

Abstract
Genetic mutations and gross structural defects in the DNA sequence permanently alter genetic loci in ways that significantly disrupt gene function. In sharp contrast, genes modified by aberrant epigenetic modifications remain structurally intact and are subject to partial or complete reversal of modifications that restore the original (i.e. non-diseased) state. Such reversibility makes epigenetic modifications ideal targets for therapeutic intervention. The epigenome of cancer cells is extensively modified by specific hypermethylation of the promoters of tumor suppressor genes relative to the extensive hypomethylation of repetitive sequences, overall loss of acetylation, and loss of repressive marks at microsatellite/repeat regions. In this review, we discuss emerging therapies targeting specific epigenetic modifications or epigenetic modifying enzymes either alone or in combination with other treatment regimens. The limitations posed by cancer treatments that elicit unintended epigenetic modifications that result in exacerbation of tumor progression are also discussed. Lastly, a brief discussion of the specificity restrictions posed by epigenetic therapies and ways to address such limitations is presented.
Keywords: Epigenome, Epigenetic, DNA Methyltransferases (DNMTs), Histone Acetyltransferases (HATs), Histone Deacetylases (HDACs), Histone Methyltransferases (HMTs), Histone Demetylases (HDMs)
INTRODUCTION
The epigenome (epi meaning above or beyond) consists of heritable modifications to histones and DNA which are independent of changes to the linear DNA sequence.1, 2 These modifications are established by transiently or stably expressed proteins that respond to developmental cues, internal stimuli and environmental factors. These modifications include DNA methylation, histone acetylation, methylation, phosphorylation, ubiquitination, citrullination, sumoylation, and ADP ribosylation. Cues established during development allow cells with identical DNA to differentiate into a wide array of cell types, a process which coupled to environmental factors, can lead monozygote twins to establish different epigenetic patterns and consequently, different profiles of gene expression. Such alterations of cellular programming can dictate differences in susceptibility to disease and therefore, represent potential targets for pharmacological intervention.3, 4
Epigenetic modifications are dynamically established by DNA methyltransfereases (DNMTs), histone acetyltranferases (HATs), histone methyltranferases (HMTs), kinases, and removed or modified by histone deacetylases (HDACs), histone demethylases (HDMs), ten eleven translocation protein 1-3 (TET1-3) activity, and phosphatases in a highly regulated manner. Such modifications alter the conformation of chromatin by effecting covalent interactions or by acting as special docking sites for reader and effector proteins.5 The interaction between chromatin modifying enzymes and reader/effector proteins regulates transcriptional activation or repression leading to varied cellular phenotypes. Epigenetic modifications are not stand alone processes, instead, are subject to considerable crosstalk among the different types of epigenetic marks. Epigenetic marks by themselves, or synergistically with other macromolecular modifications, function to either repress or activate transcription. For example, actively transcribing genes are enriched at the promoters with acetylation (AC), H3 trimethylation at lysine-4 (H3K4me3), and unmethylated CpGs. Further, enhancers are enriched with H3 methylation at lysine-1 (H3K4me1), and H3 acetylation at lysine-27 (H3K27AC), while gene bodies are enriched with H3 dimethylation at lysine-36 (H3K36me2), H3 acetylation at lysine-12 (H3K12AC) and DNA methylation (Figure 1 Top).6,7,8, 9 On the other hand, the promoters of repressed genes are enriched with DNA methylation at CpG sites, H3 trimethylation at lysine-27 (H3K27me3), H3 trimethylation at lysine-9 (H3K9me3), which can also be present in enhancer and gene bodies, as well as overall loss of acetylation (Figure 1 Bottom).8, 10, 11, 12 Unlike genetic mutations and gross structural defects that may permanently activate or inactivate genes, all epigenetic modifications identified to date are modifiable, thus conceivably allowing correction of aberrant epigenetic profiles (Figure 1).
Figure 1.

Dynamic epigenetic regulation in normal cells showing crossed talk between epigenetic modifications resulting in transcriptional activation or repression. This figure depicts different epigenetic modifications under normal conditions for both expressed and repressed genetic targets. Histone Acetyltransferases (HATs), Histone Methyltransferase (HMTs), Ubiquitin ligases (E3), Ten Eleven Translocation (TET) DNA hydroxylase (convert 5mC to 5HmC) and Kinases add transcriptionally active epigenetic marks (e.g. AC, H3K4me3, H3K4me1, H3K27Ac, H3K36me3, H3K12Ac and 5HmC) to promoters, enhancers and gene bodies which promote the formation of euchromatin and transcriptional activation (Top). DNA Methyltransferease (DNMT), Histone Methyltransferase (HMT), Ubiquitin ligase (E3), Histone Deacetyltransferase (HDAC) put repressive epigenetic marks (H3K27me3, H3K9me3 and DNA methylation) to promoters, enhancers and gene bodies promoting the formation of heterochromatin and transcriptional repression (Bottom).
Tumor cells undergo massive epigenetic reorganization, with the occurrence of oncogenic phenotypes associated with pronounced CpG-specific hypermethylation in the promoters of tumor suppressor genes, and generalized hypomethylation of the promoters of oncogenes, microsatellite regions and repetitive sequences. 13, 14 Such changes in the epigenetic landscape contribute to both initiation of tumorigenesis and progression of oncogenic phenotypes. For example, in normal cells, the promoters of tumor suppressor genes are enriched with active transcription marks, such as H3K4me3, H3K9me, H4 acetylation at lysines-5,-8,-12,-16 and 20 (H4 -K5, -K8, -K12, -K16, and -K20 ACs), while satellite regions are enriched with repressive marks, such as H3K27me3, H3K9me2 and H4K16Ac (Figure 2).15, 16 In tumor cells, the promoters of tumor suppressor genes lose nearly all acetylation, and acquire repressive marks including H3K27me3 and H3K9me (Figure 2).17 At repetitive/satellite regions, repressive marks such as DNA methylation (Me), H4K20me3 and H3K27me3 are lost leading to chromosome/microsatellite instability(Figure 2).18, 19, 20
Figure 2.

Simplified version of the dynamic plasticity of epigenetic modifications in normal and tumor cells. In normal cells, gene rich regions are dynamically enriched with active transcription marks (i.e. H4 acetylation at lysine-5,-8,-12 and −16 (H4-(K5,8,12,16)Ac), H4 acetylation at lysine-9 (H4K9Ac), H3 trimethylation at lysine-4 (H3K4me3), 5-hydroxymethylcytosine (5-HmC)) to allow the transcription of tumor suppressor genes, while repeat regions are enriched with repressive marks (H4 acetylation at lysine 16 (H4K16Ac), H4 trimethylation at lysine-20 (H3K20me3), H3 trimethylation at lysine-27(H3K27me3), H3 dimethylation at lysine-9(H3K9me2) and DNA methylation (Me)) that lead to heterochromatin formation. In tumor cells, this dynamic is reversed leading to increased repressive marks at gene rich region and active marks at repeat regions. The dotted red arrow denotes the reversal of these epigenetic modifications by epigenetic drugs to the original state or to an alternate intermediate state. The red X denotes blockage of aberrant epigenetic modification by epigenetic drugs.
In recent years, therapies targeting epigenetic modifying enzymes (e.g. DNMTs, HATs, HDACs, kinases, HMTs, and HDMs), pathways and accessory proteins have been developed to either block or reverse the aberrant epigenetic modifications (Figure 2). To date, four epigenetic drugs: 5-azacytidine (azacitidine or 5-aza-CR), 5-aza-2′-deoxycytidine (5-aza-CdR or decitabine), Vorinostat (suberoylanilide hydroxamic acid (SAHA)) and Romidepsin have been approved by the US Food and Drug Administration (FDA). Azacidine and decitabine were approved for the treatment of high risk myelodysplastic syndrome, while vorinostat and romidepsin were approved for Cutaneous T cell Lymphoma (CTCL). This review highlights some of the latest therapies targeting epigenetic enzymes, pathways and accessory proteins in cancer treatment.
Histone Acetyltransferase (HATs) and HAT Inhibitors (HATis)
Histone acetylation involves the transfer of an acetyl group from acetyl coenzyme A (acetyl-CoA) to the ε-amino group of lysine by HAT.21, 22 HATs have been grouped into Type-A and Type-B HATs based on sequence divergence of the HAT domain and intracellular localization. This nomenclature, however, was confusing and a new nomenclature has been adopted as denoted in Table 1.23 Type-A HATs are nuclear proteins that acetylate histones and other chromatin-associated proteins, while Type-B HATs are both nuclear and cytoplasmic and mainly acetylate de novo synthesized histones in the cytoplasm to promote their nuclear localization. 24, 25 Type-A HATs consist of three families: GNATs, P300/CBP, and MYST. Thus far, histone acetyltransferase-1 (HAT1/KAT1) is the only Type B HAT shown to acetylate H3 at lysines-5 or 12 (K3K5/12).22,26 The different HAT families show little sequence similarity with no homology domain, but most HATs contain a recognizable acetyl-CoA binding domain. Crystal structure analyses of all HATs have provided insight into how these enzymes interact with their substrates 27. For example, X-ray crystallography of Type-A HATs uncovered a conserved core domain consisting of three-stranded β-sheets connected to long and parallel α-helices, and this core region supports the conserved interaction of the protein with the acetyl-CoA or related substrates 27.
Table 1.
Histone Acetyltransferases: Classes, Nomenclature and Substrate Specificity
| NOMENCLATURE OF HUMAN HATs | SUBSTRATE SPECIFICITY | |
|---|---|---|
| OLD | NEW | |
| TYPE A | ||
GNAT FAMILY
|
|
|
P300/CBP FAMILY
|
|
|
MYST FAMILY
|
|
|
| TYPE B | ||
|
|
|
Despite conservation of this core domain, mutational, biochemical and enzymatic analyses showed that the different HAT families employ different mechanism to transfer the acetyl group. The GNAT family employs a one-step bi-bi ternary complex mechanism to transfer the acetyl-CoA. In this mechanism, acetyl-CoA and the substrate bind to form a ternary complex and the glutamate (Glu173 for KAT2A (GCN5) and Glu570 for KAT2B (hPCAF)) at the active site acts as a base and deprotonates the ε-amino group of lysine. This allows nucleophilic attack on the carbonyl carbon of acetyl-CoA leading to the formation of a tetrahedral intermediate which dissociates into CoA and the acetylated histone/protein.28, 29, 30, 31 The MYST family of HATs is found to either employ the one-step bi-bi ternary mechanism as reported by Berndsen et al. and Yan et al.32,33, or a two-step ping-pong mechanism in which the substrate and acetyl-CoA binds to each other allowing Cys304 at the active site to attack the acetyl moiety forming an acetylated enzyme intermediate. When the CoA is released from this intermediate, the lysine nucleophile binds the substrate and is deprotonated by Glu338 in the active site resulting in the formation of acetylated histone.31 It should be noted, however, that both Yan et al. and Berdsen et al. studied TIP60 (KAT5) in H. sapiens, with Yan et al. using a truncated KAT5 and Berdsen et al. using a full length KAT5 in the presence of accessory proteins (NuR4). In addition, substitution of Cys304 to alanine does not affect the catalytic activity of KAT5, suggesting that the GNAT family and MYST families follow similar catalytic mechanisms.33 On the other hand, the p300/CBP family employs a hit-and-run (i.e. Theorell-Chance) acetyl transfer mechanism. Mutation analysis uncovered Tyr1467 and Trp1436 which are conserved in just the p300/CBP family as the amino acid involved in the acetyl transfer mechanism. 34 Trp1436 is believed to either help orient and/or deprotonate the lysine moiety on histone allowing nucleophilic attack on the carbonyl carbon of acetyl CoA leading to the release of CoA. Tyr1467 then donates a proton to CoA such that instead of base catalysis, P300/CBP group follows acid catalysis. 34, 35
Based on the catalytic activity of HATs with acetyl CoA, HAT inhibitors (HATis) are mainly derivatives, conjugates or related compounds of acetyl-CoA and classified into synthetic peptide CoA base bisubstrate HATis, natural product HATis or small molecule HATis (Table 2). The synthetic bisubstrate HATis were the first to be identified based on the observation that polyamine-CoA conjugates can inhibit HAT activity in cell extracts 36. In particular, H3-CoA-20 and Lys-CoA specifically inhibit KAT2B (hPCAF) and KAT3B (P300) rather weakly37; however, introduction of a phenyl or methyl group between lysine and CoA improves the inhibition four-fold (Table 2).38 Most of the synthetic bisubstrate HATis work by mimicking the acetyl CoA-lysine intermediate complex in the HAT reactions. Crystal structure information for KAT2A (GCN5) and these HATis shows that KAT2A (GCN5) interacts with the pyrophosphate moiety, the pantothenic moiety and the phosphate group of CoA. The major deficiency for this class of HATis is their high degree of cellular impermeability. Unfortunately, most of the naturally occurring HATis also suffer from a similar problem. For example, anacardic acid, a drug isolated from the shell of cashewnuts, displays permeability restriction in vitro. Nevertheless, garcinol and isogarcinol were both shown to inhibit KAT3B (P300) and KAT2B (PCAF), although they were shown to be toxic (Table2).39, 40 Two derivatives of isogarcinol, LTK14 and LTK15, were shown to selectively inhibit KAT3B (P300), but not KAT2B (PCAF) without cytotoxicity, a finding that shows the great promise of these class of compounds.40 The best characterized of the naturally-occurring HATis is Curcumin, which is isolated from the Curcuma longa rhizome. Curcumin has shown high efficacy in the prevention and treatment of colorectal, prostate, kidney, lung, ovarian, breast, cervical and liver cancers. 39 A derivative of curcumin with bromine substitutions (Table 2) has been shown to inhibit KAT3B with an IC50 value of 5.0μM.42 The last group of HATis includes a number of small molecules designed to overcome challenges with permeability. These include γ-butyrolactone MB-3, quinoline and isothiazolone and their derivatives. Although in their infancy, isothiazolone has been shown to inhibit the enzymatic activity of both KAT2B (PCAF) and KAT3B (P300) leading to reductions in cell proliferation of human ovarian and colon cancer cell lines.41 λ-butyrolactone MB-3 inhibits KAT2A (GCN5) with Kd calculations showing that the affinity of λ-butyrolactone MB-3 to KAT2A is comparable to the natural substrate H3 lysine.42 A derivative of isothiazolones with nitrogen oxide and chlorine substitution at the R2 and R3 position has been shown to effectively inhibit KAT2B. Another derivative of isothiazolones generically called CCT077791 effectively inhibits both KAT2B and KAT3B44 (Table 2).
