2 hrs to monitor cell growth by optical density measurements at 600 nm. After a 48-hr incubation period, 1.5 ml of respective cultures were centrifuged at 10,000 x g for 10 mins and the supernatants were tested for the presence of toxins A and B using the enzyme-linked immunosorbent (ELISA)-based Wampole C. difficile TOX A/B II assay (TechLab, Blacksburg, VA). The culture supernatants were also tested for toxin activity using the Cdifftox activity assay [53]. Briefly, 250 µl of the culture supernatants were incubated with 5 mM of p-nitrophenyl-β-D-glucopyranoside at 37°C for 4 hrs. The cleavage of the substrate by the toxins was measured spectrophometrically at 410 nm.
Growth of Caco-2 Cells and Treatment with Taurocholate and C. difficile Toxins
The Caco-2 cell line (ATCC HTB-37), a human colonic epithelial cell line was purchased from ATCC. The cells were cultured and maintained in Dulbecco's minimal essential medium (DMEM) containing 10% fetal bovine serum in a humidified incubator with 5% CO2. No antibiotics were used in the preparation of the media. Cells were grown to confluence in 24-well culture plates (Corning, Corning, NY) in a final volume of 2 ml as described [55], prior to adding taurocholate in the presence or absence of purified C. difficile toxin A or B. The cells were visualized by light microscopy every 24 hrs over a 5-day period using an EVIS XL microscope (Advanced Microscopy Group, Bothell, WA). Cells in different treatment groups were evaluated for morphological changes including rounding, cytoskeleton disruption, and cell death resulting from exposure to the C. difficile toxins.
The Caco-2 cells were initially treated with different concentrations of taurocholate (1–25 mM) to determine the appropriate amount that could be tolerated. We determined that 5 mM taurocholate was the ideal amount, as it resulted in undetectable morphological differences when compared to the untreated control cells. Furthermore, purified toxins A and B were both tested initially for cytopathic and cytotoxic effects, but the damaging effect of toxin B was more pronounced and remarkable than toxin A. In fact, toxin B is reported to be 100-1,000-fold more potent than toxin A [5], [10], [11]. As a result, we focused the rest of the experiments on toxin B and 5 mM taurocholate.
Immunoblot Analysis
C. difficile toxins A and B were purified from the hyper-toxin producing strain VPI 10463 (ATCC 43255) according to our recently reported method [53]. Purified C. difficile toxins A and B (80 µg each) were separated on 6% polyacrylamide electrophoresis (PAGE) gels and transferred onto Immun-Blot PVDF membrane (BioRad, Hercules, CA) using a Trans-Blot cell (BioRad) transfer apparatus. The membrane was blocked overnight in 10 mM Tris buffered saline with 0.05% tween-20 (TBST) containing 5% skim milk. Following blocking, the membrane was incubated with mouse monoclonal antibodies specific for C. difficile toxins A or B (Abcam, Cambridge, MA). The Pierce ECL Western Blotting Kit (Thermo Scientific, Rockford, IL) was then used to probe the membrane for the presence of each toxin using an HRP-conjugated goat anti-mouse IgG secondary antibody, followed by incubation with the ECL substrate according to the manufacturer's instructions. The treated membrane was exposed to X-ray film (Molecular Technologies, St Louis, MO) and processed using a Konica film processor (Konica Corporation, Tokyo, Japan).
Caspase-3 Activity Assay
Confluent Caco-2 cells were incubated for 24 hrs in the presence of toxin B and 5 mM taurocholate. Following the incubation period, the cells were removed from the 24-well plates using the tip of a 1-ml pipette to scrape the attached cells from the bottom of the wells. The cells were then transferred to 1.5 ml microcentrifuge tubes and centrifuged at 6,000 x g to separate the cells from the medium. Detection of caspase-3 activity was performed as described by the manufacturer of the Caspase-3 Colorimetric Kit (Invitrogen, Carlsbad, CA). Briefly, the harvested cells were lysed on ice for 10 min using 50 µl of cell lysis buffer and centrifuged at 10,000 x g for 15 mins. For this assay, 75 µg of protein was incubated with the substrate reagent for 8 hrs at 37°C and the absorbance at 410 nm was measured using the Spectramax Plus spectrophotomer (Molecular Devices, Sunnyvale, CA). A molar extinction coefficient for p-nitrophenol of ε = 17700 M−1cm−1 was used for the calculations of caspace-3 activity [56]. Total lysate protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.).
