

Abstract
The antioxidant N-acetylcysteine (NAC) is widely used for the assessment of the role of reactive oxygen species (ROS) in various biological processes and adverse drug reactions. NAC has been found to effectively inhibit toxicity of carcinogenic metals, which was attributed to its potent ROS-suppressive properties. However, the absence of redox activity among some metals and findings from genetic models suggested a more diverse, smaller role of oxidative stress in metal toxicity. Here, we examined mechanisms of chemoprotection by NAC against Cd(II), Co(II) and Cr(VI) in human cells. We found that NAC displayed a broad-spectrum chemoprotective activity against all three metals, including suppression of cytotoxicity, apoptosis, p53 activation and HSP72 and HIF-1α upregulation. Cytoprotection by NAC was independent of cellular glutathione. NAC strongly inhibited uptake of all three metals in histologically different types of human cells, explaining its high chemoprotective potential. A loss of Cr(VI) accumulation by cells was caused by NAC-mediated extracellular reduction of chromate to membrane-impermeable Cr(III). Suppression of Co(II) uptake resulted from a rapid formation of Co(II)-NAC conjugates that were unable to enter cells. Our results demonstrate that NAC acts through more than one mechanism in preventing metal toxicity and its chemoprotective activity can be completely ROS-independent. A good clinical safety and effectiveness in Co(II) sequestration suggest that NAC could be useful for prevention of tissue accumulation and toxic effects of Co ions released by cobalt-chromium hip prostheses.
Keywords: hexavalent chromium, cadmium, cobalt, N-acetylcysteine, toxicity, chemoprevention
Introduction
Oxidative stress has been implicated in causation of many human disorders as well as in toxicity of numerous xenobiotics. However, the relative importance of oxidative mechanisms has been difficult to assess from increases in commonly measured oxidation products. Although gene expression analyses can be helpful in understanding mechanisms of cellular injury [1], oxidant-induced mRNA profiles were dominated by transactivation targets of p53 that responds to a broad variety of stressors [2]. More direct strategies for testing a role of oxidants include the use of genetic models with overexpression/deficiency in oxidative stress-protective enzymes and chemointervention with antioxidants and radical scavengers. The second approach is much more common due to its greater simplicity, the ability to time the intervention and a potential for translational applications. In general, biological effects were stronger with antioxidants than with genetic approaches, which can reflect a broader ROS specificity of chemical agents. An alternative interpretation would be that pharmacological agents have some additional activities, which contribute to their greater effectiveness.
N-acetylcysteine (NAC) is one of the most frequently used antioxidant/radical scavengers for experimental studies in tissue culture and in vivo. The antioxidant activity of NAC results from its abilities to directly inactivate reactive oxygen species (ROS) and raise levels of cellular glutathione (GSH) [3,4]. In addition to its well-known application as the treatment for acetaminophen overdosing [5], NAC has also been used as antidote in cases with acute heavy metal poisoning [3,6]. Mechanistic studies have found a very high effectiveness of NAC in preventing toxic effects of many metals, including those that are either only weakly redox-active or completely inactive. For example, NAC has been found to block toxic effects of Cd(II) [7–9] and Co(II) [10–12], which was taken as clear evidence of the central role of ROS in toxicity of both metals. Although ROS upregulation by these two metals can occur indirectly [13], it is also possible that NAC exerts some antioxidant-independent effects. Cd(II) and Co(II) show a very long retention in tissues (from a few years to >10 years) [14,15] and therefore, it is important to determine whether their adverse effects can be ameliorated with antioxidants or different approaches should be used.
Cr(VI) is a human carcinogen with documented exposures in dozens of metal-processing occupations [16]. Intracellular reduction of Cr(VI) generates variable amounts of organic radicals, Cr(V), Cr(IV) and finally yields stable Cr(III). Toxicity of Cr(VI) has been attributed to the formation of Cr(III)-DNA adducts [17] and oxidative damage by intermediate Cr forms and ROS [18,19]. Cells with deficiencies in DNA repair of oxidative damage showed a moderate sensitization to Cr(VI) [20,21], but a near complete chemoprotection by NAC argued for a much greater role of oxidative stress in Cr(VI) toxicity [22–24]. The dramatic protection by NAC is difficult to understand based solely on suppression of oxidative stress, as the importance of the adduct-mediated nonoxidative route in cyto- and genotoxicity of Cr(VI) is strongly supported by genetic studies and DNA damage measurements [25–27].
Here, we examined mechanisms of cytoprotection by NAC against Cd(II), Co(II) and Cr(VI), three toxic metals that are associated with major public health concerns due to their environmental and iatrogenic sources of exposure. We found that NAC blocked cellular uptake of all three metals, identifying its antioxidant-independent chemoprotective mechanism. Depending on the metal, uptake impairment resulted from the formation of membrane-impermeable complexes with NAC or reduction of metal. Our results demonstrate that NAC is inappropriate as a tool for investigation of metal-induced oxidative stress due to its direct reactivity with metals.