Table 2.
Specificity and Activity of Histone Acetyltranferase Inhibitors
| HATi COMPOUND | NAMES | HAT SPECIFICITY | ACTIVITY( IC50) | REF |
|---|---|---|---|---|
| SYNTHETIC HATis | ||||
|
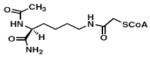
Lys-CoA | KAT3B (P300) | 3.2μM | 37 |
|
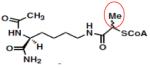
Lys-CoA-CH3 | KAT2B (PCAF) | 0.8μM | 37 |
|
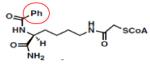
Ph-Lys-CoA | KAT3B (P300/) | 0.7μM | 38 |
| NATURAL HATis | ||||
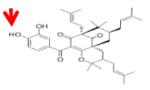
|
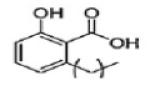
Anacardic Acid | KAT3B (P300) KAT2B (PCAF) |
8.5μM 5.0μM |
39 |
|
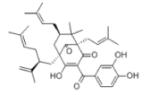
Garcinol (Toxic) |
KAT3B (P300) KAT2B (PCAF) |
7.0μM 5.0μM |
40, 41 |
|
Isogarcinol (Toxic) LTK14 (R = CH3/ -C(CH3)2) (Non-Toxic) |
KAT3B (P300) KAT2B (PCAF) KAT3B (P300) |
7.0μM 5.0μM 5.0-7.0μM |
41 |
|
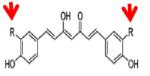
Curcumin (R = OCH3) |
KAT3B (P300) | 25μM | 40, 42 |
|
Compound-11 R=Br |
KAT3B (P300) | 5.0μM | 42 |
| SMALL MOLECULE HATis | ||||
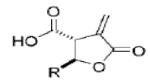
|
γ-Butyrolactone (R= nC3H7) |
KAT2A (hGCN5) | 100μM | 43 |
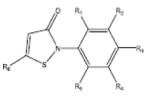
|
Isothiazolones (R1 = H, R2 = NO2, R3 = Cl, R4-6 = H) |
KAT2B (PCAF) | 1.1μM | 44 |
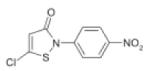
|
CCT077791 | KAT3B (P300) KAT2B (PCAF) |
3.0μM 7.3μM |
44 |
Red arrows or circles denote sites of side chain substitution
The viability of HATs as drug targets is complicated by the fact that most HATs are part of large multiprotein complexes that help to define the molecular activity of HATs.43, 44 In some instances, these complexes have been shown to restrict HAT activity from certain loci 45, while in others these complexes are assembled to orchestrate the different enzymatic functions required for transcription or alternate cellular functions. For example, the KAT 5 (TIP60) complex not only contains NUR4, but also contains ATM which is a kinase recruited during episodes of DNA damage. As such, KAT5 acetylates ATM which goes on to phosphorylate P53 and CHK2 giving rise to unintended acetylation.46 On the basis of these findings, the making of cell permeable HATis is but one step toward making these drugs viable options for cancer treatment.
Histone Deacetylases (HDACs) and HDAC inhibitors (HDACis)
Removal of acetyl groups from histones is catalyzed by a class of enzymes called histone deacetylases (HDACs). In humans, 18 isoenzymes of HDACs have been identified to date, and grouped into four classes based on their homology to yeast HDACs (Table 3). Class I (HDACs1, 2, 3, and 8) are related to yeast Rpd3 gene and are mostly located in the nuclei, except HDACs 3 and 8 which can also be cytoplasmic. Class II HDACs (HDACs 4, 5, 6, 7, 9 and 10) are related to yeast Hda1 gene and primarily located in the cytoplasm, but can also shuttle to the nucleus. Class II HDACs are further divided into two subclasses, IIa (HDAC 4, 5, 7,9) and IIb (HDAC 6, 10) based on their sequence homology and domain organization. Class III, also known as the sirtuins (sirtuins 1–7), are related to the yeast Sir2 gene and localized in the cytoplasm, mitochondria and nucleus. Class IV (HDAC 11) contains a conserved domain that is similar to the catalytic domain of class I and II HDACs. Class I, II and IV share similar structural organization and a common cofactor, Zn2+, while Class III HDACs (sirtiuns) are structurally unique and their active site is occupied by the nicotinamide adenine dinucleotide (NAD). Functionally, class II HDACs are regulated by class I HDACs and together they are involved in transcriptional silencing and genomic organization during development. Class III HDACs (sirtiuns) are involved in maintenance of acetylation, as well as gene-specific silencing.
Table 3.
Histone Deacetylases: Classes, Yeast Homologs, Cofactors and Localization
| HDAC CLASSES | HOMOLOGY TO YEAST DEACEYTLASE DOMAIN |
CO- FACTOR |
LOCALIZATION |
|---|---|---|---|
I
|
Rpd3 81, 82 % 76, 65 % |
Zn2+ Zn2+ |
Nucleus Nucleus/Cytoplasm |
IIa
|
Hda1 52, 53, 51, 51 % |
Zn2+ |
Nucleus/Cytoplasm |
IIb
|
Hda1 61 & 65 % |
Zn2+ |
Nucleus/Cytoplasm |
III
|
Sir2 |
NAD+ NAD+ NAD+ NAD+ |
Nucleus/Cytoplasm Mitochondria/Nucleus Mitochondria Nucleus |
IV
|
Rpd3 & Hda1 42 & 41% |
Zn2+ |
Nucleus |
HDAC inhibitors (HDACis) have been classified into seven categories based on their chemical structures and mode of inhibition: short chain fatty acids, benzamides, cyclic peptides, electrophilic ketones, hydroxamine-acid-derived compounds, miscellaneous compounds (e.g. Depudecin and MGCD-0103) and sirtuin inhibitors. Class I, II and IV HDACis share a common metal binding domain that serves to block Zn2+ chelation at the active site. 47 Because of the presence of NAD, a different cofactor at the active site of class III HDACs, zinc-dependent HDACis, are ineffective against class III HDACs (sirtiun). Class III HDACis are inhibited by nicotinamide, NAD+ analogues, indoles, hydroxynaphthaldehyde derivatives, splitomicins, suramins and kinase inhibitors.48 Most HDAC inhibitors work by specifically blocking the entry of cofactor into the active site. The mode of action of sirtuin (Class III) inhibitors (i.e indoles, hydroxynaphthaldehyde derivatives, splitomicins, suramins and kinase inhibitors) is not fully understood, and much less is known about their biological consequences. 49, 50, 51 In fact, sirtuin inhibitors shown to be effective in lower organisms do not work on human subtypes.42 Nevertheless, Sirtuin I and 2 specific inhibitors (SEN96 and COMPOUND 6J) are in clinical trials (Table 4)52, 53.
Table 4.
Histone Deacetylase Inhibitors: Specificity and Current Status.
| HDACi COMPOUND | NAMES | HDAC SPECIFICITY | STATUS | REF |
|---|---|---|---|---|
|
Vorinostat | HDAC1, HDAC2, HDAC3, HDAC6 |
APPROVED | 54 |
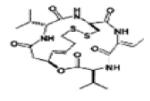
|
Romidepsin | HDAC1, HDAC2, HDAC3, HDAC8 |
APPROVED | 54 |
|
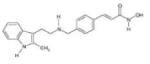
Panobinostat | HDAC1, HDAC2, HDAC3, HDAC6 |
PHASE II | 53 |
|
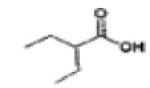
Valporic acid | CLASS I & IIa | PHASE I | 65-67 |
|
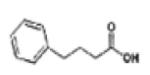
Phenyl butyrate | CLASS I & IIa | PHASE I/II | 65-67 |
|
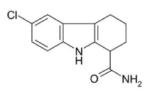
SEN196 | SIRT1 | PHASE II | 52 |
|
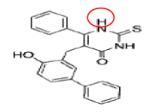
COMPOUND 6J (R= -C4H8) |
SIRT-II | IC50 = 1.0 μM | 54 |
Red circles denotes site of side chain substitution.
Zinc-dependent HDACis are established anticancer drugs, and two inhibitors (Vorinostat and Romidepsin) have been approved for cancer treatment in the United States (Table 4).54 The efficacy of these drugs for cancer treatment has been established. For example, early molecular and cellular biological studies showed that in response to these HDACis, the p21 gene is consistently upregulated in a p53-independent manner leading to G1 cell cycle arrest. 55 The upregulation of p21 correlates with increased acetylation of histones H3 and H4 near the p21 promoter.56 In addition, HDACis such as butyrate and trichostatin A have been shown to stabilize p21 mRNA56, to repress cyclins A and D, and to activate p16 and p27 culminating in cell cycle arrest. 57, 58 In other studies, HDACis have been shown to upregulate the expression of proapoptotic genes (TRAIL, DR5, Bax, Apaf-1, Bmf, Bim and TP2) and/or to downregulate the expression of antiapoptotic genes (i.e., Bcl-2, Mcl1, and XIAP).59 In contrast to other epigenetic drugs, a major advantage for HDACis is their potency, as evidenced by their ability to elicit pharmacological effects in the nano/micromolar range.60, 61
HDACis suppress angiogenesis and enhance the host immune system in cancer patients. 59, 62, 63 In combination therapies, HDACis function synergistically with a host of structurally and functionally diverse chemotherapeutic agents and biologically-active polypeptides. In this manner, HDACis increase target susceptibility and show greater promise. For example, in breast cancer therapy, the effectiveness of topoisomerase II inhibitors can be enhanced by pretreatment with vorinostat.64 Further, HDACis have been used in combination with DNA demethylating agents to reactivate silenced genes involved in tumor suppression. For example, three Phase I/II trials combining decitabine with HDAC inhibitors (phenyl butyrate or valproic acid) in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) showed both tolerability and promising efficacy.65-67 Of 93 patients treated, 14 showed complete remissions (CR), two showed partial complete remission (pCR), 4 showed partial response (PRs) and 6 showed hematologic improvements (HI) and combined established an overall response rate of 28%.