Lactate Dehydrogenase Assay
The toxicity and membrane integrity of confluent Caco-2 cells incubated with C. difficile toxin B in the presence or absence of 5 mM taurocholate was evaluated. The CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega, Madison, WI) was used to determine the activity of lactate dehydrogenase (LDH) in the spent culture medium. This assay is based on the release of cytosolic LDH from cells with damaged cellular membranes. Confluent Caco-2 cell monolayers were incubated with 4, 8, 12, and 24 µg of purified toxin B in the presence or absence of 5 mM taurocholate in a total medium volume of 2 ml in 24-well plates for 24 hrs. The supernatant was tested for LDH activity according to the protocol provided by the manufacturer. Briefly, 100 µL of the CytoTox-ONE Reagent was added to 100 µL of the culture supernatant in a 96-well microtiter plate and incubated at 22°C for 10 min. Following the incubation period, 50 µL of Stop Solution was added to each well. Fluorescence was measured using Spectromax I3 (Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 560 nm and an emission wavelength of 590 nm.
Data Analysis
All the data were analyzed and plotted using GraphPad Prism version 6.02 for Windows (GraphPad Software, San Diego, California). One-Way ANOVA and Wilcoxon-Mann-Whitney tests were used to compare differences between the samples. In all cases, statistical significance was defined as having a P value of <0.05.
Results
Purification of the C. difficile Toxins
To evaluate the effect of C. difficile toxins on confluent Caco-2 cells, native A and B toxins were purified from supernatant fluid generated from cultures of the toxin-producing strain VPI 10463 grown in dialysis bags with a 100-kDa molecular weight cut off. The purification steps consisted of DEAE-Sepharose anion exchange and Sephacryl S-300 gel filtration chromatography as previously reported [53]. Analysis of the final purified fractions by PAGE, followed by Coomassie staining confirmed the presence of two single bands in each final fraction (data not shown) corresponding to toxins A and B, respectively, as detected by immunoblot analysis (Fig. 1).
Figure 1. Immunoblot analysis of purified C. difficile toxins A and B.
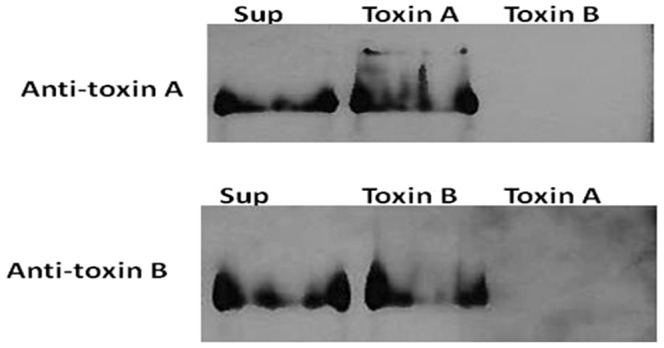
Purified proteins (80 µg each) were subjected to 6% PAGE and transferred onto PVDF membranes. Each membrane was probed using monoclonal primary antibodies specific for toxin A or B. The Pierce ECL Western Blotting Kit was used to detect the bound antibodies. The membrane was exposed to X-ray film (Molecular Technologies, St Louis, MO) and processed using a Konica film processor (Konica Corporation, Tokyo, Japan). Sup, crude culture supernatant; Toxin A, purified toxin A; Toxin B, purified toxin B.
Taurocholate and Toxin Titration Assays
The total concentration of bile salts in the human small bowel ranges from 2 to 30 mM, depending on diet and other metabolic conditions [57]. To determine the amount of taurocholate that could be tolerated by Caco-2 cells, confluent Caco-2 cells were cultured in the presence of 1 to 25 mM taurocholate, as described in the Materials and Methods. From this analysis, 5 mM of taurocholate was determined to be the optimum concentration tolerated based on cell viability. Confluent cells incubated with and without 5 mM taurocholate were indistinguishable (Fig. 2, A-B). The cells were not viable at taurocholate concentrations above 20 mM (data not shown). Confluent Caco-2 cells were also cultured in the presence of increasing amounts of the toxins (0, 4, 8, 12, 16, and 24 µg) to determine the amount of each toxin needed to elicit visible cytotoxic or cytopathic changes. Both toxins A and B caused damage to the cells. However, cell damage was more pronounced within 24 hrs of incubation with toxin B. The toxin-treated CaCo-2 cells appeared rounded, spindle-like, detached from the plate surface, and presented with an altered cytoskeleton; all of these phenotypes were consistent with previous reports describing the cytopathic and cytotoxic effects of these toxins [19], [20]. Visualization of the cell damage caused by toxin A required longer incubation times of more than 48 hrs compared to that required for toxin B (data not shown). These results confirmed the earlier reports that toxin B is more potent than toxin A [5], [10], [11]. As a consequence, we focused on the more potent toxin B for the Caco-2 protection experiments described.
Figure 2. Effect of C. difficile toxin B and taurocholate on Caco-2 Cells.

Confluent Caco-2 cell monolayers were incubated with 8 and 16 µg of toxin B in the presence or absence of 5 mM taurocholate in a total medium volume of 2 ml in 24-well plates for 24 hrs. Images were captured using an EVIS XL microscope. Magnification 10×. Tox B, purified toxin B; TC, taurocholate.
