

Abstract
Efficient metastasis is believed as the result of multiple genetic, epigenetic and/or post-translational events in the lifetime of a carcinoma. At the genetic level, these events may be categorized into those that occur during carcinogenesis, and those that occur during subclonal evolution. This review summarizes current knowledge of the genetics of pancreatic cancer from its initiation within a normal cell until the time that is has disseminated to distant organs, many features of which can be extrapolated to other solid tumor types. The implications of these findings to personalize genome analyses of an individual patient’s tumor are also discussed.
Keywords: pancreatic cancer, metastasis, clonal evolution, genetics
INTRODUCTION
Metastasis is responsible for 90% of cancer deaths from solid tumors.1 Despite this reality, the mechanisms of metastasis formation are relatively understudied compared with that of carcinogenesis, deregulation of cellular pathways or treatment resistance.2 However, what is known is that metastasis is the result of a series of biological hurdles that must be surpassed including intravasation into a functional vascular bed, survival in the circulation, extravasation from the vasculature and establishment within the newly encountered microenvironment.3 The molecular determinants of this metastatic cascade have been subjected to intense investigation revealing a host of factors that contribute to metastatic efficiency and establishment.1,4–6 Nonetheless, because patients with cancer continue to die of metastatic disease much more remains to be learned; it stands to reason that focused studies of cancer metastasis provide the opportunity to improve survival by identifying aspects that can be exploited for therapeutic development.
Pancreatic ductal adenocarcinoma is an ideal tumor type for studies of cancer metastasis. First, the genetics and molecular biology of pancreatic cancer are well described.7 Second, pancreatic cancer arises in association with well-defined precursor lesions;8–10 in the context of their genetic features, these precursor lesions define the genetic progression model of pancreatic carcinogenesis7,11 (Figure 1). Third, metastasis is a common feature of the natural history of pancreatic cancer, with up to 90% of patients having metastatic disease at death.12 Fourth, many patients with metastatic pancreatic cancer still have their primary carcinoma in situ at the time of death,13,14 thus facilitating studies of matched primary/metastasis tissues in the same patient. However, it is important to note that because pancreatic cancer is the fourth most common cause of cancer death, and is estimated to surpass breast cancer in the coming decade,15 there is an urgent medical need as well to understand the mechanisms of pancreatic cancer metastasis itself so as to improve upon its poor survival.14,16
Figure 1.
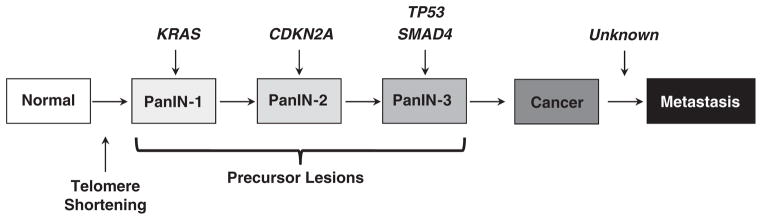
Genetic progression model of pancreatic carcinogenesis. The molecular alterations that accumulate during pancreatic carcinogenesis can be classified into early (telomere shortening and activating mutations in KRAS in PanIN-1), intermediate (inactivating mutations or epigenetic silencing of CDKN2A in PanIN-2) and late (inactivating mutations of TP53 and SMAD4 in PanIN-3) events. Mutations in additional genes may also occur during PanIN formation that are not illustrated in this example. While the genetic events that correspond to pancreatic carcinogenesis have been well described, those that occur during progression remain unknown.
GENETICS OF PANCREATIC CANCER
Pancreatic cancer arises from precursor lesions called pancreatic intraepithelial neoplasia, or PanINs17 (Figure 1). These lesions involve the small ducts of the exocrine pancreas, and depending on the extent of cytologic atypia may be classified as PanIN-1 (low-grade dysplasia), PanIN-2 (moderate dysplasia) or PanIN-3 (high-grade dysplasia) lesions.17 Microdissection of such lesions for sequencing of the most common genetic alterations in pancreatic cancer indicates that activating mutations in KRAS are a relatively early event and found in >99% of PanIN-1 lesions.18 Inactivating mutations in CDKN2A occur as early as in PanIN-2 lesions,19 and inactivating mutations in TP53 and SMAD4 in PanIN-3 lesions.20 Collectively, these observations support a genetic progression model of pancreatic carcinogenesis that leads to formation of an infiltrating cancer.11
In recent years, the pancreatic cancer research community has made tremendous strides in further understanding of the genetic basis of this disease. Jones et al.21 provided the first clues to the genetic basis of pancreatic carcinogenesis at a global level. This effort relied on high throughput dideoxy-sequencing of 24 pancreatic cancers to assess 20 661 protein coding genes representing 99.6% of the known coding genome, the most curated set of genes at that time. Copy number chips to evaluate copy number gains or deletions were also used, as well as methods to interrogate the gene expression of each sample. Among this set of genes 1562 somatic mutations were detected, most of which were accounted for by single base substitutions. Copy number analyses revealed a total of 198 homozygous deletions and 144 focal high-copy amplifications in the 24 tumors analyzed as well. Genes targeted by mutation, deletion or amplification included well-established tumor suppressors and oncogenes such as KRAS, CDKN2A, TP53 and SMAD4, and genes identified in smaller scale studies of pancreatic and other tumor types such as MLL3.22 However, many additional genes that had not been previously associated with pancreatic ductal adenocarcinoma were also found, one example being ARID1A.