In another phase I clinical trial, the efficacy of CI-994 and various other chemotherapeutic agents was examined in 54 patients. The results showed that CI-994 at doses of 4-10mg/m2/day can be safely administered to patients for 7-21 days in a 3-4 week dosing regimen.68-70 In this study, two patients with esophageal and bladder cancers, respectively, showed complete remission (CR) and 5 (3 with non-small lung carcinoma and 2 with colorectal cancer) demonstrated partial remission (PR). On a related note, some leukemias and breast cancers plagued with the expression of fusion proteins (RAR–PML, RAR–PLZF or AML–ETO chimeras) that inhibit differentiation have shown improvement when HDACis are combined with trans-retinoic acid (ATRA).67, 71, 72, 73 Another emerging area of combination therapy has been the use of HDACis with tyrosine kinase inhibitors in cancers that overexpress antiapoptotic genes. For example, vorinostat, LBH-589, LAQ-824 and romidepsin have demonstrated synergistic proapoptotic activity in combination with imatinib and other tyrosine kinase inhibitors in the treatment of both imatinib-sensitive as well as imatinib-resistant bcr-abl leukemic cells.74-77
DNA Methyltransferases (DNMTs) and DNMT Inhibitors (DNMTis)
DNA methylation involves the enzymatic transfer of a methyl group (CH3) to carbon-5 of the pyrimidine base, cytosine, by DNA methyltransferases (DNMTs). Five DNA methyltransferases (DNMTs) have been identified in higher eukaryotes: DNMT1, DNMT2, DNMT3L, DNMT3a and DNMT3b but only three (DNMT1, DMNT3a and 3b) are involved in direct DNA methylation. DNMT3L lacks DNA methylation activity78, but has been shown to colocalize and to stimulate DNMT3a and DNMT3b during maternal genomic imprinting.79 In addition, DNMT3L has been shown to recruit DNMT3a and DNMT3b to H3 histone tails to methylate DNA8. and to facilitate H3K36me3 (a repressive mark).80 DNMT2 knockout mice do not display aberrant DNA methylation81, and instead, show methylation of tRNA outside the nucleus.82 DNMT1 is a maintenance methyltransferase that recognizes and methylates hemimethylated DNA during DNA replication83, and can interact with both DNMT3a and DNMT3b to influence transcription. DNMT1 and DNMT3a have both been shown to bind to KMT1A (SUV39H1), a histone methyltransferase, to mediate repression via H3K9me84, and DNMT1 and DNMT3b were shown to interact with HDACs to repress gene expression.85,86 DNMT3a and DNMT3b primarily function as de novo methyltransferases and establish DNA methylation during embryogenesis.87 DNA methylation provides binding sites for several methyl-binding proteins including Methyl-CpG-binding domain protein 1-3 (MBD1-3) family members and CpG-methylated DNA binding complex (MeCP2) which in turn function to recruit histone modifying enzymes (e.g. HDACs, TETs, kinases, E3 ligases, HMTs).88 DNA methylation in most instances (except on gene bodies) results in the formation of heterochromatin leading to transcriptional repression.
Although previously considered irreversible, two seminal studies have shown that 5-methylcytosine (5-mC) can be oxidized to 5-hydroxymethylcytosine (5-hmC) by Ten Eleven Translocation (TET1-3) family of dioxygenases.89, 90 The role of 5-hydroxymethylcytosine (5-hmC) conversion is not fully understood since this modification is found in gene bodies, active and repressed promoters, as well as transcriptionally poised genes, suggesting that this might be an intermediate step in the demethylation process.91 In fact, other derivatives of 5-hydroxymethylcytosine such as 5-formylcytosine and 5-carboxycytosine have been uncovered more recently.92 Functionally, demethylation is a process that favors transcriptional activation, as most of the promoters of actively transcribing genes are unmethylated.
Inhibitors of DNA methylation (DNMTis) cause reactivation of silenced genes, inhibition of cell proliferation, apoptosis and enhancement of sensitivity to other cancer drugs. DNMTis are grouped into nucleoside DNMTis and non-nucleoside DNMTis based on their structure and mode of action (Table 5). Nucleoside DNMTis are analogues or derivatives of the nucleoside cytidine and they include 5-Azacytidine (5-Aza-CR), 5-Aza-2-deoxycytidine (5-Aza-CdR), Zebularine, Cytarabine and 5-Fluoro-2-deoxycytidine (FdCyd). The anticancer activity of these drugs is mediated via two mechanisms: (1) cytotoxicity that stems from the incorporation of these drugs into DNA and/or RNA, and/or (2) reactivation of tumor suppressor genes by passive demethylation of their promoter regions. These drugs do not demethylate DNA per se, but rather with continued replication, cytidines are replaced by the cytidine analogues resulting in serial dilution of methylable cytosines. Furthermore, DNMTs are trapped as covalent adducts with DNA through the incorporated cytidine analogues.
Table 5.
DNA Methyl Transferase Inhibitors: Mode of inhibition and Current Status.
| DNMTi COMPOUND | NAMES | DNMT SPECIFICITY |
STATUS | REF |
|---|---|---|---|---|
| NUCLEOSIDE ANOLOGUES | ||||
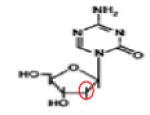
|
Azacitine (5-Aza-CR) |
DNA DNMT ADUCTs |
APPROVED | 96 |
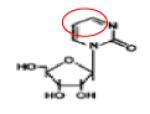
|
Decitabine (5-Aza-CdR) |
DNA/RNA DNMT ADUCTs |
APPROVED | 96 |
|
Zebularine | DNA/RNA DNMT ADUCTs |
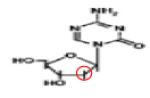
Preclinical | 102-109 |
|
5-Fluoro- deoxycytidine (FdCyd/FDAC) |
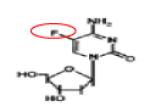
DNA/RNA DNMT ADUCTs |
Phase I/II | 111-114 |
| NON-NUCLEOSIDE ANOLOGUES | ||||
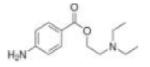
|
Procaine | DNMT1 | Phase I | 115 |
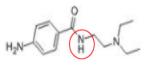
|
Procanamide | DNMT1 | Phase I | 119 |
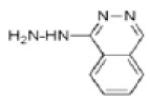
|
Hydralazine | DNMT1, DNMT3A & 3B |
Phase II/III | 116-118 |
|
RG108 | DNMT1 | IC50 = 1000μM | 121 |
Red circles denote unique atoms or side groups relative to the parent compound.
5-Aza-CR and 5-Aza-CdR are taken into the cell through the concentrated nucleoside transporter-1 (hCNT1), or equilibrative nucleoside transporters (hENT1), or the Proton-coupled oligopeptide transporters.93 Once inside the cell, these agents are monophosphorylated by Uridine-cytidine kinase (Urd-Cyd Kinase) and Deoxycytidine kinase (dCyd Kinase), respectively.94, 95 This step is followed by sequential phosphorylation by UMP-CMP kinase and Nucleoside-dCP kinase to the active triphosphates: 5-Aza-CTP and 5-Aza-dCTP. 5-Aza-CTP/-dCTP incorporated into the DNA forms covalent adducts between DNMTs and DNA which traps the DNMTs and prevents further methylation.96 In other studies, 5-Aza-dCTP was shown to be incorporated into RNA which interferes with ribosomal biogenesis and protein synthesis.94, 96 In support of this mechanism, it has been shown that 5-Aza-CR and 5-Aza-CdR hypomethylate the genome through passive dilution of cytidine97, 98. Clinical trials using Aza-CR and sodium phenylbutyrate99, Aza-CR and Valproic acid100, Aza-CR and lenalidomide101 in patients with solid tumors or MDS show greater than 40% efficacy.
Because of the cytotoxicity and instability of 5-Aza-CR and 5-Aza-CdR, DNMTis cannot be continually given to patients. For this reason, zebularine was developed as an alternative. Although this drug works in a manner similar to 5-Aza-CR and 5-Aza-CdR, it is more stable and less toxic than 5-Aza-CR and 5-Aza-CdR DNMTis.102, 103 For example, zebularine has been shown to reactivate tumor suppressor genes104, 105, enhance tumor cell response to chemotherapy and radiation sensitivity106, and exert angiostatic and antimitogenic activities.107, 108 This agent is stable for oral administration103, 109 thus, providing a significant advantage over other agents (Table 5). In addition, at low doses zebularine can be given to patients continuously without the overt cytoxicity associated with 5-Aza-CR and 5-Aza-CdR.
Another cytidine analogue, 5-fluoro-2-deoxycytidine (FdCyd) demethylates DNA in human breast and lung cancer cells (Table 5).110 In the case of FdCyd, the hydrogen atom at carbon 5 (C5), which functions as the methyl acceptor during the methylation reaction, is replaced by a fluorine atom. When FdCyd is incorporated into DNA, the β-elimination step in which DNMT transfers the methyl group to the cytidine is inhibited. At the same time, the fluorine atom traps the DNMT to prevent elimination of the FdCyd moiety.111, 112 FdCyd is currently in clinical trials for the treatment of breast and other solid tumors.113 Moreover, FdCyd, in combination with other epigenetic drugs (i.e. tetrahydrouridine and dihydro-5-azacytidine (DHAC), is being evaluated in clinical studies for the treatment of malignant mesothelioma.114
Although nucleoside DNMTis have proven effective for the treatment of cancers, their cytotoxicity remains a significant limitation. To address this shortcoming, non-nucleoside DNMTis such as procaine, L-trytophan derivatives, RG108, hydralazine, MG98, procainamide, and epigallocatechin-3-gallate (EGCG) are being evaluated. Procaine is a local anesthetic drug that can also function as a DNMTi. For example, procaine has been shown to cause global demethylation and reactivation of tumor suppressor genes in human breast cancer cells.115 Unlike the nucleoside analogues, procaine competes with DNMTs for binding to CpG rich regions.115 Procainamide and hydralazine are antiarrhythmic drugs that can also function as DNMTis, and both agents have been shown to inhibit DNA methylation through interactions between the nitrogen atom of procainamide and hydralazine with the lys-162 and Arg-240 moities in the catalytic site of DNMTs.116-118 In accord with these findings, procainamide has been shown to specifically inhibit DNMT1.119 RG108 is a small molecule inhibitor of DNMTs that inhibits free DNMTs.120 This drug works by blocking the catalytic pocket of DNMTS without the formation of covalent adducts that cause cytoxicity.94 Studies have also shown RG108 to cause demethylation and reactivation of tumor suppressor genes without affecting the methylation level of microsatellite regions in lung cancer cells121, suggesting a specificity level in RG108 that has not been seen with other DNMTis (Table 5).
Histone methyltransferases (HMTs) and HMT inhibitors
Histone methylation occurs in all three basic amino acid residues: lysine, arginine and histidine. S-adenosyl methionine (SAM) donates the methyl residue in all three cases. However, histidine methylation is rare and has not been studied extensively.122, 123 Lysine residues are mono-, di-, tri-methylated by a class of enzymes called lysine methyltransferases (KMTs)23 , which were originally categorized into the SET and the DOTL domain HMTs based on the presence or absence of the SET-domain. This unconventional and non-coherent naming of KMTs has now been replaced by a more conventional system based on the type of enzymatic activity, substrate specificity, sequence homology, structural organization of the catalytic domain and order of discovery (Table 6).23 Thus far, about fifty KMTs have been identified in humans. Arginine residues are methylated by protein arginine methyltransferases (PRMTs) which are categorized into Types-1 and 2 based on whether the methyl group is added to the guanidinyl group symmetrically (i.e. dimethyl group added to different nitrogen atoms (Type 2)) or asymmetrically (i.e. dimethyl group added to single nitrogen atom (type 1)). Eleven PRMTs have been identified in humans and classified as Type-1 PRMTs except for PRMTs-5, 7 and -9 (Table 7). In depth analysis of methyl addition to arginine residues has led to further characterization of the types of PRMTs involved, however, this is beyond the scope of this review.
Table 6.