Taurocholate Protects Caco-2 Cells from Toxin B-Mediated Toxicity
Taurocholate was shown previously in our laboratory to inhibit the substrate cleavage activity of toxins A and B in vitro in a dose-dependent manner [53]. Confluent Caco-2 cells were incubated with toxin B in the presence or absence of 5 mM taurocholate to test whether taurocholate could protect the cells from toxin B-mediated toxicity ex vivo (Fig. 2). When a lethal dose of toxin B (16 µg) was added to Caco-2 monolayers in the presence of taurocholate, no detectable cytopathic damage to the cells was apparent (Fig. 2F). A similar protective effect of taurocholate was observed when the cells were also incubated with toxin A (data not shown). These data indicate that taurocholate protects the cells from toxin-mediated damage and supports our previous report [53], which demonstrated that taurocholate inhibits the activities of toxins A and B.
Taurocholate Decreases C. difficile Toxin B-Mediated Induction of Host Cell Caspase-3 Production
During C. difficile infection, apoptosis is an important downstream effect resulting from receptor-mediated toxin endocytosis into the host cell cytoplasm and subsequent inactivation of the host GTPases. Previous reports showed that toxin A induces cell death in human epithelial cells ex vivo via the activation of caspases [58], [59], [60]. Our results indicate that various amounts of toxin B (4, 8, 12, and 24 µg) induced significant caspase-3 activity in Caco-2 cells (Fig. 3). The caspase-3 activation cascade plays a crucial role in several apoptotic mechanisms, including activation of key apoptotic mediators essential to chromatin condensation, DNA fragmentation, dismantling of the cell, and formation of apoptotic bodies [61], [62], [63]. To determine whether taurocholate protected the Caco-2 cells from toxin B damage by preventing apoptosis, caspase-3 activity was assessed in taurocholate-treated and untreated cells. Crude protein lysates prepared from confluent Caco-2 cells incubated with various amounts of toxin B in the presence or absence of taurocholate were tested for caspase-3 activity. In cells cultured without toxin B or taurocholate, no caspase-3 activity was detected. In the presence of toxin B, however, caspase-3 activity was detected in a dose-dependent manner (Fig. 3). Cells cultured with 4–24 µg of toxin B showed a 2.5-7.5-fold increase in caspase-3 activity. In contrast, cells cultured with toxin B in the presence of taurocholate had significant reduction in caspase-3 activity. Remarkably, addition of 5 mM taurocholate reduced caspase-3 activity in cells treated with 4, 8, 12, and 24 µg of toxin B by 99%, 78%, 64%, and 60%, respectively. These data demonstrated that taurocholate protected Caco-2 cells from the damaging effects of toxin B, as evidenced by the decreased levels of the pro-apoptotic protease, caspase-3.
Figure 3. Taurocholate significantly decreases C. difficile toxin B-mediated induction of caspase-3 activity in Caco-2 cells.

Confluent Caco-2 cells were incubated for 24 hrs with 0, 4, 8, 12 and 24 µg of toxin B in the presence (+) or absence (−) of 5 mM taurocholate. Cell monolayers were scraped from the 24-well plates and lysed to obtain crude protein lysates. Crude protein lysates (75 µg) were incubated with the caspase-3 colorimetric substrate reagent (Invitrogen) for 8 hrs at 37°C and absorbance at 410 nm was measured. A molar extinction coefficient for p-nitrophenol of ε = 17700 M−1cm−1 was used in the calculations of caspace-3 activity [56]. One-Way ANOVA test showed a significant difference (P<0.003) between the caspase-3 activities of the cells cultured with 5 mM taurocholate and the cells cultured without taurocholate. Error bars represents the standard deviation from three replicate experiments.
Taurocholate Impacts the Membrane Integrity of Toxin B-Treated Cells
The membrane integrity of confluent Caco-2 cells following incubation with toxin B was evaluated in the presence of taurocholate using the CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega). This assay was used to determine the activity of lactate dehydrogenase (LDH) in the medium as a result of toxin B-mediated damage to the cells. LDH is a soluble cytosolic enzyme, which is released into the extracellular medium as a consequence of damage to the cell membrane. The results showed that the membrane integrity of Caco-2 cells incubated with toxin B was compromised in a dose dependent manner, as evidenced by an increased LDH activity (Fig. 4). Interestingly, spent culture medium from confluent Caco-2 cells incubated with both toxin B and taurocholate exhibited significantly less LDH activity compared to spent medium from cells incubated with toxin B only. These results support the morphological defects and cytotoxic effects observed microscopically in the toxin B-treated cells (Fig. 1), and underscore the significance of taurocholate in protecting the cells from toxin damage.