Further categorization of the somatic alterations data generated by integrating whole exome sequencing and copy number analyses revealed that the somatic targets corresponded to at least 12 core-signaling pathways (Figure 2). Many of these pathways paralleled mechanisms already recognized as significant for pancreatic cancer formation and progression, such as DNA damage control (TP53), cell-cycle regulation (CDKN2A) and transforming growth factor β(TGFβ) signaling (SMAD4). Most importantly, while most patients’ carcinomas exhibited a single genetic alteration corresponding to each of the core pathways, the specific gene mutated for a given pathway in each patient often varied, potentially explaining the basis for tumor heterogeneity at the genetic level. Since that initial report, recurrent inactivating mutations of ARID1B, PBRM1, SMARCA2 and SMARCA4 have also been identified implicating inactivation of the SWI/SNF chromatin remodeling complex, and hence an additional core pathway, in up to a third of pancreatic cancers23 (Figure 2).
Figure 2.

Core-signaling pathways in pancreatic cancer. The fourteen pathways and processes whose component genes were genetically altered in most pancreatic cancers based on whole exome sequencing are shown in light gray,21 and the two pathways more recently identified in pancreatic cancer in dark gray.22–24 Therapeutic targeting of one or more of these pathways, rather than specific gene alterations that occur within a pathway, provides a new paradigm for treatment of pancreatic cancer. GTPase, guanosine triphosphatase; TGFβ, transforming growth factor β.
Most recently, Biankin et al.24 have reported the most comprehensive evaluation of the pancreatic cancer genome to date-based whole exome sequencing and copy number analysis of 99 pancreatic cancers. This study reaffirmed the known mutations in pancreatic cancer21,23 and revealed a multitude of novel mutated genes including those involved in chromatin modification (EPC1),25 DNA damage repair (ATM)26 and others (ZIM2, MAP2K4, NALCN, SLC16A4 and MAGEA6). Pathway-based analysis of recurrently mutated genes recapitulated clustering of mutations in core-signaling pathways in pancreatic ductal adenocarcinoma (PDAC) and identified new components in each, a likely reflection of the increased power of this sample set in finding lower frequency somatic events. Of interest, they also identified frequent and diverse somatic aberrations in genes traditionally described in association with embryonic regulation of axon guidance, particularly signaling through slit ligands and roundabout receptors (SLIT/ROBO),27 thereby identifying yet another core pathway for this disease (Figure 2).
METASTATIC EFFICIENCY OF PANCREATIC CANCER
With a thorough understanding of the global patterns of somatic alteration in pancreatic carcinogenesis comes the opportunity to understand the relationship of these changes to metastatic progression. Toward this goal, we have previously reported the features of end-stage pancreatic cancers in association with a rapid autopsy program.28 This approach revealed that not all pancreatic cancers are metastatic, a notion contrary to common perception at the time.29 Twelve percent of patients had no metastatic disease at autopsy, and an additional 18% of patients had limited metastatic burden that was not directly responsible for the patient death, leading us to define these patients as having oligometastatic disease (defined as ≤10 gross metastases) (Figure 3). Based on comprehensive postmortem evaluation in these patients death most often occurred due to complications of a bulky primary carcinoma infiltrating into surrounding vital structures. By contrast, in the remaining two thirds of patients widely disseminated metastatic disease was noted. While objectively this is defined as greater than 10 metastases, in reality for many of these patients the number of gross deposits was extensive and often exceeded 100 metastases (and in some patients 1000 s).28 Of note, these differences in metastatic efficiency were not related to clinical features at diagnosis or length of survival, suggesting an underlying biology as playing a role.
Figure 3.