Histone Lysine Methyl Transferases: Nomenclature and Substrate Specificity.
| NOMENCLATURE HMTs | SUBSTRATE SPECIFICITY | |
|---|---|---|
| NEW | OLD | |
| KMT1A | SUV39H1 | H3K9 (P53 at K373 by KMTIC) |
| KMT1B | SUV39H2 | |
| KMT1C | G9a | |
| KMT1D | GLP | |
| KMT1E | ESET/SETDB1 | |
| KMT1F | CLL8 | |
| KMT (2A-2F) | MLL (1-5) | H3K36 |
| KMT2G | hSET1A | |
| KMT2H | ASH | |
| KMT3A | SET2 | H3K36 |
| KMT3B | NSD1 | |
| KMT3C | SYMD2 | H3K36 P53 (K370), RB (K860) |
| KMT5A | PRESET7/8 | H4K20 |
| KMT5B | SUV4-20H1 | |
| KMT5C | SUV4-20H1 | |
| KMT6 | EZH2 | H3K27 |
| KMT7 | SET7/9 | H3K4/P53 |
| KMT8A | RIZ1 | H3K9 |
| KMT8B | PRDM9 | H3K4me3 |
| KMT8C | PRDM6 | H3K20 (+KMT1C) |
| KMT8D | PRDM8 | H3K9 |
| KMT8E | PRDM3 | |
| KMT8F | PRDM16 | |
| KMT4 | DOT1L | H3K79 |
Table 7.
Histone Arginine Methyltransferases: Classification and Substrate Specificity.
| HPRMTs | OTHER NAMES | TYPE | SUBSTRATE SPECIFICITY |
|---|---|---|---|
| HRMT1 | ANM1, HCP1 IR1B4, HRMT1L2 |
I | H4R3, Ewing sarcoma (EWS) gene |
| HRMT2 | MGC11137 HRMT1L1 |
I | H4 |
| HRMT3 | HRMT1L3 | I | Ribosomal Protein S2 (RPS2) |
| HRMT4 | CARM1 | I | H3R17 (H3K18ac required), p160, NF- κb. |
| HRMT5 | JBP1, SKB1, IBP72 SKB1hs,HRMT1L5 |
II | H2A, H3, H4 |
| HRMT6 | FLJ10559 HRMT1L6 |
I | H3R2, H4R3, H2A-R3 DNA polymerase B (R83, R152) |
| HRMT7 | FLJ10640 KIAA1933 |
II | H2A, H4R3 |
| HRMT8 (Brain specific) |
HRMT1L4 | I | Ewing sarcoma (EWS) gene |
| HRMT9 | FBXO11, VIT1 UBR6, FLJ12673 MGC44383 |
II | Not characterized |
| HRMT10 | LOC90826 FLJ46629 |
Unknown | Not characterized |
| HRMT11 | FBX10, FBXO10 FLJ41992 MGC149840 |
Unknown | Not characterized |
In humans, HMTs generally contain at most four domains: the SET, I-SET, Pre-SET and Post-SET domains. The core SET-domain, which is the catalytic domain, is a sequence of ~130 amino acids named after the drosophila Su(var), E(z) and Trithorax genes. Crystal structure and topological analyses of the SET-domain showed that it consists of a series of β-sheets folded into three discrete sheets surrounding a threaded-loop which is defined by signature conserved motifs ELxF/YDY and NHS/CxxPN (i.e. x is any amino acid). 124-126 All HMTs. except KMT4 (i.e. DOT1L), contain the SET-domain, however, the catalytic domain of KMT4 shares similar structural folds to the SET domain of PRMTs. 127 The Pre-SET, I-SET, and Post-SET domains vary in nature and sequence among the different HMTs and are present in different combinations with the core SET-domain. These domains act as a scaffold around the SET-domain thereby providing interacting and recognition sites for lysine or arginine substrates and cofactors for general catalysis.128,129 Within the context of the ternary structure of HMTs, the I-SET domain is packed immediately after the SET-domain to form the substrate binding groove and part of the cofactor binding pocket. The Pre-SET domain generally provides structural support by interacting with different regions of the SET-domain, while the Post-SET domain forms part of the active site by providing an aromatic residue to pack against the SET-domain, thereby constructing a hydrophobic pore.130-132 This structural arrangement forms two distinct binding pockets for the substrate and cofactor and allows them to meet at the core for catalysis to occur.
The lysines or arginines to be methylated are inserted through the hydrophobic pore into the substrate binding pocket and the catalytic addition of a methyl group(s) using an SN2 mechanism 133, 134. The lone pair of electrons on the nitrogen of lysine or arginine acts as a nucleophile that attacks the electrophilic methyl-sulphonium cation on SAM forming a penta-coordinate carbon transition complex which dissociates into a methylated-lysine/arginine and S-adenosyl-L-homocysteine (SAH). For each round of methylation, the nitrogen atom has to be deprotonated before a methyl group can be added. The mechanism by which the initial or multiple rounds of nitrogen deprotonation occurs is still debated. The general base theory135, 136 which regards the conserved tyrosine residues (e.g., Tyr-335 in KMT7, Tyr-283in DIM-5, Tyr-287 in Rubisco LSMT, Tyr-336 in KMT5A) in the active site as the general bases that deprotonate the nitrogen residue were found to be too far from the active site to participate in general base catalysis. Another deficiency of this theory is that it does not explain repeated deprotonation of nitrogen residues for di- and tri-methylation. Other findings have shown that tyrosine residues function by providing hydrogen bonding sites to the substrate which function to restrain and align the lone pair of electrons to the scissile bond of the sulfonium group. 137-141 Alternatively, the bulk solvent theory explains many of the short comings of the general base theory.142 In this model, HMTs bind a substrate and lose a proton to a bulk solvent (e.g. H2O) followed by methyl transfer through an S2N mechanism. In the case of mono-methylation, the HMTs dissociate from the substrate to complete the methylation cycle. However, for the di- or tri-methylated state, the lysine or arginine residues are sequentially deprotonated by the bulk solvent for the addition of the second and third methyl-groups. Bulk solvent deprotonation was challenged by current findings on the ternary structure of KMT1D and KMT7 which revealed that lysine inserts into the narrow groove at the junction of SET, Pre-SET and Post-SET domain. 143 In this conformation, the lysine substrate is shielded from the solvent, thus arguing against bulk solvent deprotonation.
Unlike DNA methylation which generally represses transcription (except at gene bodies), or acetylation which generally activates transcription, the functional impact of histone methylation is contextual and can lead to both transcriptional activation and repression. A possible explanation for this conundrum lies on the timing and manner in which the HMTs are recruited to their target destination, the protein composition of HMT complexes, the occurrence of other epigenetic marks, the degree of histone methylation, and the contribution of small non-coding RNAs (sncRNAs) and long non-coding RNAs (lncRNAs). The influence of most of these factors is explained by the fact that histone methylation does not alter the charge on histone tails. Instead, it influences the basicity and affinity of different reader proteins to the methylated site (s). For example, histone methylation reader proteins (i.e. PHD finger domain, WD40 repeats, PWWP domains, chromo- and bromo-domain proteins, royal-family domain) have been shown to recruit specific HMTs to their target sites. In particular, H3K4 methylation by KMT2A (i.e. the mixed lineage leukemia (MLL)) is followed by recruitment of bromo-domain-containing complexes which result in transcriptional activation.144-146 KMT6 (i.e. EZH2) methylation at H3K27 leads to recruitment of chromo domain-containing polycomb complexes resulting in silencing of homeotic genes and KMT1A (i.e. SUV39h1) trimethylation at H3K9 of the promoters of cell cycle control genes, recruitment of chromo domain-containing heterochromatin protein 1 (HP1) and transcriptional repression.147-149 Other factors such as tissue-specific expression of HMTs, lncRNAs and sncRNAs are beyond the scope of this review.
Histone methylation and associated chromatin regulators play an important role in tumorigenesis. For example, aberrant H3K9 methylation by KMT1C has been associated with chromosomal instability and epithelial to mesenchymal transition, with recurrent translocation and/or mutation of KMT2, KMT3A, and KMT6 found in several cancers.150, 151 Loss of KMT1 is associated with decreased viability, genomic instability and increase tumor risk in mice.152 In addition, frequent somatic mutation involving the conversion of Tyr to Phe in the active site of KMT6 has been reported in lymphomas.132 Tyr to Phe mutation converts KMT6 from a multifunctional mono-, di- and tri-methylase into a dominant tri-methylase.153 KMT4 which methylates H3K79 has been shown to be mutated, or its gene translocated or fused to other genes (i.e. AF4, AF5, AF9, ENL and AF10) in many leukemias resulting in ectopic histone methylation.154, 155
The direct involvement of HMTs in tumorigenesis has led to the development of two types of HMT inhibitors (HMTi) namely the SET-domain inhibitors and non-SET-domain inhibitors. The SET-domain inhibitors include analogs of SAM such as SAH, and sinefungin, a natural inhibitor isolated from Streptomyces, small molecule inhibitors such as Chaetocin, BIX-01294 and BIX-01294 derivatives (i.e. UNC0224, UNC0631 and UNC0638), and arginine methyltransferase inhibitors 1-9 (i.e. AMI1-9) (Table 8).156-159 SAH has limited effects against HMTs because its affinity for HMTs is less than, or comparable to SAM. Sinefungin is about 3-10 fold more potent than SAM, but found to be non-selective toward most KMTs and to target DNA methyltransferase or other enzymes that use SAM as a co-factor. 159 Chaetocin, a fungal mycotoxin, was shown to be a competitive inhibitor of SAM and displays specificity toward H3K9-specific KMTs such as KMT1, KMT1A, KMT1C, but its drug-ability is dampened by significant cytotoxicity.160,161
Table 8.
Histone Methyl Transferase Inhibitors: Specificity and Activity.
| HMTi COMPOUND | NAMES | HMT SPECIFICITY |
ACTIVITY IC50 |
REF |
|---|---|---|---|---|
| HISTONE METHYLTRANSFERASE INHIBITORS (HMTis) | ||||
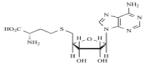
|
SAH | Non-Specific | - | 154 |
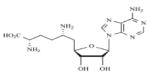
|
Sinefungin | Non-Specific | - | 157 |
|
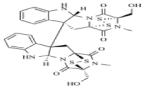
Chaetocin | KMT1 KMT1A KMT1C |
- | 157,158 |
|
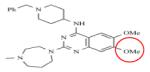
BIX01294 | KMT1C KMT1D |
1.7nM 0.7nM |
159 |
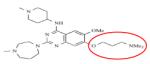
|
UNC-0624 | KMT1C KMT1D |
1.7nM 1.9nM |
160 |
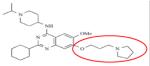
|
UNC-0638 | KMT1C KMT1D |
15nM 19nM |
160 |
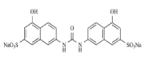
|
AMI-1 | Non-Specific | 9μM | 165 |
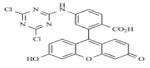
|
AMI-6 | PRMT1 | 1.4μM | 165 |
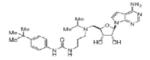
|
EPZ00477 | KMT4 | 0.4nM | 166 |
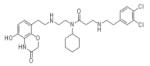
|
AZ505 | KMT3C | 0.12μM | 172 |
Red circles denote position where side groups (R) are substituted.
BIX-01294 is a selective inhibitor of a structurally similar KMT1C and KMT1D with IC50 values of 1.7μM and 0.7μM, respectively, over KMT1A and PRMT1 (Table 8).162-165 BIX-01294 is made of a central quinazoline ring connected to a seven-membered diazepane and to a benzylated six-membered piperidine. Crystal structure analysis of BIX-01294 in complex with KMT1D and SAM show that BIX-01294 occupies the substrate binding site with the quinozoline moiety interacting with the four aspartate residues on the acidic surface of SET-domain.162 This interaction blocks the insertion and binding of the substrate to the substrate binding pocket. Because of the selectivity and potency of BIX-01294, several derivatives of this drug have been developed such as UNCO224.163,165 In UNCO224, the methoxy group on carbon 5 of the quinazoline ring was replaced by a 7-aminoalkoxy side chain to increase the interaction at the substrate binding site. Accordingly, this switch increases the potency 7-fold with IC50 values of 1.7nM and 1.9nM for KMT1C and KMT1D, and more than 1000-fold selectivity for KMT1C over KMT5A and KMT7 in in-vitro assays.162 Other analogues with cyclic amino groups such as pyrrolidine and piperidine substituted at the 5-methoxy position have been shown to be more potent than UNCO224 because these cyclic side chains pack more efficiently in the substrate binding groove. However, their potency is dampened by their impermeability.163,164 To address permeability deficits, other derivatives such as UNC0638 have been developed. In the case of UNC0638, the nitrogen on the piperidine ring was substituted by isopropyl group, the diazepane ring with cyclohexyl group and the methoxy group on carbon-5 of the quinazoline with a *********propyl-pyrrolidin group.165 In vitro testing of this compound showed increased permeability, a potency with IC50 values of 15nM and 19nM for KMT1C and KMT1D and non-activity against KMT1, KMT6, KMT4, KMT7, KMT5A, HRMT1 and HRDM3. 165 Although still in their infancy, BIX-01294 and its derivatives have shown great potential for the treatment of cancers that involve dysregulation of KMTs.