Figure 4. Taurocholate significantly decreases toxin B-mediated Caco-2 cell membrane integrity.

Confluent Caco-2 cells were incubated for 24 hrs with 0, 4, 8, 12 and 24 µg of toxin B in the presence (+) or absence (−) of 5 mM taurocholate. The spent culture supernatant fluid (100 µL) was tested for lactate dehydrogenase activity using the CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega). Fluorescence was measured using Spectromax I3 (Molecular Devices) at an excitation wavelength of 560 nm and an emission wavelength of 590 nm. One-Way ANOVA test showed a significant difference (P<0.001) between the lactate dehydrogenase activities of toxin B-treated Caco-2 cells cultured with 5 mM taurocholate and the cells cultured without taurocholate. Error bars represents the standard deviation from three replicate experiments.
Taurocholate Inhibits C. difficile Toxin Activity with no Significant Effect on Growth or Toxin Production
In addition to their role in fat digestion and absorption, bile salts also inhibit bacterial overgrowth in the small intestine [25], [26], which is a major site of nutrient and metabolite absorption. Some enteric bacteria such as E. coli and Salmonella produce various bile salt hydrolases capable of modifying bile salts and rendering them non-toxic to bacteria. Analysis of the C. difficile genome revealed the presence of homologues of bile salt hydrolases similar to those characterized in classic enteric bacteria. To examine whether C. difficile could grow in the presence of the physiologic taurocholate concentration used, different toxigenic C. difficile strains were cultured in the presence of 5 mM taurocholate. Some strains appeared to grow better than others when cultured with taurocholate, however, none of the strains tested exhibited viability defects (Fig. 5).
Figure 5. Taurocholate has no significant effect on the growth of C. difficile toxin A- and B-producing strains.

Overnight cultures (OD 600 nm = 1.4) of each strain of C. difficile tested were diluted 1∶100 in 30 ml of fresh medium with or without 5 mM of taurocholate (TC) and incubated anaerobically at 37°C for 24 hrs. Portions (2 ml) of the cultures were removed at the indicated times for OD600 nm measurement. Strain designations: 630, ATCC BAA-1382; 432, ATCC 43255; 1805, ATCC BAA-1805; 1875, ATCC BAA-1875. The error bars represent the standard deviation from three different experiments. Mann-Whitney test showed no significant difference between the C. difficile cells growth in the presence or absence of 5 mM taurocholate.
To assess the effect of taurocholate on toxin production and toxin activity, an ELISA-based assay was used to analyze toxin production and the Cdifftox activity assay [53] was used to determine toxin activity. As shown in Figure 6A, the presence of taurocholate did not affect toxin production. However, toxin activity was significantly lower (P<0.05) in the supernatant fluids of the strains cultured in the presence of taurocholate (Fig. 6B). The addition of 5 mM taurocholate to the C. difficile culture medium reduced the total toxin activity differently, depending on the strain: 91% (ATCC BAA-1382), 85% (ATCC 700057), 70% (ATCC 43255), 67% (ATCC BAA-1875), and 61% (ATCC BAA-1805). These results indicate that taurocholate specifically inhibits toxin activity without affecting toxin production, and suggests that bile salts play an important role in the pathogenesis of the C. difficile toxins. Furthermore, the presence of genes that encode bile salt hydrolases in the C. difficile genomes did not appear to affect the inhibition of C. difficile toxin activity by taurocholate.
Figure 6. Taurocholate has no effect on C. difficile toxin production (A), but inhibits C. difficile toxin activity (B).

Overnight cultures (OD600 nm = 1.4) of each strain of C. difficile tested were diluted 1∶100 in 30 ml of fresh medium with or without 5 mM of taurocholate and incubated anaerobically at 37°C for 48 hrs. The culture supernatant fluids were tested for the presence of toxins by ELISA using the Wampole C. difficile TOX A/B II assay and toxin activity using the Cdifftox activity assay [53]. Strain designations: 630, ATCC BAA-1382; 432, ATCC 43255; 1805, ATCC BAA-1805; 057, ATCC 700057; 1875, ATCC BAA-1875. The error bars represent the standard deviation from three different experiments. TC = taurocholate. Mann-Whitney test showed no significant difference [P = 0.333 (630) P = 0.332 (432); P = 0.667 (1805); P = 0.333 (057); P = 0.999 (1875)] in the amount of toxin produced by C. difficile cells cultured either with 5 mM taurocholate or without taurocholate. However, toxin activity detected in the culture supernatant fluids of C. difficile cells cultured with taurocholate was significantly lower than those cultured without taurocholate.