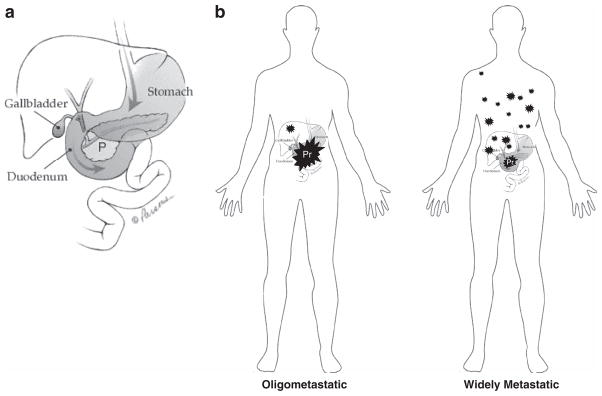
Patterns of metastatic failure. (a) Representation of the anatomic structures surrounding the pancreas (P). Image provided courtesy of the Johns Hopkins Pancreatic Cancer Home page (www.path.jhu.edu/pancreas/). (b) At autopsy, patients with. oligometastatic pancreatic cancer have few metastases (0 to 10), and the cause of death is most often due to the large primary carcinoma that invades into adjacent vital structures such as the duodenum, stomach or large vessels. By contrast, approximately two thirds of patients have widely metastatic pancreatic cancer that is defined as >10 distant metastases. In reality, the number of metastases in these patients is in the tens to hundreds of deposits.
Given the genetic features known for pancreatic cancer at the time, a logical first experiment was to determine the extent to which the genetic status of the most frequently altered known driver genes correlated with these observations. Loss of Smad4 protein expression in the primary and metastatic cancer tissues in these patients, indicating a deletion or mutation of the gene,20 was highly correlated with the presence of widespread metastatic disease. Inactivating mutations of the TP53 tumor suppressor gene in these same tissues also correlated with these patterns, although not to the extent of Smad4. Thus, pancreatic cancer appears to be represented by two phenotypes that differ not in their morphologic differences at diagnosis but in their metastatic efficiencies for which inactivation of SMAD4, and as a result loss of Smad4 protein expression, is a marker.20 This is consistent with the findings of Blackford et al.30 in which genetic inactivation of SMAD4, whether by deletion or mutation, was associated with a worse overall survival compared with patients in which this gene is intact within their resected carcinoma tissues. A similar conclusion was previously shown by Tascilar et al.31 based on Smad4 protein expression.
How might Smad4 loss promotes metastatic efficiency? Smad4 is a central mediator of both the TGFβ and BMP signaling pathways.32 In pancreatic cancer cell lines, SMAD4 alterations universally impair TGFβ stimulated transcriptional responses at DNA sequences containing Smad Binding Elements, suggesting that Smadmediated transcription is a feature that is targeted for disruption by SMAD4 loss.33,34 Loss of Smad4-mediated transcription leads in part to a loss of growth inhibition due to loss of p21 and p15 expression.35–38 The consequences of disruption of BMP signaling in pancreatic cancer metastasis, similarly inactivated by Smad4 loss, are less well studied. However, given their implications in metastasis in other tumor types39–41 dysregulated BMP signaling likely also contributes to pancreatic cancer aggressiveness.
To more fully explore the relationship of these specific genes with metastatic efficiency, Yachida et al.42 documented the extent to which the common somatic alterations KRAS, CDKN2A, TP53 and SMAD4 that may be coexistent in an individual patient’s pancreatic cancer including their correlation to these patterns of metastatic failure. Genetic inactivation of TP53 or SMAD4 was each significantly correlated with high metastatic efficiency, similarly to prior observations.28 However, as inactivation of these two genes is often coexistent in the same carcinoma, these relationships were further evaluated and revealed two interesting and previously unrecognized relationships. First, SMAD4 loss of function almost always occurred in association with genetic inactivation of TP53, whereas TP53 inactivation did not, indicating SMAD4 alterations are selected for in association with TP53 genetic alterations. This relationship also suggests that SMAD4 inactivation occurs later than TP53 inactivation in the genetic progression model of pancreatic carcinogenesis (Figure 1). Second, the types of TP53 alterations coexistent with SMAD4 loss were highly enriched for missense mutations, whereas TP53 inactivation in association with wild-type SMAD4 was highly enriched for null mutations (nonsense, deletion or frameshift) that abolish gene expression, suggesting that SMAD4 loss is selected in part for its ability to remove residual cytostatic or apoptotic functions of TP53 missense mutant proteins. Such a conclusion has been demonstrated in mouse models of pancreatic cancer based on Trp53 mutations with differing abilities to induce p21 activation and growth arrest.43 Consistent with this possibility, these two genetic subtypes, that is, TP53 null mutant versus TP53 missense mutant in association with SMAD4/SMAD4 loss, were both highly correlated with high metastatic efficiency. Of interest, Oshima et al.44 also recently reported that coexistent genetic alterations of these two genes are strongly associated with malignant behavior of PDACs in a Japanese population of patients.