The first inhibitors of HRMTs are binaphthylureas, symmetrical sulfonated ureas, and their derivatives, named arginine methyltransferase inhibitors 1-9 (i.e. AMI1-9) 157, 166, 168. Cell culture studies using RNA binding fusion protein GFP-Npl3 as a target showed that AMI-1 decreases methylation of GFP-Np13 by inhibiting PRMT1 with IC50 values of 9μM (Table 8). 164 AMI-1 was shown to be a competitive inhibitor of the arginine substrate and to occupy the substrate binding site of PRMTs. 157 Because of the structural similarity between AMI-1 and sirtuin inhibitors (i.e. surnamin), AMI-1 has been shown to inhibit sirtuin-1 (SIRT-1) with an IC50 of 32μM.166 To increase specificity, several derivatives have been developed using AMI-1 and AMI-5 as templates. 165 In AMI-6, these investigators substituted bromines at positions 2, 4, 5, and 7 on the xanthene ring resulting in greater specificity for PRMT1 with an IC50 of 1.4μM (Table 8).157
The so called non-SET domain inhibitors targeting KMT4 are regarded as analogs of SAM in spite of the absence of the SET domain because the protein uses SAM as a cofactor. The first of these drugs, EPZ004777, was synthesized by replacing the methionine amino acid moiety in SAM with urea (Table 8).168 EPZ004777 was found to be greater than 1000-fold selective for KMT4 over CARM1, PRMT1, PRMT5, PRMT8, KMT1D, KMT6, and KMT7. This agent showed antiproliferative, antidifferentiation and apoptotic activity toward cells harboring MLL fusion.168 EPZ004777 was also shown to prolong survival in a mouse model of mixed-lineage leukemia.168 This molecule has poor permeability across the membrane due to the presence of 6 hydrogen bonding sites. Other derivatives have been developed to address this limitation.
The value of focusing on HMTs as viable targets for therapeutic intervention in cancer stems from their high specificity for lysine and arginine methylation.169 For example, KMTA/1B preferentially trimethylates H3K9 compared to its mono-methylated state and KMT1C has shown preference for the mono-methylated compared to the dimethylated state H3K9.170 Nevertheless, the specificity of HMTs and their potential as therapeutic targets is dampened by the fact that they also methylate non-histone proteins and can be redundant with regard to the number of HMTs that methylate the same residue. For example, KMT1C and PRMT1 have been shown to specifically methylate H3K9 and H4R3, respectively, but other KMTs (i.e. KMT1, KMT1B, KMT1C) and PRMTs (i.e. PRMT6a, PRMT5, PRMT7) have been shown to methylate the same lysine and arginine residues, respectively.172, 173 Recently, Astrazeneca developed a KMT specific drug, AZ505, which specifically inhibits KMT3C.174 KMT3C has been shown to methylate H3K36 as well as K370 and K860 in p53 and RB, respectively. K370 methylation prevents p53 from binding to the promoters of target genes and methylation at K860 in RB has been shown to create an epitope that selectively recognizes the transcriptional repressor L3MBTL1.175-178 Binding studies show that AZ505 is a competitive inhibitor of the lysine substrate and shows selectivity toward KMT3C with an IC50 value of 0.12μM compared to greater than 83.3μM IC50 values for KMT4, KMT6, KMT1D, KMT1C and KMT7. KMT1C, which is inhibited by BIX-01294 and its derivatives, has also been shown to dimethylate K373 in p53 leading to inactivation (Table 8). 174 As such, drugs targeting aberrant methylation on histones and non-histone proteins are being developed to combat cancer.
Histone demethylase (HDMs) and HDM inhibitors
Two classes of enzymes are responsible for removing methyl groups from histones: lysine-specific demethylase (KDM) and arginine-specific demethylase (RDM). KDM includes amine oxidase-like (AOL) domain-containing demethylases179 and Jumonji C (JmjC) domain-containing demethylases.180 RDM includes peptidyl-arginine deiminase 4 (PAD4) (Table 9).181 PAD4 is the only known RDM and it does not demethylate arginine, instead it converts methylarginine to citrulline.181 Citrullination has been described as a unique epigenetic modification because PAD4 can covert unmethylated arginines to citrulline.182 This modification implicates citrullination as a mechanism to deplete methylable arginines.
Table 9.
Histone Demetylases: Nomenclature and Substrate Specificity.
| NOMENCLATURE HDMs | SUBSTRATE | |
|---|---|---|
| NEW | OLD | |
| KDM1A | LSD1/AOF2 | H3K4me2/me1; H3K9me2/me (AR) |
| KDM1B | LSD2/AOF1 | H3K4me2/me1 |
| KDM2A | FBXL11A, JHDM1A | H3K36me2/me1 |
| KDM2B | FBXL10B, JHDM1B | H3K36me2/me1; H3K4me3 |
| KDM3A | JMJB1A, JHDM2A | H3K9me3/me1 |
| KDM3B | JMJB1B, JHDM2B | |
| KDM4A | JMJB2A, JHDM3A | H3K9me3/me2 H3K36me3/me2 |
| KDM4B | JMJD2B | |
| KDM4C | JMJD2C, GASC1 | |
| KDM4D | JMJD2D | H3K9me3/me2/me1; H3K36me3/me2 |
| KDM4E | JMJD2E | H3K9me3/me2 |
| KDM5A | JARID1A, RBP2 | H3K4me3/me2 |
| KDM5B | JARID1B, PLU1 | |
| KDM5C | JARID1C, SMCX | |
| KDM5D | JARID1D, SMCY | |
| KDM6A | UTX, MGC141941 | H3K27me3/me2 |
| KDM6B | JMJD3, KIAA1111 | H3K27me3/me2; H3K9me2/me1 H4K20me1 |
| KDM7 | KIAA1718 | H3K4me2/me1 H3K27m2/me1 |
| KDM8 | JMJD5/ FLJ13798 | H3K36me2 |
Thus far, KDM1A (LSD1) and KDM1B (LSD2) are the only AOL domain-containing demethylases.179, 183 Structurally both enzymes consist of three domains: a common SWIRM (SWI3P, RSC8p and Moira) domain, an AOL domain interrupted by a spacer domain (i.e. the so called tower domain) in KDM1A only, and a CW-type zinc-finger domain present only in KDM1B. The SWIRM domain is required to stabilize the enzyme and through interactions with the AOL domain form a wide cleft that provides an interface for substrate interaction184. The tower domain is believed to provide an allosteric interacting surface that directs the specificity of the enzyme.185 The AOL domain is homologous to the convention FAD-dependent binding domain and consists of two subdomains: the FAD binding and substrate binding subdomains; the interface of the two subdomains form the catalytic site.
KDM1A and KDM1B specifically catalyze the removal of mono/di- methyl groups from H3K4; however they have been shown to change substrate specificity when in complex with different accessory proteins. For example, KDM1A in complex with androgen receptor is able to demethylate H3K9.186 In contrast, KDM1A when in complex with silencer corepressor of RE1 silencing transcription factor (CoREST) can demethylate histones within nucleosome substrates.187,187 The catalytic removal of methyl groups is initiated by the formation of an imine intermediate through oxidative protonation of the lone pair of electrons on the nitrogen atom.179 The iminium intermediate is then hydrolyzed to a carbinolamine intermediate that degrades spontaneously into formaldehyde and demethylated H3K4. The reduced cofactor, FADH2, is reoxidized by O2 to FAD and H2O2. Because the initial step requires a lone pair of electrons on the nitrogen atom for protonation, KDM1 family members cannot demethylate tri-methylated residues.
About 30 members of the Jumonji C (JmjC) domain-containing demethylases have been discovered in humans and divided into 7-groups based on phylogenetic and structural analysis. Structurally they consist of a minimum of a core Jmj domain which has been shown to be sufficient to perform the catalytic demethylation.180 The additional domains are believed to enhance demethylation by facilitating substrate and/or protein-protein interactions. The catalytic demethylation of JmjC domain containing enzymes is different from the KDM family members in that a lone pair of electrons is not needed to initiate catalysis; hence they can demethylate all three states of methylation.188 Rather, the catalysis depends on the two cofactors: Fe2+ and α-ketoglularate.
In the unbound state, the Fe2+ at the active site is coordinated by three molecules of water, two histidine residues and one glutamate residue.189 It is believed that in the initial step of the demethylation, molecular dioxygen and α-ketoglularate coordinate to Fe2+ by displacing the water molecules, and this is followed by a single electron transfer from Fe2+ to the dioxygen creating reactive peroxide radical. The peroxide radical attacks the C2 bond of α-ketoglularate creating a bond between α-ketoglularate and Fe3+. In the next step, α-ketoglularate decarboxylates into succinate and CO2 with the generation of Fe3+-OXO radical in the catalytic core. The highly reactive hydroxyl radical group then activates the C-H bond in the substrate methyl-lysine and transfers oxygen to the methyl group forming hydroxymethyllysine which is an unstable hemiaminal. The hemiaminal readily decomposes into formaldehyde and a lysine residue lacking a methyl group.180, 188 The coordination of the three water molecules to the Fe2+ regenerates the initial catalytic species which can undergo a second and third round of demethylation.190-192
Dysregulation of demethylases have been reported in several cancers. For example, KDM1A overexpression is a predictive biomarker of prostate cancer193 and JMD2C and KDM1A have been identified as coactivators of the androgen receptor through demethylation of the repressive mark H3K9 leading to expression of androgen dependent genes.184,193 Overexpression of the H3K9 methylation mark is associated with higher risk of relapse in prostate cancer. In addition, the microenvironment of tumors is depleted of oxygen which leads to the activation of hypoxia inducible factor (HIF) which in turn targets the JMJD1 and JMJD2 family of demethylases.194-197 Also, the overexpression of KDM5A has been implicated in drug resistant cancers. 198 Therefore, inhibitors have been developed for both classes of demethylases and their potency toward the different HDMs tested.
Three different classes of inhibitors have been developed to specifically target KDM1A: Mono Amine Oxidase Inhibitors, Substrate-based Inhibitors and Bisquanidine analogues (Table 10). The monoamine oxidase inhibitors were originally designed to target Mono Amine Oxidase A & B (MAOA & MAOB) in the treatment of depression and neurological disorders.199-201 While the MAO activity removes the amine groups of neurotransmitters in an FAD dependent manner, sequence and structural analyses showed that the MOA domain is similar to the AOL domain of KDM1As. As such, tranylcypromine (PCPA), while itself not effective against KDM1A (Ki = 357μM), served as the lead to design potent inhibitors of KDM1A.202 A homoserine derivative of PCPA have been shown to be 100-fold more potent than PCPA and is selective against MAOA & MAOB (Table 10). Methylation analysis showed recovery of H3K4 dimethylation at a concentration of 6-67μM in HEK293 cells.203 Another derivative, S2101 increased dimethylation at H3K4 at concentrations of 1μM with a Ki value of 0.6μM against KDM1A.204 Crystal structure analyses determined that these drugs inhibit the activity of KDM1A by forming a covalent adduct with the FAD cofactor.202
Table 10.