Discussion
Clostridium difficile is the etiological agent of antibiotic-associated colitis, including pseudomembranous colitis, and the leading definable cause of nosocomial diarrhea. Treatment of C. difficile infections (CDI) has been hampered by recurrence of the infection, emergence of strains with increased virulence, sporulation, multi-drug resistance, lack of drugs with superior functional activity in the colon, and the lack of drugs that restore or preserve the colonic microbiota following antibiotic treatment. As a result, there is new interest in finding alternative treatments, either as stand-alone therapies or therapies designed to augment the efficacy of currently used antibiotic regimens. A novel treatment approach would be to inhibit the activities of toxins A and B, which are directly responsible for the intestinal damage and subsequent inflammation associated with CDI. An approach, which targets the toxins without affecting cell growth, may be ideal since it is unlikely to impose selective pressure on C. difficile, thereby minimizing the risk of developing resistance. In our search for compounds that inhibit toxin activity we developed the Cdifftox assay [53], which detects the ability of toxins A and B to cleave a chromogenic substrate that is stereochemically similar to their native substrate (UDP-glucose). Using this Cdifftox assay, we identified taurocholate as a compound that inhibits in vitro the substrate cleavage activity of these toxins [53].
In this report, we demonstrated that a physiologic concentration of taurocholate (5 mM) protected ex vivo confluent human colonic epithelial Caco-2 cells from C. difficile toxin B-mediated damage. Taurocholate also protected the cells from toxin A damage (data not shown). When taurocholate and toxin B (16 µg) were added simultaneously to confluent Caco-2 cell monolayers, toxin-mediated cytopathic effects were prevented (Fig. 2). One of the mechanisms by which C. difficile toxins mediate cell damage is by inducing apoptosis. Specifically, toxin A has been reported to induce cell death in human epithelial cells ex vivo by activating caspases [58], [59], [60]. We demonstrate here for the first time that toxin B induces caspase-3 production in Caco-2 cells in a dose-dependent manner (Fig. 3). Furthermore, toxin B also induced membrane damage in a dose-dependent manner, as evidenced by the elevated lactate dehydrogenase activity in the culture medium (Fig. 4). Remarkably, both caspase-3 and lactate dehydrogenase activities were significantly reduced when toxin B-treated confluent Caco-2 cells were cultured with physiologic concentration of taurocholate. The protective effect of taurocholate was apparent even when the cells were treated with lethal doses of toxin B.
The concentration of taurocholate used in this study did not affect the growth or toxin production of the C. difficile strains tested (Fig. 5, 6A). However, it significantly decreased the total toxin activity in the supernatant fluids of all the strains tested (Fig. 6B). We noted that the lowest percent inhibition of toxin activity was observed in the hypervirulent C. difficile strains that produce high levels of the toxins, suggesting that more taurocholate may be necessary to neutralize the cells.
The mechanism of taurocholate-mediated inhibition of C. difficile toxin activity remains to be determined. Brandes et al.[64] reported that tauroursodeoxycholic acid, a modified conjugated bile acid, induced phosphorylation of Rac1/Cdc42 leading to inhibition of C. difficile toxin B-mediated monoglucosylation of this GTPase. Taurocholate may function through hydrophobic interactions to saturate the Caco-2 cell membranes, thereby inhibiting toxin entry and/or toxin activity. Other possible inhibitory mechanisms may involve direct effects of taurocholate on the toxins, such as a structural alteration with subsequent loss of toxin activity, or binding of taurocholate to the toxins leading to the prevention of entry into the host cell. Further research is on-going to identify the mechanism of taurocholate action.
One limitation of this ex vivo study was the inability to co-culture C. difficile with the Caco-2 cells in the presence of taurocholate; C. difficile does not grow under aerobic conditions, and Caco-2 cells do not grow under anaerobic conditions. Thus, an in vivo C. difficile animal model is required to provide more insight into how these findings may be exploited to develop a treatment intervention against C. difficile infections.
It is important to note that the majority of nutrient absorption in the gastrointestinal tract occurs in the small intestine, where bile salts are at much higher concentrations compared to the colon. This difference in bile salts concentration is due to the reabsorption of more than 95% of the total human bile via the enterohepatic circulation in the ileum [65], which is directly upstream of the colon. Clearly, only a small amount of bile salts enter the colon where C. difficile most frequently colonizes. An intriguing explanation for why CDI pathology is mostly limited to the bile salt-deficient colon, and not the bile salt-rich small intestine, is that toxin activity may be inhibited in the small intestine by the high bile salt concentrations. Our data suggest that the C. difficile toxins are active in the colon because of its low bile salt concentrations. The therapeutic benefits of bile salts are well documented; they prevent hepatocyte injury and cholestasis [66], [67], [68], drug-induced cholestasis [69], and endotoxin absorption [70], [71]. Our findings highlight the point that bile salt concentration represents a host-mediated mechanism that naturally protects the absorptive surfaces of the small intestine from deleterious microbial products and also acts to inhibit bacterial growth. Thus, the lack of bile salts in the small intestine in diseased states (such as cirrhosis of the liver) may lead to bacterial overgrowth and result in a competition for the essential nutrients required for normal human growth and function.