While additional genes not evaluated in these studies undoubtedly also have a role in aggressiveness of pancreatic cancer, these observations lend strong support to the notion that the genetic features of a patient carcinoma selected for during carcinogenesis contribute to differences in metastatic propensity. How this information can be translated in a meaningful way to improve the management of patients with this disease is a work in progress.45 However, based on these and others observations46 plans for a national Phase II randomized clinical trial are underway to evaluate the significance of Smad4 status in patients with unresectable pancreatic cancer who undergo high versus standard dose radiation or systemic chemotherapy (www.rtog.org). It is the hope that such a trial will indicate the extent to which determining the genetic status of pancreatic cancers at diagnosis can be used to tailor therapies for each patient.
SUBCLONAL EVOLUTION OF PANCREATIC CANCER
Multiple genetic, epigenetic and post-translational events are required for efficient metastasis to occur.3,47 These mechanisms include epithelial–mesenchymal transition, upregulation of specific miRNAs that activate oncogenic signaling pathways, the formation of prometastatic niches by the microenvironment, or immune suppression.48–53 Hence, the genetic changes discussed thus far represent initial events that occur during carcinogenesis upon which additional prometastatic events may be superimposed. However, until recently the extent to which genetic alterations specifically continue to accumulate and promote metastatic progression beyond carcinogenesis was unknown.
To address this question, Yachida et al. relied on data generated from whole exome sequencing of seven metastases included in the pancreatic cancer genome sequencing effort.21,54 These data provided the framework for initial characterizations of the genetics of pancreatic cancer from its initiation from a normal cell until the time that is has disseminated to distant organs. Importantly, the genetic features of these seven metastases to the 17 surgically resected carcinomas also sequenced by Jones et al.21 indicated that they did not differ in their total number of genetic alterations, nor did they differ in the spectrum of mutations seen. Thus, based on whole exome sequencing the overall genetic features of advanced stage pancreatic cancers are similar to those seen in surgical resection specimens.
Comparative lesion sequencing is a simple yet powerful method to evaluate the clonal relatedness of different carcinoma samples taken from within a single individual based on the extent of shared genetic alterations.55,56 Thus, for each of the seven patients all mutations identified in the index metastasis lesion were assessed for their presence or absence in multiple additional samples obtained from the same patients primary carcinoma and matched metastases. Two categories of mutations were identified by this approach. The first category corresponded to those mutations present in all samples analyzed from a given patient (founder mutations). Because these mutations were shared by both the primary carcinoma and the matched metastases they likely accumulated within the PanIN that ultimately gave rise to that infiltrating pancreatic cancer. Thus, these mutations are present in the majority, if not all, of the cells of the tumor and are genetic markers of the most common ancestral clone of cells (the parental clone) that formed that carcinoma. The second category corresponded to those mutations present in a subset of the samples analyzed for each patient (progressor mutations). Because founder mutations are present in all samples analyzed for a patient, but progressor mutations are present in only a subset of those samples, progressor mutations occurred later than founder mutations and thus represent subclonal evolution beyond the parental clone. A comparison of the features of founder versus progressor gene alterations revealed that the majority of genetic alterations present in a pancreatic carcinoma were contained within the parental clone of cells that initiated the infiltrating neoplasm. Founder mutations also represented the majority of deleterious genetic alterations attributed to pancreatic cancer including activating mutations in KRAS and inactivating mutations in CDKN2A, TP53 and SMAD4, consistent with the progression model of pancreatic carcinogenesis.11 Overall, these findings indicate that the parental clone most closely represents the initial population of cells that formed the infiltrating carcinoma upon which subclonal evolution of the cancer genome occurs during clinical tumor progression (Figure 4).
Figure 4.

Clonal evolution of pancreatic carcinogenesis and progression. Red arrows indicate the lineage of the index metastasis from its origin in a normal cell. Carcinogenesis, and time T1, begins with an initiating alteration (M) in a normal cell that provides a selective advantage. Over time, waves of clonal expansion occur in association with the acquisition of additional mutations in genes such as CDKN2A, TP53 or SMAD4, corresponding to the progression model of PanIN. This clonal expansion is expected to generate more than one subclone within a PanIN, one of which will give rise to the founder cell (blue clone) that will eventually become the parental clone and hence initiate the infiltrating carcinoma. The birth of this cell corresponds to the beginning of time T2. Following additional waves of clonal expansion from the parental clone, subclones are again generated within the infiltrating carcinoma leading to genetic heterogeneity. The birth of the cell within the primary carcinoma that will become the metastatic subclone (green clone) corresponds to the start of time T3. Whether a single metastatic subclone generates all metastases in a patient is currently unknown.