Materials and Methods
Materials
Cobalt(II) chloride hexahydrate (BioReagent purity), cadmium(II) chloride hemipentahydrate (A.C.S. reagent), potassium chromate (99% purity, A.C.S. reagent) N-acetyl-L-cysteine (≥99% purity), L-cysteine (≥98% purity), L-glutathione reduced (≥98% purity), L-buthionine-sulfoximine (≥97% purity) and nitric acid (>99.999% purity) were obtained from Sigma-Aldrich (St. Louis, MO, USA). All other salts and buffers were also from Sigma-Aldrich.
Cell culture and exposures
All cells were obtained from the American Type Culture Collection (ATCC) (Manassas, Virginia, USA). H460 human lung and Daudi human lymphoblastoma cells were maintained in RPMI-1640 medium (Gibco 11875-093) supplemented with 10% fetal bovine serum. A549 human lung and 293T human kidney cells were grown in 10% serum-supplemented F-12K (Corning Cellgro 10-025-CV) and DMEM (Gibco 12430-062) media, respectively. IMR90 normal human fibroblasts were propagated in 10% serum-containing DMEM medium (Gibco 12430-062). Media for all cells was additionally supplemented with penicillin (100 Units/ml) and streptomycin (100 μg/ml). H460, A549, 293T and Daudi cells were grown at 37°C in 95% air/5% CO2, whereas IMR90 cells were maintained in the atmosphere containing 5% O2 and 5% CO2. Cells were seeded at approximately 50% confluency and treated with metals the next day. Stock solutions of CoCl2, CdCl2, K2CrO4 and NAC were freshly prepared in deionized water and filter-sterilized before use. The pH of the NAC was adjusted to 7.0.
Cytotoxicity
Cytotoxic effects of metals were examined by measurements of metabolically active cells using the CellTiter-Glo luminescent cell viability kit (Promega, Madison, WI, USA) and the SpectraMax M5 microplate reader. H460 cells were seeded into black 96-well optical bottom cell culture plates (4000 cells/well) and grown overnight prior to the addition of test chemicals. In experiments assessing the role of GSH, cells were grown for 24 hrs in the presence of 0.2 mM L-buthionine-sulfoximine (BSO), a potent inhibitor of γ-glutamylcysteine synthetase. Caspase 3/7 activity was measured by a luminescence-based kit from Promega. In clonogenic survival experiments, H460 cells were seeded onto 60-mm dishes (400 cells/dish), allowed to attach overnight and then treated with metals and NAC for 48 hr. At 7–8 days post-exposure, colonies were fixed with methanol and stained with a Giemsa solution. The detection limit for the clonogenic survival assay was 0.1%.
Western blotting
Adherent and floating cells were collected by scraping, washed twice with ice-cold PBS and centrifuged at 1100×g for 5 min at 4°C. Cellular proteins were extracted using ice-cold lysis buffer [50 mM Tris (pH 8.0), 250 mM NaCl, 1% NP40, 0.1% SDS, 5 mM EDTA] supplemented with Halt Protease and Phosphatase Inhibitors (Thermo Scientific, Rockford, IL, USA). Cells were incubated with the lysis buffer for 15 min on ice and proteins lysates were collected by centrifugation at 10000×g for 10 min at 4°C. Proteins were separated by standard SDS-PAGE and electrotransferred onto PVDF membranes (Bio-Rad, Hercules, California, USA). After the transfer, PVDF membranes were blocked in 5% w/v nonfat dry milk, 1×TBS, 0.1% Tween-20 for 1 hr at room temperature with gentle shaking. The following primary antibodies were used: mouse monoclonal anti-HIF-1α (1:500; BD Biosciences, San Jose, CA, USA), rabbit polyclonal anti-PARP (1:1000; Cell Signaling Technology, Danvers, MA), rabbit polyclonal antibodies against cleaved caspase-3 (1:1000; Cell Signaling Technology), mouse monoclonal anti-γ-tubulin (1:2000; Sigma-Aldrich), rabbit polyclonal anti-phospho-p53 (Ser-15) (1:1000; Cell Signaling Technology), mouse monoclonal anti-p53 (DO-1) (1:1000; Santa Cruz, CA, USA), rabbit polyclonal anti-HSP72 (Enzo Life Sciences, Farmingdale, NY, USA). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Cell Signaling Technology). Protein bands were developed using a western blot detection kit from Thermo Scientific (Rockford, IL, USA).
Cellular uptake of metals
Cellular concentrations of metals were measured by graphite furnace atomic absorption spectroscopy (GF-AAS) [20]. Cells were seeded into 6-well plates (H460 and 293T - 3x105 cells/well, A549 - 2x105 cells/well, Daudi - 7.5x105 cells/well, IMR90 - 2x105 cells/well) and treated with metals the next day. After removal of metal-containing media, cell monolayers were washed twice with warm PBS. Attached cells were collected by trypsinization (by centrifugation for suspension cultures of Daudi cells), washed twice with ice-cold PBS and resuspended in 50 μl of ice-cold deionized water followed by the addition of 50 μl of 10% nitric acid. Samples were subjected to one cycle of freezing/thawing (−80°/37°C), heated for 60 min at 50°C followed by incubation on ice for 30 min. Metal-containing extracts were collected by centrifugation at 10000×g for 10 min at 4°C. The supernatants were diluted with water to give 2% nitric acid prior to metal measurements by GF-AAS (AAnalyst600 Atomic Absorption Spectrometer, Perkin-Elmer). Twenty microliters of undiluted or diluted extracts per injection were used. Metal-extracted cell pellets were washed twice with ice-cold 5% nitric acid, centrifuged at 10000×g for 5 min at 4°C and solubilized in 100 μl of 0.5 M NaOH by incubation for 30 min at 37°C. The dissolved pellets were used for protein measurements, which were necessary for normalization of metal uptake.