We suggest that uncovering a mechanism to deliver higher concentrations of bile salts and/or their derivatives, perhaps in conjunction with antibiotics, into the colon of individuals suffering from recurrent CDI may help protect the colon from the damaging effects of the C. difficile toxins and facilitate clearance of the pathogen. This line of research may result in a novel treatment of C. difficile infections; an option likely to maintain intestinal homeostasis and minimize the risk of drug resistance.
Acknowledgments
We thank Dr. Alex Therien (Merck & Co) for helpful discussions.
Funding Statement
This work was supported in part, by discretionary funds from The University of Texas School of Public Health, Texas Medical Center Digestive Diseases Center (Public Health Service Grant DK56338), and United Negro College Fund-MERCK Science Initiative (University of Michigan School of Information). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. DuPont HL (2011) The search for effective treatment of Clostridium difficile infection. N England J Med 364: 473–475. [DOI] [PubMed] [Google Scholar]
- 2. Lyerly DM, Saum KE, MacDonald DK, Wilkins TD (1985) Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 47: 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rupnik M, Brazier JS, Duerden BI, Grabnar M, Stubbs SL (2001) Comparison of toxinotyping and PCR ribotyping of Clostridium difficile strains and description of novel toxinotypes. Microbiol 147: 439–447. [DOI] [PubMed] [Google Scholar]
- 4. Geric B, Rupnik M, Gerding DN, Grabnar M, Johnson S (2004) Distribution of Clostridium difficile variant toxinotypes and strains with binary toxin genes among clinical isolates in an American hospital. J Med Microbiol 53: 887–894. [DOI] [PubMed] [Google Scholar]
- 5. Voth DE, Ballard JD (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18: 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, et al. (2010) The role of toxin A and toxin B in Clostridium difficile infection. Nature 467: 711–713. [DOI] [PubMed] [Google Scholar]
- 7. Dillon ST, Rubin EJ, Yakubovich M, Pothoulakis C, LaMont JT, et al. (1995) Involvement of Ras-related Rho proteins in the mechanisms of action of Clostridium difficile toxin A and toxin B. Infect Immun. 63: 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, et al. (1995) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 375: 500–503. [DOI] [PubMed] [Google Scholar]
- 9. Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, et al. (1995) The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270: 13932–13936. [DOI] [PubMed] [Google Scholar]
- 10. Von Eichel-Streiber C, Boquet P, Sauerborn M, Thelestam M (1996) Large clostridial cytotoxins--a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol 4: 375–382. [DOI] [PubMed] [Google Scholar]
- 11. Just I, Gerhard R (2004) Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol 152: 23–47. [DOI] [PubMed] [Google Scholar]
- 12. Ho JG, Greco A, Rupnik M, Ng KK (2005) Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci U S A. 102: 18373–18378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dingle T, Wee S, Mulvey GL, Greco A, Kitova EN, et al. (2008) Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile . Glycobiol 18: 698–706. [DOI] [PubMed] [Google Scholar]
- 14. Hofmann F, Busch C, Prepens U, Just I, Aktories K (1997) Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem 272: 11074–11078. [DOI] [PubMed] [Google Scholar]
- 15. Egerer M, Giesemann T, Jank T, Satchell KJ, Aktories K (2007) Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 282: 25314–25321. [DOI] [PubMed] [Google Scholar]
- 16. Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, et al. (2007) Autocatalytic cleavage of Clostridium difficile toxin B. Nature. 446: 415–419. [DOI] [PubMed] [Google Scholar]
- 17. Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, et al. (2003) Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 278: 44535–44541. [DOI] [PubMed] [Google Scholar]
- 18. Rupnik M, Pabst S, von Eichel-Streiber C, Urlaub H, Soling HD (2005) Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiol 151: 199–208. [DOI] [PubMed] [Google Scholar]
- 19. Genth H, Dreger SC, Huelsenbeck J, Just I (2008) Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol 40: 592–597. [DOI] [PubMed] [Google Scholar]
- 20. Huelsenbeck SC, May M, Schmidt G, Genth H (2009) Inhibition of cytokinesis by Clostridium difficile toxin B and cytotoxic necrotizing factors--reinforcing the critical role of RhoA in cytokinesis. Cell Motil Cytoskeleton 66: 967–975. [DOI] [PubMed] [Google Scholar]
- 21. Moser SA, Savage DC (2001) Bile salt hydrolase activity and resistance to toxicity of conjugated bile salts are unrelated properties in lactobacilli. Appl Environ Microbiol 67: 3476–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47: 241–259. [DOI] [PubMed] [Google Scholar]