Based on the patterns of somatic mutation and the locations of individual metastatic deposits evolutionary maps were constructed for each patient’s carcinoma (Yachida et al.42). This indicated that the clones that give rise to distant metastases were genetically distinct from the parental clones, the result of subclonal evolution, yet these metastatic subclones were also present within the primary carcinoma. Thus, primary carcinomas are mixtures of numerous subclones, each of which has independently expanded to constitute a large number of cells and some of which may have a greater potential to seed distant metastases than others.
Others have also described such patterns. For example, Campbell et al.57 showed by massively parallel paired-end sequencing that the majority of rearrangements within the pancreatic cancer genome were shared among the primary and metastases, indicating that they occurred before the development of metastatic disease. The types and frequency of specific rearrangements differed from those observed in breast cancer, with a notable prevalence of ‘fold-back inversions’ implicating failure in G1/S transition with intact G2/M checkpoints as occurs with loss of CDKN2A during PanIN progression and the formation of parental clones.58 Additional rearrangements were noted in metastases that accumulated during subclonal progression, analogous to progressor mutations seen by whole exome sequencing. Similarly, high-resolution copy number analyses of metastatic prostate cancers indicated that they maintain a unique signature copy number pattern of the parent cancer cell while also accumulating a variable number of separate subclonally sustained changes;59 related observations have been made in breast cancer or leukemic cells using similar techniques.60–63 Most recently, next genome sequencing of matched primary and metastatic renal cell carcinomas, breast cancers and melanomas also documented a wide spectrum of copy number alterations and mutations were present in both the primary tumors and metastases upon which additional clonal events were found that were metastasis specific.64–66
While these studies illustrate the presence of genetic heterogeneity in pancreatic cancers arising during subclonal evolution, they do not indicate the mechanisms by which genetic heterogeneity arises. For example, while genetic heterogeneity in pancreatic cancer may arise from waves of clonal evolution in association with the stochastic accumulation of genetic alterations, it has also been suggested that heterogeneity may arise from preexistent small populations of cancer stem cells that continuously give rise to genetically diverse populations of non-tumorigenic cells.67 The latter scenario is supported by the demonstration of small populations of highly tumorigenic CD44 +CD24 +ESA + cells within infiltrating pancreatic cancers or metastases with the ability for self-renewal and repopulation of all phenotypic progeny present within the neoplasm;68 however, given the finding that cells that undergo epithelial–mesenchymal transition themselves can generate cells with the classical properties of stem populations,48,69 and epithelial–mesenchymal transition is a common and robust feature that arises during pancreatic cancer progression,28 the existence of classical stem cells for this disease remains unclear. Future studies that rely on deep sequencing of tumorigenic and non-tumorigenic populations, or those with and without features of epithelial–mesenchymal transition, should clarify the extent to which these mechanisms occur in a given patients carcinoma, and their respective contributions to the genetic diversity in a given tumor.70
COMPUTATIONAL MODELS OF PANCREATIC CANCER PROGRESSION
Is the poor prognosis of pancreatic cancer due to its late diagnosis, or because it metastasizes early during clonal evolution? To address this important question, the estimated timeline of clonal evolution during PDAC development and progression was calculated based on a computational model that incorporated the number of somatic alterations, driver versus passenger events, founder versus progressor alterations and the relative proliferation rates of cells as they progress from normal to cancerous and finally to metastatic cells.54,71 This model proposed an average of 11.7 years from the initiating event that began pancreatic carcinogenesis until development of the parental clone and a further 6.8 years to the development of metastatic subclones within the primary cancer, with patients dying an average of 2.7 years later (Figure 4). Unfortunately, most patients with PDAC are diagnosed well toward the end of this 21-year average time span, indicating that the poor prognosis is likely due to late diagnosis in the natural history of the disease. Nonetheless, PDAC is similar to other types that have a long latency from initiation to patient death that is on the order of decades.55,65 The significance of this finding is vast and suggests that there is a long latency to development of infiltrating pancreatic cancer and thus a large window of opportunity for early diagnosis and cure. Of course, much more work will be required to determine the optimal timing and modes of early detection for patients at risk.72,73