Histone Demethylase Inhibitors: Specificity and Activity.
| HDMis COMPOUND | NAMES | HDM SPECIFICITY |
ACTIVITY IC50 |
REF |
|---|---|---|---|---|
| HISTONE DEMETHYLRASE INHIBITORS (HDMis) | ||||
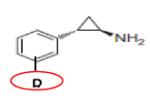
|
TRANYLCYPROMINE (PCPA) (R = H) |
KMD1A | Ki = 357μM Non-Specific |
200 |
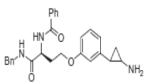
|
Compound 2 | KMD1A | 1.9μM | 201 |
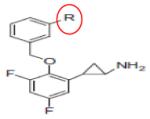
|
S2101 (R = H) |
KMD1A | 0.6μM | 203 |
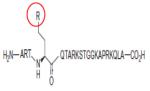
|
Compound 22 (R= -CH2CH2NHNH2) |
KMD1A | 4.4nM | 203 |
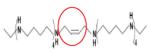
|
PG11140 (Cis-Form) PG11150 (Trans-Form) |
KMD1A KMD1A |
1.0μM 5.0μM |
202 |
|
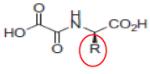
N-OXALYLGLYCINE (R = H) |
JMJD2E | 78μM 28μM (30mins) |
215 |
|
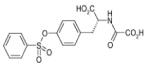
N-OXALYLTYROSINE | JMJD2E | 5.4μM | 215 |
|
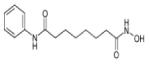
SAHA | JMJD2E | 520μM 14μM (30mins) |
213 |
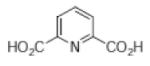
|
Pyridine-2-4-dicarboxylic acid (3,4-PDCA) |
JMJD2E | 1.4μM | 210 |
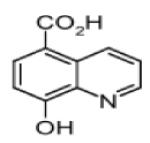
|
Hydroxyquinoline Carboxylic acid (8-HQ-5-COOH) |
JMJD2A | 2.0μM | 214 |
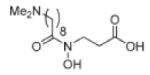
|
HYDROXAMINE | JMJD2C JMJD2A |
1.0μM 3.0μM |
215 |
Red circles denote position where side groups (R) are substituted or where conformation stereoisomer originate (i.e. PG11144 and PG11150).
Substrate base inhibitors are designed from the last 21 amino acid peptide sequence of the histone-3 tail with the addition of chemical subgroups at lysine-4 (K4) of the peptide sequence.205 The most potent of this class of drugs is a compound in which a dimethyl-hydrazine group was substituted at the K4 position.202 Compared to the lead substrate (methionine substitution at K4), this compound displays an inhibition rate in the nanomolar range (Ki = 4.4nM) making it the most potent inhibitor of KDM1A identified thus far. 202 It is believed that substrate based inhibitors act through suicide inactivation by permanently trapping the enzyme thereby preventing it from acting on another substrate. Substrate based inhibitors are promising in the sense that they are structurally similar to the natural substrates and with smart design can remain inert until acted on by the KDM1A enzyme.
Bisguanidine analogues were developed based on the observation that KDM1A shares considerable homology with FAD-dependent polyamine oxidases and that biguanidines can inhibit polyamines oxidases.206-208 Thus far, two analogues (PG-1150 and PG-11144) have shown more than 50% inhibition of KDM1A at a concentration of 1μM and a noncompetitive inhibition profile of purified KDMIA enzymes at a concentration of 2.5μM.206, 209 Exposure of HCT116 human colon carcinoma cells to these compounds significantly increased mono-/dimethylation at H3K4 followed by reexpression of previously silenced genes.206, 209 An interesting finding in these studies is that the bisguanidine anologues exhibit noncompetitive inhibition despite the structural similarity between the bisguanidine anologues and the natural substrate.
Thus far, three classes of inhibitors have been identified for JmjC HDMs: N-oxalylgycine analogues, HDAC inhibitors and pyridine carboxylate derivatives. N-oxalylgycine analogues were originally designed to target Fe2+ and α-ketoglutarate dependent oxygeneses such as Hypoxia Inducible Factor (HIF), prolyl hydroxylase PHD2 (Ki = 8μM) and Asparaginyl hydroxylase factor-inhibiting −HIF (Ki = 1.2mM).209-211 N-oxalyl glycine, the lead compound in this group, was tested against JMJD2E and showed an IC50 of 78μM which decreased to 28μM with a 30 minute preincubation.212 Other derivatives with substitution of the benzyl or ethylphenyl group at the N-oxalylated D-amino acid, namely, N-Oxalyl-D-phenylalanine and N-oxalyl-D-homophenylalanine, showed IC50 values of 320 and 100 μM, respectively.212 The most effective in this group is the N-oxalyltyrosine derivative which showed an IC50 of 5.4μM against JMJD2E (Table 10).213
HDACs inhibitors (e.g. SAHA or Vorinostat) are defined in part by a hydroxamic acid group and a lipophilic stretch that allows them to coordinate to Zn2+ in the cofactor binding pocket of HDACs. Since the catalytic mechanism employed by JmjC involves coordination to Fe2+, it was thought that HDAC inhibitors such as SAHA could also inhibit JmjC HDMs. In addition, some of the JmjC HDMs such as JMJD2 family members (A-E) contain a zinc binding site.214 As such, HDAC inhibitors such as trichostatin A (TSA), SAHA and butyric acid were tested against JMJD3E.212 Butyric acid was ineffective against JMJD2E, and TSA could not be assayed because it inhibited formaldehyde dehydrogenese (FDH) which was used in the assay to oxidize the product of demethylation, formaldehyde to formate.212 SAHA on the other hand was moderately effective against JMJD2E with an IC50 value of 540μM which significantly decreased to 14μM with a 30 minute preincubation.212 In comparison, SAHA has been shown to inhibit HDAC1 with an IC50 value of 48nM which is more than 1000-fold more effective.215 Nevertheless, the advantage of JmjC HDAC specific inhibitors such SAHA and TSA is that these drugs are already in the market and enzyme kinetic studies have shown mixed competitive inhibition for SAHA against JMJD2E, suggesting that the way SAHA inhibits JmjC HDMs is different from HDACs.
The pyridine carboxylate derivatives were originally developed to inhibit collagen prolyl hydroxylases (e.g. collagen prolyl-4-hydroxylases) with Fe2+ as a cofactor. The most effective of these inhibitors are pyridine-2,4-dicarboxylic acid (2,4-PDCA) with an IC50 of 1.4μM for JMJD2E212, 8-hydroxyquinoline-5-carboxylic acid (8-HQ-5-COOH) with an IC50 of 2.0 μM for JMJD2A216 and internal hydroxyamic acid with an IC50 of 2 μM for JMJ2A and 1.0 μM for JMJ2C.217 To be effective, JmjC inhibitors should be nonspecific against other Fe2+/2α-ketoglutarate dependent oxygeneses (i.e. HIF asparaginyl hydroxylase, factor-inhibiting HIF (FIH), and the human HIF prolyl hydroxylase domain 2 (PHD2) whose activities have been shown to control a wide array of genes. In this context, small molecules that target different regions of these JmjC enzymes will be the most promising candidates to remedy the problem of specificity.
Closing Remarks and Perspectives
Despite the promise of epigenetic therapies, in most cases available therapies lack specificity. Epigenetic drugs targeting DNA methylation or DNMTs show considerable cytotoxicity because these drugs cause global demethylation by passive demethylation or trapping of DNMTs. Such unintended consequences have limited the use of these drugs for prolong periods of time. Because HDACs and HATs are part of macromolecular protein complexes, targeting them can also lead to unintended consequences. The lack of specificity is in keeping with the fact that epigenetic modifications are not stand alone processes, with synergistic interactions between and within marks increasing complexity of regulatory control. The inhibitors designed to target HMTs and HDMs mainly target cofactors and/or cofactor binding sites, leading to a considerable degree of non-specificity in light of the vast array of enzymes using the same co-factors to catalyze multiple processes. Thus, a more comprehensive structural analysis is needed to identify unique domain(s) and residues critical to the catalytic mechanisms used by these enzymes. Increased understanding of the histone code and the macromolecular protein complexes involved in epigenetic regulation will help refine the targeting of epigenetic therapies. The will involve the design of so called smart drugs that will target only specific epigenetic modifications, either alone or in combination with other marks. Thus, unless our understanding of the epigenome continues to improve and the key regulators of epigenetic control are identified, therapies targeting epigenetic modifications within the human genome will remain of limited value and restricted for widespread dissemination in the clinical setting.
Footnotes
Publisher's Disclaimer: This is a PDF le of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its nal citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Waddington CH. Selection of the genetic basis for an acquired character. Nature. 1952;169:625–6. doi: 10.1038/169625b0. [DOI] [PubMed] [Google Scholar]
- 2.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes & development. 2009;23:781–3. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteller M. Epigenetics in cancer. The New England journal of medicine. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 5.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–11. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 7.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature genetics. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 8.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC genomics. 2009;10:143. doi: 10.1186/1471-2164-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, Khorasanizadeh S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. The EMBO journal. 2001;20:5232–41. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 12.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Human molecular genetics. 2007;16(Spec No 1):R50–9. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 14.Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, Wheeler DA, Gibbs RA, Kucherlapati R, Lee C, Kharchenko PV, Park PJ. Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967–71. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20:1170–7. doi: 10.1093/bioinformatics/bth059. [DOI] [PubMed] [Google Scholar]
- 17.i A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature genetics. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 19.Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene. 2008;27:404–8. doi: 10.1038/sj.onc.1210631. [DOI] [PubMed] [Google Scholar]
- 20.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews. Cancer. 2010;10:457–69. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleff S, Andrulis ED, Anderson CW, Sternglanz R. Identification of a gene encoding a yeast histone H4 acetyltransferase. The Journal of biological chemistry. 1994;270:24674–7. doi: 10.1074/jbc.270.42.24674. [DOI] [PubMed] [Google Scholar]
- 23.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 24.Allis CD, Chicoine LG, Richman R, Schulman IG. Deposition-related histone acetylation in micronuclei of conjugating Tetrahymena. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:8048–52. doi: 10.1073/pnas.82.23.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz-Carrillo A, Wangh LJ, Allfrey VG. Processing of newly synthesized histone molecules. Science. 1974;190:117–28. doi: 10.1126/science.1166303. [DOI] [PubMed] [Google Scholar]
- 26.Parthun MR, Widom J, Gottschling DE. The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell. 1996;87:85–94. doi: 10.1016/s0092-8674(00)81325-2. [DOI] [PubMed] [Google Scholar]
- 27.Yuan H, Marmorstein R. Histone Acetyltransferases: Rising Ancient Counterparts to Protein Kinases. Biopolymers. 2012;99(2):98–111. doi: 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. The Journal of biological chemistry. 1999;274:18157–60. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 29.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. The Journal of biological chemistry. 2000;275:22048–55. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 30.Tanner KG, Langer MR, Denu JM. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry. 2000;39:11961–9. doi: 10.1021/bi001272h. [DOI] [PubMed] [Google Scholar]
- 31.Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nature structural biology. 2002;9:862–9. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 32.Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Cote J. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes & development. 2003;17:1415–28. doi: 10.1101/gad.1056603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berndsen CE, Albaugh BN, Tan S, Denu JM. Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry. 2007;46:623–9. doi: 10.1021/bi602513x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–50. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 35.Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. The Journal of biological chemistry. 2001;276:33721–9. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 36.Cullis PM, Wolfenden R, Cousens LS, Alberts BM. Inhibition of histone acetylation by N-[2-(S-coenzyme A)acetyl] spermidine amide, a multisubstrate analog. The Journal of biological chemistry. 1982;257:12165–9. [PubMed] [Google Scholar]
- 37.Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, Cole PA. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. The Journal of biological chemistry. 2000;275:21953–9. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- 38.Sagar V, Zheng W, Thompson PR, Cole PA. Bisubstrate analogue structure-activity relationships for p300 histone acetyltransferase inhibitors. Bioorganic & medicinal chemistry. 2004;12:3383–90. doi: 10.1016/j.bmc.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 39.Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, Kundu TK. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. The Journal of biological chemistry. 2004;279:33716–26. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- 40.Mantelingu K, Reddy BA, Swaminathan V, Kishore AH, Siddappa NB, Kumar GV, Nagashankar G, Natesh N, Roy S, Sadhale PP, Ranga U, Narayana C, Kundu TK. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chemistry & biology. 2007;14:645–57. doi: 10.1016/j.chembiol.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, Bavetsias V, Gorsuch S, Jarman M, Bannister A, Kouzarides T, McDonald E, Workman P, Aherne GW. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Molecular cancer therapeutics. 2005;4:1521–32. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- 42.Biel M, Kretsovali A, Karatzali E, Papamatheakis J, Giannis A. Design, synthesis, and biological evaluation of a small-molecule inhibitor of the histone acetyltransferase Gcn5. Angewandte Chemie. 2004;43:3974–6. doi: 10.1002/anie.200453879. [DOI] [PubMed] [Google Scholar]
- 43.Murr R, Vaissiere T, Sawan C, Shukla V, Herceg Z. Orchestration of chromatin-based processes: mind the TRRAP. Oncogene. 2007;26:5358–72. doi: 10.1038/sj.onc.1210605. [DOI] [PubMed] [Google Scholar]
- 44.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–57. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13182–7. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Molecular and cellular biology. 2007;27:8502–9. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. Journal of medicinal chemistry. 2003;46:5097–116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 48.Schemies J, Sippl W, Jung M. Histone deacetylase inhibitors that target tubulin. Cancer letters. 2009;280:222–32. doi: 10.1016/j.canlet.2009.01.040. [DOI] [PubMed] [Google Scholar]