- 23. Begley M, Hill C, Gahan CG (2006) Bile salt hydrolase activity in probiotics. Appl Environ Microbiol 72: 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hofmann AF (1999) The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 159: 2647–2658. [DOI] [PubMed] [Google Scholar]
- 25. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, et al. (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 103: 3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sung JY, Shaffer EA, Costerton JW (1993) Antibacterial activity of bile salts against common biliary pathogens. Effects of hydrophobicity of the molecule and in the presence of phospholipids. Dig Dis Sci 38: 2104–2112. [DOI] [PubMed] [Google Scholar]
- 27. Lillienau J, Borgstrom B (1991) Bacterial deconjugation and enterohepatic circulation of norursocholic acid conjugates in rats. Am J Physiol 261: G1065–1071. [DOI] [PubMed] [Google Scholar]
- 28. Tanaka H, Doesburg K, Iwasaki T, Mierau I (1999) Screening of lactic acid bacteria for bile salt hydrolase activity. J Dairy Sci 82: 2530–2535. [DOI] [PubMed] [Google Scholar]
- 29. Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR (2008) Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A 105: 13580–13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huijghebaert SM, Mertens JA, Eyssen HJ (1982) Isolation of a bile salt sulfatase-producing Clostridium strain from rat intestinal microflora. Appl Environ Microbiol 43: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Eldere J, Robben J, De Pauw G, Merckx R, Eyssen H (1988) Isolation and identification of intestinal steroid-desulfating bacteria from rats and humans. Appl Environ Microbiol 54: 2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Smet I, Van Hoorde L, Vande Woestyne M, Christiaens H, Verstraete W (1995) Significance of bile salt hydrolytic activities of Lactobacilli. J Appl Bacteriol 79: 292–301. [DOI] [PubMed] [Google Scholar]
- 33. Pothoulakis C, Lamont JT (2001) Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol 280: G178–183. [DOI] [PubMed] [Google Scholar]
- 34. Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, et al. (2012) Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathogens 8: e1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aronsson B, Mollby R, Nord CE (1985) Antimicrobial agents and Clostridium difficile in acute enteric disease: epidemiological data from Sweden, 1980–1982. The J Infect Dis 151: 476–481. [DOI] [PubMed] [Google Scholar]
- 36. Bartlett JG (2009) New antimicrobial agents for patients with Clostridium difficile infections. Curr Infect Dis Rep 11: 21–28. [DOI] [PubMed] [Google Scholar]
- 37. Bartlett JG (1992) Antibiotic-associated diarrhea. Clin Infect Dis 15: 573–581. [DOI] [PubMed] [Google Scholar]
- 38. Bartlett JG (2002) Clinical practice. Antibiotic-associated diarrhea. N Engl J Med 346: 334–339. [DOI] [PubMed] [Google Scholar]
- 39. Bartlett JG, Gerding DN (2008) Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis 46 Suppl 1S12–18. [DOI] [PubMed] [Google Scholar]
- 40. Bishara J, Bloch Y, Garty M, Behor J, Samra Z (2006) Antimicrobial resistance of Clostridium difficile isolates in a tertiary medical center, Israel. Diagn Microbiol Infect Dis 54: 141–144. [DOI] [PubMed] [Google Scholar]
- 41. Aspevall O, Lundberg A, Burman LG, Akerlund T, Svenungsson B (2006) Antimicrobial susceptibility pattern of Clostridium difficile and its relation to PCR ribotypes in a Swedish university hospital. Antimicrob Agents Chemother 50: 1890–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shubeita HE, Sambrook JF, McCormick AM (1987) Molecular cloning and analysis of functional cDNA and genomic clones encoding bovine cellular retinoic acid-binding protein. Proc Natl Acad Sci U S A 84: 5645–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pelaez T, Alcala L, Alonso R, Rodriguez-Creixems M, Garcia-Lechuz JM, et al. (2002) Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob Agents Chemother 46: 1647–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aas J, Gessert CE, Bakken JS (2003) Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin Infect Dis 36: 580–585. [DOI] [PubMed] [Google Scholar]
- 45. Brandt LJ (2012) Fecal transplantation for the treatment of Clostridium difficile infection. Gastroenterol Hepatol (N Y) 8: 191–194. [PMC free article] [PubMed] [Google Scholar]
- 46. Mattila E, Uusitalo-Seppala R, Wuorela M, Lehtola L, Nurmi H, et al. (2012) Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterol 142: 490–496. [DOI] [PubMed] [Google Scholar]
- 47. Kelly CR, de Leon L, Jasutkar N (2012) Fecal microbiota transplantation for relapsing Clostridium difficile infection in 26 patients: methodology and results. J Clin Gastroenterol 46: 145–149. [DOI] [PubMed] [Google Scholar]