Reactivity of metals with NAC
Solutions of metals and NAC were freshly prepared in deionized water and the pH of the NAC stock was adjusted to 7.0. Reactions contained 100 mM Tris-HCl (pH 7.5), 2 mM Co(II), 200 μM Cd(II), 100 μM Cr(VI) with or without 20 mM NAC. Mixtures were incubated for 1 hr at 37°C, diluted 1:10 with complete media and added to H460 and IMR90 cells for 2 and 6 hr, respectively. Collection of cells and metal measurements by GF-AAS were as described above. Reduction of Cr(VI) was monitored spectroscopically as previously described [22]. The pH of freshly prepared solutions of NAC, Cys and GSH was adjusted to 7.0. Reaction mixtures contained 50 mM HEPES (pH 7.0), 100 mM NaCl, 100 μM K2CrO4 and 0–10 mM NAC, Cys or GSH.
Cellular GSH
Cellular amounts of GSH were measured by HPLC as described previously [20]. In brief, cells were resuspended in 40 mM methanesulfonic acid and lysed by two cycles of freezing/thawing (−80°/37°C). Soluble extracts were recovered at 12000g for 10 min at 4°C and reacted with the thiol-specific dye monobromobimane. The fluorescent GSH conjugates were detected by HPLC using the Ultrasphere ODS column (5 μm, 250 × 4.6 mm).
Statistics
P-values were calculated using two-tailed, unpaired t-test.
Results and Discussion
We chose H460 human lung epithelial cells as our main biological model, since lung is the most common target of carcinogenic and other adverse health effects that are found among individuals occupationally exposed to toxic metals [14–16]. H460 and primary human lung cells showed similar stress signaling and activation of the redox-sensitive transcriptional factor p53 by ionizing radiation [28], Cr(VI) [29] and carbonyl stress [30]. A utility of H460 for studies of oxidative stress is further supported by responsiveness of these cells to pharmacological and shRNA-based manipulations of antioxidant levels and oxidative DNA damage repair [21,31]. H460 cells also displayed a very efficient uptake of metals [29,32], which obviates a need for the use of excessively high metal concentrations that can cause undesirable extracellular effects and direct plasma membrane damage. We tested 2–10 mM NAC, which is a typical dose range for experiments with cultured cells.
Impact of NAC on metal cytotoxicity
We first examined protective properties of NAC against Cr(VI), which can be reduced to Cr(III) intra- and extracellularly. This reduction process is responsible for geno- and cytotoxicity of Cr(VI) inside the cells but it is protective outside the cells due to the inability of Cr(III) to cross the plasma membrane [17]. The induction of oxidative stress by Cr(VI) results from direct and indirect oxidation of biomolecules by Cr(V,IV) intermediates [18,19]. We found that 48 hr-long incubations of H460 cells with 3 and 10 μM Cr(VI) alone caused severe cytotoxicity, as assessed by measurements of total ATP which reflects the number of cells and their metabolic activity (Fig. 1A). Addition of 10 mM NAC resulted in a nearly complete rescue of cell viability even for the lethal dose of 10 μM Cr(VI) (Fig. 1A). The determination of cellular ATP is a very versatile cytotoxicity assay that is responsive to various modes of cell death. Because Cr(VI)-treated H460 cells are known to die via apoptosis [29], we next assessed the impact of NAC on this dominant form of cell death by Cr(VI). Consistent with the ATP viability results, co-incubation of 5 mM NAC with 3 μM Cr(VI) completely eliminated apoptotic responses, such as the production of the active form of executioner caspase 3 and apoptotic cleavage of PARP (Fig. 1B). A strong suppressive effect of NAC on apoptotic markers was also observed for lethal 10 μM Cr(VI). A more complete rescue of 10 μM Cr(VI)-induced cytotoxicity based on the ATP measurements was due to the use of a higher concentration of NAC (10 mM vs. 5 mM in apoptosis studies). Similarly to the results with Cr(VI), the addition of 10 mM NAC had a fully protective effect against cytotoxic effects of Cd(II), as measured by both the ATP-based viability and apoptotic cleavage of PARP (Fig. 1C,D). NAC was also able to preserve viability of H460 cells in the presence of Co(II) ions (Fig. 1E). Co(II) acts as a chemical hypoxia mimic by stabilizing the hypoxia-sensitive transcriptional factor HIF-1α [33]. We found that the presence of NAC led to a dose-dependent inhibition of HIF-1α stabilization by Co(II) (Fig. 1F). In addition to blocking cytotoxic responses, NAC was also very effective in suppressing activation of the stress-responsive transcriptional factor p53 as evidenced by loss of its protein accumulation and Ser15 phosphorylation for all three metals (Fig. 1B,D,F). Cd(II) is known as a strong inducer of the heat shock proteins and we found that NAC also eliminated HSP72 upregulation in Cd-treated H460 cells (Fig. 1D). Finally, we determined that blocking of cell death was not limited to early apoptosis and cytotoxicity as NAC was also very effective in rescuing cell viability in clonogenic survival experiments (Fig. 1G). Overall, the ability of NAC to block diverse cytotoxic effects of metals, including redox-independent heat shock and hypoxic responses, suggests that it could possess some antioxidant-unrelated protective properties.