Recently, Haeno et al.74 investigated PDAC progression by utilizing a mathematical framework of metastasis formation together with comprehensive data of 228 patients, 101 of whom had autopsies in association with our autopsy program. The rationale for that study was that, until early detection becomes routine and pancreatic cancer is resected in the curative stage, the need will remain to understand the growth dynamics of pancreatic cancer so as to optimize the efficacy of available treatments. This stochastic mathematical model revealed that all patients are expected to harbor metastasis-enabled cells in the primary tumor at the time of diagnosis, even when the size of the primary tumor is small. A patient with a primary tumor of 1 cm diameter has a probability of 28% of harboring metastases at the time of diagnosis; as the primary size increases to 2 and 3 cm, the risk of harboring metastases increases to 73 and 94%, respectively. Such findings are in keeping with those of the rates of distant metastases after surgery based on tumor size of resected PDACs.75 Interestingly, chemotherapeutic agents that efficiently reduced the growth rate of cells earlier in the course of treatment were predicted to be superior to upfront tumor resection, suggesting that neoadjuvant treatment of resectable PDACs may be more effective than adjuvant treatment. This concept is in keeping with the emerging clinical trend for managing pancreatic cancer patients.76
IMPLICATIONS AND FUTURE DIRECTIONS
There are two major implications of these observations in light of current interests in sequencing of human tumors for identification of therapeutic targets or biologic pathways of significance.77–79 First, while sequencing of a single region of a carcinoma may be sufficient for identification of genes altered during carcinogenesis it will not accurately capture the extent of genetic heterogeneity within that neoplasm, a reflection of subclonal evolution.54,64 For example, MLL3 is a newly identified tumor suppressor gene in pancreatic cancer,21,22 found in a subset of carcinomas based on whole exome or targeted sequencing. We noted that MLL3 mutations were present in all samples analyzed by comparative lesion analysis from two different patients, indicating that the MLL3 mutations arose during carcinogenesis and were contained within the parental clone. Thus, if confirmed on a larger scale, future studies of MLL3 should focus on understanding how its loss promotes pancreatic carcinogenesis; should a therapeutic consequence of MLL3 loss of function be found, it would then become a viable target given its loss of function is present in all cells of that neoplasm. By contrast, in the same study we noted somatic OVCH1 mutations in two patients, and based on comparative lesion sequencing both were progressor mutations indicating that they arose during subclonal evolution. Targeting of mutant OVCH1 protein, assuming it is druggable, would not be fruitful as only a subclone of the carcinoma would be affected. Thus, the timing of development of mutations can thus provide important insight not only into their roles in pancreatic cancer biology, but also their therapeutic consequences and this cannot be determined based on a single sample (Figure 5). Targeting of somatic mutations based on a single region biopsy may also permit expansion of resistant subclonal populations already present in the patient. This is the basis for colorectal cancer resistance to anti-EGFR therapies80,81 and indicates multiple subclones in the patient, each with potentially differing therapeutic vulnerabilities, will need to be targeted for effective management of cancer and to avoid this scenario.64
Figure 5.

Clinical implications of subclonal evolution in pancreatic cancer. As discussed, pancreatic carcinogenesis follows the accumulation of both driver and passenger mutations culminating in the formation of the founder cell (blue clone) that will become the parental clone of the carcinoma. The parental clone that initiates the infiltrating carcinoma will continue to undergo clonal evolution, leading to the formation of subclones that differ in the presence of newly acquired mutations in the hypothetical genes α, β, γ and Δ. Should a therapy that targets the subclone with mutant β effectively clear all cancer cells containing that mutation, over time the remaining subclones will continue to grow and new subclones (for example, ε) will emerge.
Of course, with this reality comes the need for a practical manner to categorize genetic events in a given patient tumor into founder and passenger events. Fortunately, with next-generation sequencing methods the technical needs for microdissection or xenografting of cancer tissues can be bypassed by high-resolution sequencing of a patients tumor to identify genetic events of significance. In time, it is even conceivable that these methods can be further optimized to identify founder and progressor events based on their frequency of mutant alleles within a single specimen.65 Until then, multiregional sampling of a patients tumor, provided it can be done safely, would be most beneficial toward identifying potential therapeutic vulnerabilities.
Genetic studies of pancreatic cancer progression have been fruitful in efforts to understand how metastasis forms. However, a variety of new questions now present themselves. First, what is the extent to which specific genetic events that arise during subclonal evolution promote metastatic dissemination? To date, the majority of genes that are deemed metastasis suppressors show features of haploinsufficiency; few to no examples exist of specific genetic alterations that change protein function and consequently contribute specifically to metastatic dissemination.82 Second, to what extent does the immune system have a role in metastasis of pancreatic cancer? An influx of both myeloid-derived suppressor cells and FoxP3 T-regulatory lymphocytes has been shown in pancreatic cancer that corresponds to an immuno-suppressed microenvironment.83–85 In breast cancer models, loss of TGFβ signaling is linked to an influx of myeloid-derived suppressor cells into the stroma that have metastasis promoting activity;86 it is intriguing to consider that a similar mechanism occurs in the pancreas. Passive features of the microenvironment may also have a role, such as the extent to which the infiltrating neoplasm has a functional vasculature for the cancer cells to intravasate into and escape the primary site, the degree of intratumoral hypoxia or of interstitial fluid pressures.87,88 Ultimately, many or all of these mechanisms are likely at play in a given pancreatic cancer.