- 49.North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome biology. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends in biochemical sciences. 2007;32:555–60. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Fatkins DG, Zheng W. Substituting N(epsilon)-thioacetyl-lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacetylases. International journal of molecular sciences. 2008;9:1–11. doi: 10.3390/ijms9010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Molecular and cellular biology. 26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature reviews. 2006;1:287–99. doi: 10.1038/nrd772. Drug discovery. [DOI] [PubMed] [Google Scholar]
- 53.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medda F, Russell RJ, Higgins M, McCarthy AR, Campbell J, Slawin AM, Lane DP, Lain S, Westwood NJ. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. Journal of medicinal chemistry. 52:2673–82. doi: 10.1021/jm8014298. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hirsch CL, Bonham K. Histone deacetylase inhibitors regulate p21WAF1 gene expression at the post-transcriptional level in HepG2 cells. FEBS letters. 2004;570:37–40. doi: 10.1016/j.febslet.2004.06.018. 2009. [DOI] [PubMed] [Google Scholar]
- 55.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. British journal of cancer. 2000;83:817–25. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wharton W, Savell J, Cress WD, Seto E, Pledger WJ. Inhibition of mitogenesis in Balb/c-3T3 cells by Trichostatin A. Multiple alterations in the induction and activation of cyclin-cyclin-dependent kinase complexes. The Journal of biological chemistry. 2000;275:33981–7. doi: 10.1074/jbc.M005600200. [DOI] [PubMed] [Google Scholar]
- 57.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews. Drug discovery. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 58.Espino PS, Drobic B, Dunn KL, Davie JR. Histone modifications as a platform for cancer therapy. Journal of cellular biochemistry. 2005;94:1088–102. doi: 10.1002/jcb.20387. [DOI] [PubMed] [Google Scholar]
- 59.Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:3578–88. [PubMed] [Google Scholar]
- 60.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:3971–93. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 61.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. Journal of cellular biochemistry. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 62.Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. Journal of cellular biochemistry. 2004;92:223–37. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- 63.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek MA, Zhao M, Smith BD, Manning J, Jiemjit A, Dover G, Mays A, Zwiebel J, Murgo A, Weng LJ, Herman JG. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer research. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 64.Maslak P, Chanel S, Camacho LH, Soignet S, Pandolfi PP, Guernah I, Warrell R, Nimer S. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2006;20:212–7. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 65.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–9. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nemunaitis JJ, Orr D, Eager R, Cunningham CC, Williams A, Mennel R, Grove W, Olson S. Phase I study of oral CI-994 in combination with gemcitabine in treatment of patients with advanced cancer. Cancer journal. 2003;9:58–66. doi: 10.1097/00130404-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 67.Undevia SD, Kindler HL, Janisch L, Olson SC, Schilsky RL, Vogelzang NJ, Kimmel KA, Macek TA, Ratain MJ. A phase I study of the oral combination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2004;15:1705–11. doi: 10.1093/annonc/mdh438. [DOI] [PubMed] [Google Scholar]
- 68.Pauer LR, Olivares J, Cunningham C, Williams A, Grove W, Kraker A, Olson S, Nemunaitis J. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer investigation. 2004;22:886–96. doi: 10.1081/cnv-200039852. [DOI] [PubMed] [Google Scholar]
- 69.Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, Germing U, Gattermann N. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004;104:1266–9. doi: 10.1182/blood-2003-12-4333. [DOI] [PubMed] [Google Scholar]
- 70.Pilatrino C, Cilloni D, Messa E, Morotti A, Giugliano E, Pautasso M, Familiari U, Cappia S, Pelicci PG, Lo Coco F, Saglio G, Guerrasio A. Increase in platelet count in older, poor-risk patients with acute myeloid leukemia or myelodysplastic syndrome treated with valproic acid and all-trans retinoic acid. Cancer. 2005;104:101–9. doi: 10.1002/cncr.21132. [DOI] [PubMed] [Google Scholar]
- 71.Raffoux E, Chaibi P, Dombret H, Degos L. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica. 2005;90:986–8. [PubMed] [Google Scholar]
- 72.Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101:3236–9. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 73.Wittmann S, Bali P, Donapaty S, Nimmanapalli R, Guo F, Yamaguchi H, Huang M, Jove R, Wang HG, Bhalla K. Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer research. 2003;63:93–9. [PubMed] [Google Scholar]
- 74.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102:3765–74. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- 75.Fiskus W, Pranpat M, Bali P, Balasis M, Kumaraswamy S, Boyapalle S, Rocha K, Wu J, Giles F, Manley PW, Atadja P, Bhalla K. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl-expressing human leukemia cells. Blood. 2006;108:645–52. doi: 10.1182/blood-2005-11-4639. [DOI] [PubMed] [Google Scholar]
- 76.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 77.Chen ZX, Mann JR, Hsieh CL, Riggs AD, Chedin F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. Journal of cellular biochemistry. 2005;95:902–17. doi: 10.1002/jcb.20447. [DOI] [PubMed] [Google Scholar]
- 78.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. The Journal of biological chemistry. 2010;285:26114–20. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okano M, Xie S, Li E. Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic acids research. 1998;26:2536–40. doi: 10.1093/nar/26.11.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–8. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- 81.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic acids research. 1999;27:2291–8. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic acids research. 2003;31:2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature genetics. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 84.Geiman TM, Sankpal UT, Robertson AK, Zhao Y, Robertson KD. DNMT3B interacts with hSNF2H chromatin remodeling enzyme, HDACs 1 and 2, and components of the histone methylation system. Biochemical and biophysical research communications. 2004;318:544–55. doi: 10.1016/j.bbrc.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 85.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 86.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in biochemical sciences. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 87.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews. Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rius M, Stresemann C, Keller D, Brom M, Schirrmacher E, Keppler D, Lyko F. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Molecular cancer therapeutics. 2009;8:225–31. doi: 10.1158/1535-7163.MCT-08-0743. [DOI] [PubMed] [Google Scholar]
- 91.Huber-Ruano I, Pastor-Anglada M. Transport of nucleoside analogs across the plasma membrane: a clue to understanding drug-induced cytotoxicity. Current drug metabolism. 2009;10:347–58. doi: 10.2174/138920009788499030. [DOI] [PubMed] [Google Scholar]
- 92.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. International journal of cancer. Journal international du cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 93.Issa JP, Kantarjian HM. Targeting DNA methylation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3938–46. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Momparler RL, Rossi M, Bouchard J, Vaccaro C, Momparler LF, Bartolucci S. Kinetic interaction of 5-AZA-2′-deoxycytidine-5′-monophosphate and its 5′-triphosphate with deoxycytidylate deaminase. Molecular pharmacology. 1984;25:436–40. [PubMed] [Google Scholar]
- 95.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Azadeoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Molecular and cellular biology. 2005;25:4727–41. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96.Kuo HK, Griffith JD, Kreuzer KN. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer research. 2007;67:8248–54. doi: 10.1158/0008-5472.CAN-07-1038. [DOI] [PubMed] [Google Scholar]
- 97.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–8. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 98.Voso MT, Santini V, Finelli C, Musto P, Pogliani E, Angelucci E, Fioritoni G, Alimena G, Maurillo L, Cortelezzi A, Buccisano F, Gobbi M, Borin L, Di Tucci A, Zini G, Petti MC, Martinelli G, Fabiani E, Fazi P, Vignetti M, Piciocchi A, Liso V, Amadori S, Leone G. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clinical cancer research, 2009; an official journal of the American Association for Cancer Research. 2009;15:5002–7. doi: 10.1158/1078-0432.CCR-09-0494. [DOI] [PubMed] [Google Scholar]
- 99.Sekeres MA, List AF, Cuthbertson D, Paquette R, Ganetzky R, Latham D, Paulic K, Afable M, Saba HI, Loughran TP, Jr., Maciejewski JP. Phase I combination trial of lenalidomide and azacitidine in patients with higher-risk myelodysplastic syndromes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:2253–8. doi: 10.1200/JCO.2009.26.0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou DC, Kim SH, Ding W, Schultz C, Warrell RP, Jr., Gallagher RE. Frequent mutations in the ligand-binding domain of PML-RARalpha after multiple relapses of acute promyelocytic leukemia: analysis for functional relationship to response to all-trans retinoic acid and histone deacetylase inhibitors in vitro and in vivo. Blood. 2002;99:1356–63. doi: 10.1182/blood.v99.4.1356. [DOI] [PubMed] [Google Scholar]
- 101.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. Journal of the National Cancer Institute. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 102.Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, Plass C, Niemeyer CM, Lubbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2009;23:1019–28. doi: 10.1038/leu.2008.397. [DOI] [PubMed] [Google Scholar]
- 103.Billam M, Sobolewski MD, Davidson NE. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast cancer research and treatment. 2010;120:581–92. doi: 10.1007/s10549-009-0420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dote H, Cerna D, Burgan WE, Camphausen K, Tofilon PJ. ErbB3 expression predicts tumor cell radiosensitization induced by Hsp90 inhibition. Cancer research. 2005;65:6967–75. doi: 10.1158/0008-5472.CAN-05-1304. [DOI] [PubMed] [Google Scholar]
- 105.Balch C, Yan P, Craft T, Young S, Skalnik DG, Huang TH, Nephew KP. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Molecular cancer therapeutics. 2005;4:1505–14. doi: 10.1158/1535-7163.MCT-05-0216. [DOI] [PubMed] [Google Scholar]
- 106.Hellebrekers DM, Jair KW, Vire E, Eguchi S, Hoebers NT, Fraga MF, Esteller M, Fuks F, Baylin SB, van Engeland M, Griffioen AW. Angiostatic activity of DNA methyltransferase inhibitors. [DOI] [PubMed]
- 107.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. Journal of molecular biology. 2002;321:591–9. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beumer JH, Parise RA, Newman EM, Doroshow JH, Synold TW, Lenz HJ, Egorin MJ. Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU) Cancer chemotherapy and pharmacology. 2008;62:363–8. doi: 10.1007/s00280-007-0603-8. [DOI] [PubMed] [Google Scholar]
- 109.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 110.Reither S, Li F, Gowher H, Jeltsch A. Catalytic mechanism of DNA-(cytosine-C5)-methyltransferases revisited: covalent intermediate formation is not essential for methyl group transfer by the murine Dnmt3a enzyme. Journal of molecular biology. 2003;329:675–84. doi: 10.1016/s0022-2836(03)00509-6. [DOI] [PubMed] [Google Scholar]
- 111.Gowher H, Jeltsch A. Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer biology & therapy. 2004;3:1062–8. doi: 10.4161/cbt.3.11.1308. [DOI] [PubMed] [Google Scholar]
- 112.Kratzke RA, Wang X, Wong L, Kratzke MG, Green MR, Vokes EE, Vogelzang NJ, Kindler HL, Kern JA. Response to the methylation inhibitor dihydro-5-azacytidine in mesothelioma is not associated with methylation of p16INK4a: results of cancer and leukemia group B 159904. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2008;3:417–21. doi: 10.1097/JTO.0b013e318168da0a. [DOI] [PubMed] [Google Scholar]
- 113.Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer research. 2003;63:4984–9. [PubMed] [Google Scholar]
- 114.Singh N, Duenas-Gonzalez A, Lyko F, Medina-Franco JL. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem. 2009;4:792–9. doi: 10.1002/cmdc.200900017. [DOI] [PubMed] [Google Scholar]
- 115.Mund C, Brueckner B, Lyko F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: basic concepts and clinical applications. Epigenetics: official journal of the DNA Methylation Society. 2006;1:7–13. doi: 10.4161/epi.1.1.2375. [DOI] [PubMed] [Google Scholar]
- 116.Song Y, Zhang C. Hydralazine inhibits human cervical cancer cell growth in vitro in association with APC demethylation and re-expression. Cancer chemotherapy and pharmacology. 2009;63:605–13. doi: 10.1007/s00280-008-0773-z. [DOI] [PubMed] [Google Scholar]
- 117.Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. The Journal of biological chemistry. 2005;280:40749–56. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer research. 2005;65:6305–11. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- 119.Suzuki T, Tanaka R, Hamada S, Nakagawa H, Miyata N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorganic & medicinal chemistry letters. 2010;20:1124–7. doi: 10.1016/j.bmcl.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 120.Gershey EL, Haslett GW, Vidali G, Allfrey VG. Chemical studies of histone methylation. Evidence for the occurrence of 3-methylhistidine in avian erythrocyte histone fractions. The Journal of biological chemistry. 1969;244:4871–7. [PubMed] [Google Scholar]