Figure 1. Cytotoxicity of Cr(VI), Cd(II) and Co(II) with and without NAC.

(A,C,E) Viability of H460 cells treated with metals for 48 hr in the absence or presence of 10 mM NAC. Data are means±SD (n=3). (B,D,F) Western blots of protein lysates from H460 cells treated with metals and NAC for 24 hr. Control cells received equivalent volumes of water. (G) Clonogenic survival of H460 cells treated with metals for 48 hr in the absence or presence of 10 mM NAC. Data are means±SD.
NAC effects in GSH-depleted cells
Elevation of GSH levels is one of the mechanisms accounting for antioxidant effects of NAC [3,4]. It is currently unknown whether GSH is involved in NAC-induced protection of cells against carcinogenic metals. To test a role of GSH in cytoprotective effects of NAC, we examined metal toxicity in H460 cells grown in the presence of the GSH synthase inhibitor BSO. Cells were pre-treated with 0.2 mM BSO for 24 hr prior to the addition of metals, which resulted in a nearly complete loss of cellular GSH (1.0±0.4% remaining GSH based on HPLC measurements). BSO was also present during treatments with metals to prevent restoration of GSH synthesis. As expected, we found that BSO caused a strong sensitization of H460 cells to Cd(II) cytotoxicity (Fig. 2A, two left panels vs. two right panels), providing a functional confirmation of GSH deficiency. NAC fully retained its ability to protect H460 cells against Cd(II) even in the absence of GSH. Depletion of GSH also increased cytotoxicity of Cr(VI) but the addition of 10 mM NAC completely rescued cell viability in both control and GSH-deficient cells (Fig. 2B). Consistent with cytotoxicity measurements, GSH depletion enhanced activation of the main executioner caspases 3 and 7 by Cr(VI) and NAC fully blocked this apoptotic response irrespective of the ability of cells to synthesize GSH (Fig. 2C). NAC also totally abrogated activation of caspases 3/7 by Co(II) in both normal and GSH-depleted H460 cells (Fig. 2D). Thus, cytoprotective effects of NAC against carcinogenic Cd(II), Co(II) and Cr(VI) were not mediated by cellular GSH in our experimental system.
Figure 2. Cytoprotective effects of NAC in H460 cells with depleted GSH.

Data are means±SD (n=3). GSH was depleted by pre-treatments with 0.2 mM BSO for 24 hr. BSO was also present during 48 hr metal treatments. Equivalent volumes of water served as a solvent control. (A) Cytotoxicity of Cd(II) in H460 cells with normal (two left panels) and depleted GSH (two right panels). (B) Cytotoxicity of Cr(VI) in control (left panel) and GSH-depleted cells (right panel). (C) Caspase activity in H460 cells treated with Cr(VI) for 48 hr in the absence (left panel) or presence of BSO (right panel). (D) Caspase activity in H460 cells treated with Co(II) for 48 hr in the absence or presence of BSO.
Metal accumulation by cells
The observed dramatic sensitization of GSH-depleted cells to Cd(II) and the complete rescue of its cytotoxicity by NAC are difficult to explain by purely antioxidant mechanisms, as Cd(II) is not redox-active and its pro-oxidant effects are indirect [13]. Therefore, we next explored a possibility that NAC could impact cellular levels of toxic metals. We found that for all three metals tested, NAC caused a dose-dependent inhibition of their accumulation in H460 cells during 24 hr-long incubations (Fig. 3A–C). Consistent with its highly effective protection against cytotoxicity (Fig. 1), 10 mM concentration of NAC severely diminished levels of all three metals in H460 cells: 96.8, 94.4 and 87.2% decreases for Cd, Co and Cr, respectively. The ability of NAC to suppress accumulation of metals was also found in human Daudi lymphoblasts that grow in suspension (Fig. 3D–F). Cd(II) and NAC have recently been shown to form a complex [34], which can explain a suppressed uptake of this metal.
Figure 3. Cellular accumulation of metals in the presence of NAC.

Background-subtracted values are shown (means±SD, n=3, *-p<0.05, **-p<0.01, ***-p<0.001 relative to metal-treated cells without NAC). (A–C) H460 cells were treated with metals (5 μM Cd, 300 μM Co, 3 μM Cr) in the presence of 0–10 mM NAC for 24 hr. Control dishes received the same concentrations of NAC but no metals. (A) Cellular levels of Cd, (B) Co and (C) Cr. (D–F) Daudi cells were incubated for 6 hr in the presence of NAC and metals (5 μM Cd, 200 μM Co, 2 μM Cr). (D) Uptake of Cd, (E) Uptake of Co and (F) Uptake of Cr.