- 48. Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, et al. (2010) Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 362: 197–205. [DOI] [PubMed] [Google Scholar]
- 49. McFarland LV, Surawicz CM, Greenberg RN, Fekety R, Elmer GW, et al. (1994) A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 271: 1913–1918. [PubMed] [Google Scholar]
- 50. Borriello SP, Barclay FE (1985) Protection of hamsters against Clostridium difficile ileocaecitis by prior colonisation with non-pathogenic strains. J Med Microbiol 19: 339–350. [DOI] [PubMed] [Google Scholar]
- 51. Wilson KH, Sheagren JN (1983) Antagonism of toxigenic Clostridium difficile by nontoxigenic C. difficile . J Infect Dis 147: 733–736. [DOI] [PubMed] [Google Scholar]
- 52. Sambol SP, Merrigan MM, Tang JK, Johnson S, Gerding DN (2002) Colonization for the prevention of Clostridium difficile disease in hamsters. J Infect Dis 186: 1781–1789. [DOI] [PubMed] [Google Scholar]
- 53. Darkoh C, Kaplan HB, DuPont HL (2011) Harnessing the glucosyltransferase activities of Clostridium difficile for functional studies of toxins A and B. J Clin Microbiol. 49: 2933–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Darkoh C, DuPont HL, Kaplan HB (2011) Novel one-step method for detection and isolation of active-toxin-producing Clostridium difficile strains directly from stool samples. J Clin Microbiol 49: 4219–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brown EL, Xue Q, Jiang ZD, Xu Y, DuPont HL (2010) Pretreatment of epithelial cells with rifaximin alters bacterial attachment and internalization profiles. Antimicrob Agents Chemother 54: 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shikita M, Fahey JW, Golden TR, Holtzclaw WD, Talalay P (1999) An unusual case of 'uncompetitive activation' by ascorbic acid: purification and kinetic properties of a myrosinase from Raphanus sativus seedlings. Biochem J 341 (Pt 3): 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Northfield TC, McColl I (1973) Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14: 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gerhard R, Nottrott S, Schoentaube J, Tatge H, Olling A, et al. (2008) Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J Med Microbiol 57: 765–770. [DOI] [PubMed] [Google Scholar]
- 59. Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, et al. (2002) Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis 186: 1438–1447. [DOI] [PubMed] [Google Scholar]
- 60. Carneiro BA, Fujii J, Brito GA, Alcantara C, Oria RB, et al. (2006) Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro . Infect Immun 74: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Porter AG, Janicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell death and differentiation 6: 99–104. [DOI] [PubMed] [Google Scholar]
- 62. Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P (2007) Caspases in cell survival, proliferation and differentiation. Cell death and differentiation 14: 44–55. [DOI] [PubMed] [Google Scholar]
- 63. Yuan J, Horvitz HR (2004) A first insight into the molecular mechanisms of apoptosis. Cell 116: S53–56. [DOI] [PubMed] [Google Scholar]
- 64.Brandes V, Schelle I, Brinkmann S, Schulz F, Schwarz J, et al. (2011) Protection from C. difficile Toxin B-catalysed Rac/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac/Cdc42 phosphorylation. Biol Chem. [DOI] [PubMed]
- 65. Dowling RH (1973) The enterohepatic circulation of bile acids as they relate to lipid disorders. J Clin Pathol Suppl (Assoc Clin Pathol) 5: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Poupon RE, Balkau B, Eschwege E, Poupon R (1991) A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Eng J Med 324: 1548–1554. [DOI] [PubMed] [Google Scholar]
- 67. Heuman DM, Pandak WM, Hylemon PB, Vlahcevic ZR (1991) Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: in vitro studies in rat hepatocytes and human erythrocytes. Hepatol 14: 920–926. [DOI] [PubMed] [Google Scholar]
- 68. Heuman DM, Mills AS, McCall J, Hylemon PB, Pandak WM, et al. (1991) Conjugates of ursodeoxycholate protect against cholestasis and hepatocellular necrosis caused by more hydrophobic bile salts. In vivo studies in the rat. Gastroenterol 100: 203–211. [DOI] [PubMed] [Google Scholar]
- 69. Queneau PE, Bertault-Peres P, Mesdjian E, Durand A, Montet JC (1993) Diminution of an acute cyclosporin-induced cholestasis by tauroursodeoxycholate in the rat. Transplantation 56: 530–534. [DOI] [PubMed] [Google Scholar]
- 70. Bailey ME (1976) Endotoxin, bile salts and renal function in obstructive jaundice. British J Surgery 63: 774–778. [DOI] [PubMed] [Google Scholar]
- 71. Gouma DJ, Coelho JC, Fisher JD, Schlegel JF, Li YF, et al. (1986) Endotoxemia after relief of biliary obstruction by internal and external drainage in rats. Am J Surgery 151: 476–479. [DOI] [PubMed] [Google Scholar]