SUMMARY
Genomic methods have been invaluable in studies of pancreatic cancer progression. Only with this knowledge is ‘metastasis genetics’ possible by allowing study of the genes, pathways and mechanisms that are specifically dysregulated after invasive ability develops. This is important to know for the ultimate goal of devising therapies that target the metastatic phenotype and that take tumor heterogeneity into account.
Acknowledgments
This study was supported by NCI CA140599 (CID).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Nguyen DX, Massagué J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 4.Fidler IJ. The biology of cancer metastasis. Semin Cancer Biol. 2011;21:71. doi: 10.1016/j.semcancer.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Langley RR, Fidler IJ. The seed and soil hypothesis revisited--the role of tumorstroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comen E, Norton L, Massagué J. Clinical implications of cancer self-seeding. Nat Rev Clin Oncol. 2011;8:369–377. doi: 10.1038/nrclinonc.2011.64. [DOI] [PubMed] [Google Scholar]
- 7.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matthaei H, Schulick RD, Hruban RH, Maitra A. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8:141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci USA. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macgregor-Das AM, Iacobuzio-Donahue CA. Molecular pathways in pancreatic carcinogenesis. J Surg Oncol. 2013;107:8–14. doi: 10.1002/jso.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 12.Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009;133:413–422. doi: 10.5858/133.3.413. [DOI] [PubMed] [Google Scholar]
- 13.Sohn TA, Yeo CJ, Cameron JL, Koniaris L, Kaushal S, Abrams RA, et al. Resected adenocarcinoma of the pancreas-616 patients: results, outcomes, and prognostic indicators. J Gastrointest Surg. 2000;4:567–579. doi: 10.1016/s1091-255x(00)80105-5. [DOI] [PubMed] [Google Scholar]
- 14.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 15.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 16.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 17.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Kanda M, Matthaei H, Wu J, Hong S-M, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia 2012. Gastroenterology. 142:730–733. e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740–4744. [PubMed] [Google Scholar]
- 20.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156:37–43. doi: 10.1016/S0002-9440(10)64703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, et al. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67:3545–3550. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- 23.Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci USA. 2012;109:E252–E259. doi: 10.1073/pnas.1114817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, et al. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem. 2005;280:1199–1208. doi: 10.1074/jbc.M412509200. [DOI] [PubMed] [Google Scholar]
- 26.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 27.Ballard MS, Hinck L. A roundabout way to cancer. Adv Cancer Res. 2012;114:187–235. doi: 10.1016/B978-0-12-386503-8.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–1813. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 30.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674–4679. doi: 10.1158/1078-0432.CCR-09-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115–4121. [PubMed] [Google Scholar]
- 32.Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, et al. Targeted deletion of Smad4 shows it is required for transforming growth factor beta and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 35.Grau AM, Zhang L, Wang W, Ruan S, Evans DB, Abbruzzese JL, et al. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res. 1997;57:3929–3934. [PubMed] [Google Scholar]
- 36.Dai JL, Bansal RK, Kern SE. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA. 1999;96:1427–1432. doi: 10.1073/pnas.96.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai JL, Turnacioglu KK, Schutte M, Sugar AY, Kern SE. Dpc4 transcriptional activation and dysfunction in cancer cells. Cancer Res. 1998;58:4592–4597. [PubMed] [Google Scholar]
- 38.Peng B, Fleming JB, Breslin T, Grau AM, Fojioka S, Abbruzzese JL, et al. Suppression of tumorigenesis and induction of p15(ink4b) by Smad4/DPC4 in human pancreatic cancer cells. Clin Cancer Res. 2002;8:3628–3638. [PubMed] [Google Scholar]
- 39.Murakami M, Suzuki M, Nishino Y, Funaba M. Regulatory expression of genes related to metastasis by TGF-beta and activin A in B16 murine melanoma cells. Mol Biol Rep. 2010;37:1279–1286. doi: 10.1007/s11033-009-9502-x. [DOI] [PubMed] [Google Scholar]