NAC interactions with Cr(VI)
Diminished levels of cellular Cr found after 24 hr-long incubations of H460 cells with NAC can potentially be caused by either lower uptake of Cr(VI) or accelerated efflux of Cr. Rapid reduction of Cr(VI) inside the cells produces Cr(III) that avidly binds to intracellular macromolecules, which traps the majority of Cr(III) for very long periods of time [17]. Therefore, we focused on the possibility that reaction of NAC with Cr(VI) inhibits cellular uptake of this metal. We found that pre-reaction of Cr(VI) with 20 mM NAC for 1 hr at 37°C almost completely eliminated its uptake measured after 2 hr incubation of H460 cells with 1:10 media-diluted metal-NAC solutions (Fig. 4A). A dramatically diminished uptake of NAC-preincubated Cr(VI) was also found in IMR90 normal human fibroblasts (Fig. 4B). More than 95% of vehicle- and Cr/NAC-treated IMR90 cells excluded Trypan Blue stain (Fig. 4C), arguing against a significant leakage of cellular Cr. Extracellular reduction of Cr(VI) produces membrane-impermeable Cr(III) [16], which led us to test Cr(VI)-reducing properties of NAC. We found that NAC was capable of Cr(VI) reduction at physiologically relevant conditions (Fig. 4D), with the rate of reduction exhibiting a nearly linear dependence on NAC concentration. Incubation with 20 mM NAC for 1 hr at 37°C left only 5.9% unreduced Cr(VI), which closely corresponds to the residual uptake of Cr(VI) after pre-reaction with this concentrations of NAC (5.5% uptake in Fig. 4A). Measurements of reaction kinetics showed that NAC was a slower reducer of Cr(VI) than biological thiols GSH and especially, Cys (Fig. 4E). The main difference in Cr(VI) reduction by NAC in comparison to GSH was the absence of the initial fast reduction component, which is believed to involve a two-electron transfer reaction [20]. Slower rates of Cr(VI) reduction by NAC relative to Cys could be attributed to the loss of the positive charge on the acetylated amino group, with the resulting inhibition of the initial interactions between the SH-group of the negatively charged NAC and chromate anion (CrO42−).
Figure 4. Interactions of NAC with Cr(VI).

(A) H460 uptake of Cr(VI) pre-incubated with or without NAC for 60 min at 37°C. The final concentrations of Cr(VI) and NAC in cell culture media were 10 μM and 2 mM, respectively. Control cells were treated with 10-fold media-diluted 100 mM Tris-HCl (pH 7.5). H460 cells were incubated with Cr(VI) for 2 hr. Data are means±SD (n=3, ** - p<0.01 relative to Cr-treated cells without NAC). (B) Uptake of Cr(VI) by IMR90 fibroblasts after 6 hr incubation. Cr(VI) was pre-reacted with NAC or buffer alone for 60 min at 37°C prior to addition to cells. The final concentrations of Cr(VI) and NAC in media were 10 μM and 2 mM, respectively. Data are means±SD (n=3, *** - p<0.001 relative to Cr-treated cells without NAC). (C) Percentage of Trypan Blue-negative (live) IMR90 cells collected for Cr(VI) uptake in B. (D) Reduction of 100 μM Cr(VI) by different concentrations of NAC. (E) Reduction of 100 μM Cr(VI) by 2 mM Cys, GSH or NAC.
Overall, our results indicate that chemoprotection against Cr(VI) toxicity by NAC under co-exposure conditions can almost entirely be attributed to the irreversible extracellular reduction of Cr(VI), which prevents its cellular uptake. Although pre-treatments of cells with NAC followed by addition of Cr(VI) would avoid this problem, cytoplasmic NAC and its hydrolysis product Cys will be able to take part in the intracellular reduction of Cr(VI), shifting a larger portion of its metabolism to one-electron reduction from a predominantly two-electron reduction by ascorbate and GSH [35]. As a result, NAC-preloaded cells are expected to generate different yields of Cr(V) and Cr(IV) intermediates, making it difficult to offer a clear mechanistic interpretation of cellular responses. Elevation of cellular concentrations of Cys and GSH by NAC is also expected to increase the formation of Cys-Cr-DNA and GSH-Cr-DNA crosslinks [36,37] that are less mutagenic than ascorbate-Cr-DNA crosslinks [38]. Thus, genotoxicity-suppressive effects of intracellular NAC may result from changes in the spectrum of Cr-DNA adducts and not necessarily due to lower oxidative DNA damage.