- 121.Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Archives of biochemistry and biophysics. 1969;148:558–67. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 122.Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome biology. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qian C, Wang X, Manzur K, Sachchidanand, Farooq A, Zeng L, Wang R, Zhou MM. Structural insights of the specificity and catalysis of a viral histone H3 lysine 27 methyltransferase. Journal of molecular biology. 2006;359:86–96. doi: 10.1016/j.jmb.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 124.Xiao B, Wilson JR, Gamblin S. JSET domains and histone methylation. Current opinion in structural biology. 2003;13:699–705. doi: 10.1016/j.sbi.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 125.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–23. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 126.Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell. 2002;111:105–15. doi: 10.1016/s0092-8674(02)00964-9. [DOI] [PubMed] [Google Scholar]
- 127.Taylor WR, Xiao B, Gamblin SJ, Lin K. A knot or not a knot? SETting the record ‘straight’ on proteins. Computational biology and chemistry. 2003;27:11–5. doi: 10.1016/s1476-9271(02)00099-3. [DOI] [PubMed] [Google Scholar]
- 128.Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–6. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 129.Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell. 2002;111:105–15. doi: 10.1016/s0092-8674(02)00964-9. [DOI] [PubMed] [Google Scholar]
- 130.Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–27. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, Wilson JR. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes & development. 2005;19:1444–54. doi: 10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hu P, Zhang Y. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: an ab initio QM/MM-FE study with multiple initial structures. Journal of the American Chemical Society. 2006;128:1272–8. doi: 10.1021/ja056153+. [DOI] [PubMed] [Google Scholar]
- 133.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Molecular cell. 2003;12:177–85. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qian C, Zhou MM. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cellular and molecular life sciences. 2006;63:2755–63. doi: 10.1007/s00018-006-6274-5. CMLS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annual review of biophysics and biomolecular structure. 2005;34:267–94. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kwon T, Chang JH, Kwak E, Lee CW, Joachimiak A, Kim YC, Lee J, Cho Y. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet. The EMBO journal. 2003;22:292–303. doi: 10.1093/emboj/cdg025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu H, Moshkina N, Min J, Zeng H, Joshua J, Zhou MM, Plotnikov AN. Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. PNAS. 2012;109:8925–30. doi: 10.1073/pnas.1114117109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo HB, Guo H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. PNAS. 2007;104:8797–802. doi: 10.1073/pnas.0702981104. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, Selker EU, Shinkai Y, Cheng X. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. JBC. 2005;280:5563–70. doi: 10.1074/jbc.M410483200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochimica et biophysica acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu H, Chen X, Xiong J, Li Y, Li H, Ding X, Liu S, Chen S, Gao S, Zhu B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell research. 2011;21:365–7. doi: 10.1038/cr.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Molecular cell. 2002;10:1107–17. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 143.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer research. 2005;65:11367–74. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 144.Huang H, Zhang J, Shen W, Wang X, Wu J, Shi Y. Solution structure of the second bromodomain of Brd2 and its specific interaction with acetylated histone tails. BMC structural biology. 2007;7:57. doi: 10.1186/1472-6807-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 146.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 147.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–18. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 148.Fang JY, Lu YY. Effects of histone acetylation and DNA methylation on p21( WAF1) regulation. World journal of gastroenterology:WJG. 2002;8:400–5. doi: 10.3748/wjg.v8.i3.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013;32:1351–62. doi: 10.1038/onc.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. PNAS. 2010;107:20980–5. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Molecular cell. 2003;12:177–85. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sanders DS, Muntean AG, Hess JL. Significance of AF4-MLL reciprocal fusion in t(4;11) leukemias? Leukemia research. 2011;35:299–300. doi: 10.1016/j.leukres.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 153.Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–8. doi: 10.1182/blood-2008-12-197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vedel M, Robert-Gero M. Comparative effect of S-adenosyl-homocysteine (SAH) and Sinefungin on tRNA-base methylation in whole cells and in vitro. FEBS letters. 1981;128:87–9. doi: 10.1016/0014-5793(81)81086-1. [DOI] [PubMed] [Google Scholar]
- 155.Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. JBC. 2004;279:23892–9. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 156.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nature chemical biology. 2005;1:143–5. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 157.Vedel M, Lawrence F, Robert-Gero M, Lederer E. The antifungal antibiotic sinefungin as a very active inhibitor of methyltransferases and of the transformation of chick embryo fibroblasts by Rous sarcoma virus. Biochemical and biophysical research communications. 1978;85:371–6. doi: 10.1016/s0006-291x(78)80052-7. [DOI] [PubMed] [Google Scholar]
- 158.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity. Journal of the American Chemical Society. 2010;132:4078–9. doi: 10.1021/ja101280p. 2010. [DOI] [PubMed] [Google Scholar]
- 159.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nature structural & molecular biology. 2009;16:312–7. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. Journal of medicinal chemistry. 2009;52:7950–3. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong A, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin J. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. Journal of medicinal chemistry. 2010;53:5844–57. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He Y, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. Journal of medicinal chemistry. 2011;54:6139–50. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nature chemical biology. 2011;7:566–74. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins) ChemMedChem. 2007;2:1419–31. doi: 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- 165.Ragno R, Simeoni S, Castellano S, Vicidomini C, Mai A, Caroli A, Tramontano A, Bonaccini C, Trojer P, Bauer I, Brosch G, Sbardella G. Small molecule inhibitors of histone arginine methyltransferases: homology modeling, molecular docking, binding mode analysis, and biological evaluations. Journal of medicinal chemistry. 2007;50:1241–53. doi: 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- 166.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–28. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–37. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 169.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes & development. 2002;16:1779–91. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Molecular cell. 2003;12:1577–89. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 171.Sun L, Wang M, Lv Z, Yang N, Liu Y, Bao S, Gong W, Xu RM. Structural insights into protein arginine symmetric dimethylation by PRMT5. PNAS. 2011;108:20538–43. doi: 10.1073/pnas.1106946108. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, Cheung T, Garcia-Arenas R, Cowen S, Wu J, Godin R, Chen H, Keen N. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19:1262–73. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 173.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–60. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 174.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–32. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 175.Saddic LA, West LE, Aslanian A, Yates JR, 3rd, Rubin SM, Gozani O, Sage J. Methylation of the retinoblastoma tumor suppressor by SMYD2. The Journal of biological chemistry. 2010;285:37733–40. doi: 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate lysine 373 in the tumor suppressor p53. The Journal of biological chemistry. 2010;285:9636–41. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 178.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 179.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–83. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 180.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–53. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 181.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. The Journal of biological chemistry. 2009;284:17775–82. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nature structural & molecular biology. 2006;13:626–32. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 183.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) PNAS. 2006;103:13956–61. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–9. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 185.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–5. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 186.Trewick SC, McLaughlin PJ, Allshire RC. Methylation: lost in hydroxylation? EMBO reports. 2005;6:315–20. doi: 10.1038/sj.embor.7400379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Critical reviews in biochemistry and molecular biology. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 188.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nature reviews. Genetics. 2006;7:715–27. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 189.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–11. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 190.Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes & development. 2006;20:1557–62. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R, Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer research. 2006;66:11341–7. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 192.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nature cell biology. 2006;9:347–53. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 193.Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, Buhrer C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochemical and biophysical research communications. 2008;372:892–7. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 194.Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, Ratcliffe PJ. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. The Biochemical journal. 2008;416:387–94. doi: 10.1042/BJ20081238. [DOI] [PubMed] [Google Scholar]
- 195.Yang J, Ledaki I, Turley H, Gatter KC, Montero JC, Li JL, Harris AL. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Annals of the New York Academy of Sciences. 2009;1177:185–97. doi: 10.1111/j.1749-6632.2009.05027.x. [DOI] [PubMed] [Google Scholar]
- 196.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Burger A, Yost WL. Arylcycloalkylamines, 1.2-Phenylcyclopropylamine. Journal of the American Chemical Society. 1948;70(6):2198. [Google Scholar]
- 198.Tedeschi RE, Tedeschi DH, Ames PL, Cook L, Mattis PA, Fellows EJ. Some pharmacological observations on tranylcypromine (SKF trans-385), a potent inhibitor of monoamine oxidase. Proceedings of the Society for Experimental Biology and Medicine. 1959;102:380–1. doi: 10.3181/00379727-102-25256. Society for Experimental Biology and Medicine. [DOI] [PubMed] [Google Scholar]
- 199.West ED, Dally PJ. Effects of iproniazid in depressive syndromes. British medical journal. 1959;1:1491–4. doi: 10.1136/bmj.1.5136.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. Journal of the American Chemical Society. 2010;132:3164–76. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. Journal of the American Chemical Society. 2009;131:17536–7. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 202.Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry. 2010;49:6494–503. doi: 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- 203.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry. 2007;46:6892–902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. PNAS. 2007;104:8023–8. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bi X, Lopez C, Bacchi CJ, Rattendi D, Woster PM. Novel alkylpolyaminoguanidines and alkylpolyaminobiguanides with potent antitrypanosomal activity. Bioorganic & medicinal chemistry letters. 2006;16:3229–32. doi: 10.1016/j.bmcl.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 206.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clinical cancer research. 2009;15:7217–28. doi: 10.1158/1078-0432.CCR-09-1293. Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. The Journal of biological chemistry. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 208.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, Kaelin WG., Jr. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. PNAS. 2002:13459–64. doi: 10.1073/pnas.192342099. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.McDonough MA, McNeill LA, Tilliet M, Papamicael CA, Chen QY, Banerji B, Hewitson KS, Schofield CJ. Selective inhibition of factor inhibiting hypoxia-inducible factor. Journal of the American Chemical Society. 2005;127:7680–1. doi: 10.1021/ja050841b. [DOI] [PubMed] [Google Scholar]
- 210.Rose NR, Ng SS, Mecinovic J, Lienard BM, Bello SH, Sun Z, McDonough MA, Oppermann U, Schofield CJ. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. Journal of medicinal chemistry. 2008;51:7053–6. doi: 10.1021/jm800936s. [DOI] [PubMed] [Google Scholar]
- 211.Rose NR, Woon EC, Kingham GL, King ON, Mecinovic J, Clifton IJ, Ng SS, Talib-Hardy J, Oppermann U, McDonough MA, Schofield CJ. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. Journal of medicinal chemistry. 2010;53:1810–8. doi: 10.1021/jm901680b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tiainen P, Pasanen A, Sormunen R, Myllyharju J. Characterization of recombinant human prolyl 3-hydroxylase isoenzyme 2, an enzyme modifying the basement membrane collagen IV. The Journal of biological chemistry. 2008;283:19432–9. doi: 10.1074/jbc.M802973200. [DOI] [PubMed] [Google Scholar]
- 213.Belvedere S, Witter DJ, Yan J, Secrist JP, Richon V, Miller TA. Aminosuberoyl hydroxamic acids (ASHAs): a potent new class of HDAC inhibitors. Bioorganic & medicinal chemistry letters. 2007;17:3969–71. doi: 10.1016/j.bmcl.2007.04.089. [DOI] [PubMed] [Google Scholar]
- 214.King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PloS one. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorganic & medicinal chemistry letters. 2009;19:2852–5. doi: 10.1016/j.bmcl.2009.03.098. [DOI] [PubMed] [Google Scholar]