- 40.Nishimori H, Ehata S, Suzuki HI, Katsuno Y, Miyazono K. Prostate cancer cells and bone stromal cells mutually interact with each other through bone morphogenetic protein-mediated signals. J Biol Chem. 2012;287:20037–20046. doi: 10.1074/jbc.M112.353094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park C-Y, Kim D-K, Sheen YY. EW-7203, a novel small molecule inhibitor of transforming growth factor-β (TGF-β) type I receptor/activin receptor-like kinase-5, blocks TGF-β1-mediated epithelial-to-mesenchymal transition in mammary epithelial cells. Cancer Sci. 2011;102:1889–1896. doi: 10.1111/j.1349-7006.2011.02014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yachida S, White C, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long term survivors. Clin Cancer Res. 2012;18:6339–6347. doi: 10.1158/1078-0432.CCR-12-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oshima M, Keiichi O, Shinobu M, Reiji H, Takashi M, Yasuyuki S, et al. Immunohistochemically detected expression of three major genes (CDKN2A/p16, TP53 and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg. 2013 doi: 10.1097/SLA.0b013e3182827a65. in press. [DOI] [PubMed] [Google Scholar]
- 45.Crane CH, Iacobuzio-Donahue CA. Keys to personalized care in pancreatic oncology. J Clin Oncol. 2012;30:4049–4050. doi: 10.1200/JCO.2012.45.1799. [DOI] [PubMed] [Google Scholar]
- 46.Crane CH, Varadhachary GR, Yordy JS, Staerkel GA, Javle MM, Safran H, et al. Phase II trial of cetuximab, gemcitabine, and oxaliplatin followed by chemoradiation with cetuximab for locally advanced (T4) pancreatic adenocarcinoma: correlation of Smad4(Dpc4) immunostaining with pattern of disease progression. J Clin Oncol. 2011;29:3037–3043. doi: 10.1200/JCO.2010.33.8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oskarsson T, Acharyya S, Zhang XH-F, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. doi: 10.1038/onc.2012.578. (e-pub ahead of print 10 December 2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones S, Chen W-D, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2012;61:1085–1094. doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larsson L-G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol. 2011;21:367–376. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B, Kendall JT, et al. Novel genomic alterations and clonal evolution in chronic lymphocytic leukemia revealed by representational oligonucleotide microarray analysis (ROMA) Blood. 2009;113:1294–1303. doi: 10.1182/blood-2008-05-158865. [DOI] [PubMed] [Google Scholar]
- 63.Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060–1066. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turajlic S, Furney SJ, Lambros MB, Mitsopoulos C, Kozarewa I, Geyer FC, et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res. 2012;22:196–207. doi: 10.1101/gr.125591.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 69.Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, Ryschich E, et al. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS ONE. 2012;7:e46391. doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gisselsson D. Intratumor diversity and clonal evolution in cancer--a skeptical standpoint. Adv Cancer Res. 2011;112:1–9. doi: 10.1016/B978-0-12-387688-1.00001-6. [DOI] [PubMed] [Google Scholar]
- 71.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci USA. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shin EJ, Canto MI. Pancreatic cancer screening. Gastroenterol Clin North Am. 2012;41:143–157. doi: 10.1016/j.gtc.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Canto MI, Hruban RH, Fishman EK, Kamel IR, Schulick R, Zhang Z, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142:796–804. doi: 10.1053/j.gastro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–375. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Egawa S, Takeda K, Fukuyama S, Motoi F, Sunamura M, Matsuno S. Clinicopathological aspects of small pancreatic cancer. Pancreas. 2004;28:235–240. doi: 10.1097/00006676-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 76.Warshaw AL, Lillemoe KD, Fernandezdel Castillo C. Pancreatic surgery for adenocarcinoma. Curr Opin Gastroenterol. 2012;28:488–493. doi: 10.1097/MOG.0b013e3283567f2c. [DOI] [PubMed] [Google Scholar]
- 77.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 78.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shoushtari AN, Szmulewitz RZ, Rinker-Schaeffer CW. Metastasis-suppressor genes in clinical practice: lost in translation? Nat Rev Clin Oncol. 2011;8:333–342. doi: 10.1038/nrclinonc.2011.65. [DOI] [PubMed] [Google Scholar]
- 83.Goedegebuure P, Mitchem JB, Porembka MR, Tan MCB, Belt BA, Wang-Gillam A, et al. Myeloid-derived suppressor cells: general characteristics and relevance to clinical management of pancreatic cancer. Curr Cancer Drug Targets. 2011;11:734–751. doi: 10.2174/156800911796191024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Porembka MR, Mitchem JB, Belt BA, Hsieh C-S, Lee H-M, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61:1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3 + regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 86.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1 + CD11b + myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Provenzano PP, Cuevas C, Chang AE, Goel VK, Hoff Von DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]