Reactivity of NAC with Co(II)
To determine a mechanism by which NAC suppressed cellular accumulation of Co(II), we examined uptake of this metal after incubation of cells with pre-reacted NAC-Co(II) solutions (1 hr at 37°C, pH 7.0). We found that the pre-reaction with NAC resulted in a dramatically lower entry of Co(II) in H460 cells (Fig. 5A). A strongly diminished uptake of NAC-prereacted Co was also observed in IMR90 normal human fibroblasts (Fig. 5B). Staining with Trypan Blue showed no changes in plasma membrane integrity of IMR90 cells collected for metal analyses, excluding any significant metal leakage from cells (Fig. 5C). Co(II) is believed to enter cells in the form of Co2+ ions [39]. Therefore, binding of Co(II) by NAC would eliminate membrane-permeable Co2+ and consequently, suppress cellular uptake of this metal. The formation of NAC-Co(II) conjugates was evident from the instantaneous appearance of red-brown color after mixing of colorless solutions of Co(II) and NAC (Fig. 5D). Electronic spectra of 1 hr-preincubated and freshly mixed solutions of Co(II) and NAC were almost identical (Fig. 5E), further pointing to a very rapid binding of Co(II) by NAC and the presence of similar products. A poor uptake of NAC-bound Co(II) was not unique to H460 and IMR90 cells, as NAC also suppressed Co(II) accumulation by 293T human kidney and A549 human lung cells (Fig. 5F,G). A more complete uptake inhibition for 300 μM versus 500 μM points to the importance of high NAC/metal ratios for effective sequestration of Co(II), suggesting that chelation of low-level Co in human plasma might not require high NAC doses.
Figure 5. Interactions of NAC with Co(II).
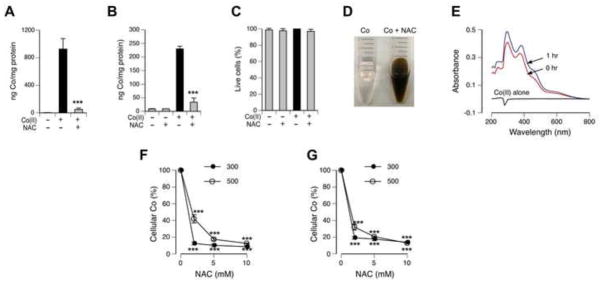
(A) H460 uptake of Co(II) pre-reacted with and without NAC for 60 min at 37°C. Cells were incubated for 2 hr with Co(II) and Co(II)+NAC at final concentrations of 200 μM and 2 mM for Co(II) and NAC, respectively. Control cells were treated with 10-fold media-diluted 100 mM Tris-HCl (pH 7.5). Data are means±SD (n=3), ***- p<0.001 relative to Co-treated cells without NAC. (B) Uptake of Co(II) by IMR90 fibroblasts after 6 hr incubation. Co(II) was pre-reacted with NAC as in A. Data are means±SD (n=3), ***- p<0.001 relative to Co-treated cells without NAC. (C) Percentage of Trypan Blue-negative IMR90 cells in samples analyzed for metal uptake in B. (D) Reactivity of Co(II) with NAC immediately after mixing (room temperature). Reaction mixtures contained 100 mM Tris-HCl (pH 7.5), 2 mM Co(II) with or without of 20 mM NAC. (E) Representative electronic spectra of 0.2 mM Co(II) alone (black line) and solutions of 0.2 mM Co(II) with 2 mM NAC immediately after mixing (0 hr, red line) and following 1 hr incubation at 37°C (1 hr, blue line). Spectra of NAC alone did not have significant absorbance in the selected wavelength range and not shown for clarity. (F) Metal accumulation by 293T human kidney cells after 6 hr incubation with 300 and 500 μM Co(II) in the presence of 0–10 mM NAC. Equivalent volumes of water were added to cells as a solvent control. Data are means±SD, n=3, ***- p<0.001 relative to 0 mM NAC. (G) Metal accumulation by A549 human lung cells after 6 hr incubation with 300 and 500 μM Co(II) in the presence of 0–10 mM NAC. Data are means±SD, n=3, ***- p<0.001 relative 0 mM NAC.
The most common source of human exposure to Co is the internal release of this metal during corrosion of cobalt-containing hip implants, which has been recognized as a particularly significant problem for the recently introduced metal-on-metal type of cobalt-chromium alloy hip prostheses. Hip implant-released Co has been associated with a number of severe adverse health effects, including cardiomyopathy, thyroid toxicity, blindness, deafness and neurotoxicity [40,41]. Animal studies showed that a portion of Co is retained in tissues for several years [15]. Chelation therapy for toxic metals trapped inside the cells typically has not been very effective due stability of metal binding with cellular proteins and difficulties in designing cell-permeable metal-specific chelators. Our results showed that NAC was a very effective chelator of Co(II), preventing cellular uptake of this metal in the serum-containing biological media. Thus, NAC can be potentially used for interception of Co(II) ions in the systemic circulation after hip replacement surgery, particularly during the post-operative times with the most significant increases in blood concentrations of Co. NAC has already been approved for the clinical use in humans and it appears to be safe, especially when administered via ingestion [4]. Although at low ratios to metal, EDTA and DTPA were the most effective in protection of mice following intraperitoneal co-injections with large doses of Co(II) [42], the use of these multidentate nonspecific chelators is better suited for cases of acute poisoning. NAC offers an important advantage in chronic applications by avoiding depletion of essential divalent metals, such as Ca and Mg, due to its lack of binding with metal ions exhibiting hard Lewis acid properties.
Highlights.
N-acetylcysteine effectively protected human lung cells against carcinogenic metals chromium(VI), cadmium(II) and cobalt(II).
Cytoprotection by N-acetylcysteine was fully effective in the absence of GSH synthesis.
N-acetylcysteine inhibited cellular uptake of all three metals.
Direct reactivity with metals makes N-acetylcysteine unsuitable as a tool for assessment of oxidative stress by toxic metals.
Acknowledgments
This work was supported by grants ES008786 and ES013660 from the National Institute of Environmental Health Sciences. The sponsor had no role in the study design, interpretation of the results or preparation of the manuscript.
Abbreviations
- BSO
buthionine-sulfoximine
- GF-AAS
graphite furnace atomic absorption spectroscopy
- GSH
glutathione
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dickinson DA, Warnes GR, Quievryn G, Messer J, Zhitkovich A, Rubitski E, Aubrecht J. Differentiating DNA-reactive and non-reactive genotoxic mechanisms using gene expression profile analysis. Mutat Res. 2004;549:29–41. doi: 10.1016/j.mrfmmm.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Desaint S, Luriau S, Aude JC, Rousselet G, Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2. J Biol Chem. 2004;279:31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- 3.Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev. 1998;3:114–127. [PubMed] [Google Scholar]
- 4.Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8:1955–1962. doi: 10.1517/14728220802517901. [DOI] [PubMed] [Google Scholar]
- 5.Dargan PI, Jones AL. Management of paracetamol poisoning. Trends Pharmacol Sci. 2003;24:154–157. doi: 10.1016/S0165-6147(03)00053-1. [DOI] [PubMed] [Google Scholar]
- 6.Lin CC, Wu ML, Yang CC, Ger J, Tsai WJ, Deng JF. Acute severe chromium poisoning after dermal exposure to hexavalent chromium. J Chin Med Assoc. 2009;72:219–221. doi: 10.1016/S1726-4901(09)70059-0. [DOI] [PubMed] [Google Scholar]
- 7.Wang SH, Shih YL, Kuo TC, Ko WC, Shih CM. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol Sci. 2009;108:124–131. doi: 10.1093/toxsci/kfn266. [DOI] [PubMed] [Google Scholar]
- 8.Fang X, Huang T, Zhu Y, Yan Q, Chi Y, Jiang JX, Wang P, Matsue H, Kitamura M, Yao J. Connexin43 hemichannels contribute to cadmium-induced oxidative stress and cell injury. Antioxid Redox Signal. 2011;14:2427–2439. doi: 10.1089/ars.2010.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang KC, Hsu CC, Liu SH, Su CC, Yen CC, Lee MJ, Chen KL, Ho TJ, Hung DZ, Wu CC, Lu TH, Su YC, Chen YW, Huang CF. Cadmium induces apoptosis in pancreatic β-cells through a mitochondria-dependent pathway: the role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS One. 2013;8:e54374. doi: 10.1371/journal.pone.0054374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan A, Liao X, Mo L, Yang C, Yang Z, Wang X, Hu F, Chen P, Feng J, Zheng D, Xiao L. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One. 2011;6:e25921. doi: 10.1371/journal.pone.0025921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan R, Mo Y, Feng L, Chien S, Tollerud DJ, Zhang Q. DNA damage caused by metal nanoparticles: involvement of oxidative stress and activation of ATM. Chem Res Toxicol. 2012;25:1402–1411. doi: 10.1021/tx200513t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Wang C, Wang Y, Ma Z, Xiao J, McClain C, Li X, Feng W. Cobalt chloride decreases fibroblast growth factor-21 expression dependent on oxidative stress but not hypoxia-inducible factor in Caco-2 cells. Toxicol Appl Pharmacol. 2012;264:212–221. doi: 10.1016/j.taap.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartwig A. Metal interaction with redox regulation: an integrating concept in metal carcinogenesis? Free Radic Biol Med. 2013;55:63–72. doi: 10.1016/j.freeradbiomed.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Järup L, Akesson A. Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol. 2009;238:201–208. doi: 10.1016/j.taap.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Simonsen LO, Harbak H, Bennekou P. Cobalt metabolism and toxicology-a brief update. Sci Total Environ. 2012;432:210–215. doi: 10.1016/j.scitotenv.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic and chromium. Chem Res Toxicol. 2008;21:28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhitkovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- 18.Sugden KD, Stearns DM. The role of chromium(V) in the mechanism of chromate-induced oxidative DNA damage and cancer. J Environ Pathol Toxicol Oncol. 2000;19:215–230. [PubMed] [Google Scholar]
- 19.Myers JM, Antholine WE, Myers CR. The intracellular redox stress caused by hexavalent chromium is selective for proteins that have key roles in cell survival and thiol redox control. Toxicology. 2011;281:37–47. doi: 10.1016/j.tox.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messer J, Reynolds M, Stoddard L, Zhitkovich A. Causes of DNA single-strand breaks during reduction of chromate by glutathione in vitro and in cells. Free Radic Biol Med. 2006;40:1981–1992. doi: 10.1016/j.freeradbiomed.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds M, Armknecht S, Johnston T, Zhitkovich A. Undetectable role of oxidative DNA damage in cell cycle, cytotoxic and clastogenic effects of Cr(VI) in human lung cells with restored ascorbate levels. Mutagenesis. 2012;27:437–443. doi: 10.1093/mutage/ger095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge X, Liu Z, Qi W, Shi X, Zhai Q. Chromium (VI) induces insulin resistance in 3T3-L1 adipocytes through elevated reactive oxygen species generation. Free Radic Res. 2008;42:554–563. doi: 10.1080/10715760802155113. [DOI] [PubMed] [Google Scholar]
- 25.Quinteros FA, Machiavelli LI, Miler EA, Cabilla JP, Duvilanski BH. Mechanisms of chromium (VI)-induced apoptosis in anterior pituitary cells. Toxicology. 2008;249:109–115. doi: 10.1016/j.tox.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Xiao F, Feng X, Zeng M, Guan L, Hu Q, Zhong C. Hexavalent chromium induces energy metabolism disturbance and p53-dependent cell cycle arrest via reactive oxygen species in L-02 hepatocytes. Mol Cell Biochem. 2012;371:65–76. doi: 10.1007/s11010-012-1423-7. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes DNA phosphate-chromium adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 28.Peterson-Roth E, Reynolds M, Quievryn G, Zhitkovich A. Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol Cell Biol. 2005;25:3596–3607. doi: 10.1128/MCB.25.9.3596-3607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynolds MF, Peterson-Roth EC, Johnston T, Gurel VM, Menard HL, Zhitkovich A. Rapid DNA double-strand breaks resulting from processing of Cr-DNA crosslinks by both MutS dimers. Cancer Res. 2009;69:1071–1079. doi: 10.1158/0008-5472.CAN-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds M, Zhitkovich A. Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis. 2007;28:1613–1620. doi: 10.1093/carcin/bgm031. [DOI] [PubMed] [Google Scholar]
- 32.Wong VC, Cash HL, Morse JL, Lu S, Zhitkovich A. S-phase sensing of DNA-protein crosslinks triggers TopBP1-independent ATR activation and p53-mediated cell death by formaldehyde. Cell Cycle. 2012;11:2526–2537. doi: 10.4161/cc.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietruska JR, Johnston T, Zhitkovich A, Kane AB. XRCC1 deficiency sensitizes human lung epithelial cells to genotoxicity by crocidolite asbestos and Libby amphibole. Environ Health Perspect. 2010;118:1707–1713. doi: 10.1289/ehp.1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pietruska JR, Liu X, Smith A, McNeil K, Weston P, Zhitkovich A, Hurt R, Kane AB. Bioavailability, intracellular mobilization of nickel, and HIF-1α activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol Sci. 2011;124:38–148. doi: 10.1093/toxsci/kfr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279:40337–40344. doi: 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- 35.Jalilehvand F, Amini Z, Parmar K, Kang EY. Cadmium(II) N-acetylcysteine complex formation in aqueous solution. Dalton Trans. 2011;40:12771–12778. doi: 10.1039/c1dt11705j. [DOI] [PubMed] [Google Scholar]
- 36.Zhitkovich A. Chromium in drinking water: sources, metabolism and cancer risks. Chem Res Toxicol. 2011;24:1617–1629. doi: 10.1021/tx200251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhitkovich A, Voitkun V, Costa M. Glutathione and free amino acids form stable adducts with DNA following exposure of intact mammalian cells to chromate. Carcinogenesis. 1995;16:907–913. doi: 10.1093/carcin/16.4.907. [DOI] [PubMed] [Google Scholar]
- 38.Voitkun V, Zhitkovich A, Costa M. Cr(III)-mediated crosslinks of glutathione or amino acids to the DNA phosphate backbone are mutagenic in human cells. Nucleic Acids Res. 1998;26:2024–2030. doi: 10.1093/nar/26.8.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quievryn G, Peterson E, Messer J, Zhitkovich A. Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry. 2003;42:1062–1070. doi: 10.1021/bi0271547. [DOI] [PubMed] [Google Scholar]
- 40.Simonsen LO, Harbak H, Bennekou P. Passive transport pathways for Ca2+ and Co2+ in human red blood cells. 57Co2+ as a tracer for Ca2+ influx. Blood Cells Mol Dis. 2011;47:214–225. doi: 10.1016/j.bcmd.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Catalani S, Rizzetti MC, Padovani A, Apostoli P. Neurotoxicity of cobalt. Hum Exp Toxicol. 2012;31:421–437. doi: 10.1177/0960327111414280. [DOI] [PubMed] [Google Scholar]
- 42.Llobet JM, Domingo JL, Corbella J. Comparison of antidotal efficacy of chelating agents upon acute toxicity of Co(II) in mice. Res Commun Chem Pathol Pharmacol. 1985;50:305–308. [PubMed] [Google Scholar]