

Abstract
Integrins are critical in thrombosis and hemostasis1. Antagonists of the platelet integrin αIIbβ3 are potent anti-thrombotic drugs, but also have the life-threatening adverse effect of bleeding2,3. It is thus desirable to develop new antagonists that do not cause bleeding. Integrins transmit signals bidirectionally4,5. Inside-out signaling activates integrins via a talin-dependent mechanism6,7. Integrin ligation mediates thrombus formation and outside-in signaling8,9, which requires Gα13 and greatly expands thrombi. Here we show that Gα13 and talin bind to mutually exclusive, but distinct sites within the integrin β3 cytoplasmic domain in opposing waves. The first talin binding wave mediates inside-out signaling and also “ligand-induced integrin activation”, but is not required for outside-in signaling. Integrin ligation induces transient talin dissociation and Gα13 binding to an ExE motif, which selectively mediates outside-in signaling and platelet spreading. The second talin binding wave is associated with clot retraction. An ExE motif-based inhibitor of Gα13-integrin interaction selectively abolishes outside-in signaling without affecting integrin ligation, and suppresses occlusive arterial thrombosis without affecting bleeding time. Thus, we have discovered a novel mechanism for the directional switch of integrin signaling and, based on this mechanism, we designed a potent new anti-thrombotic that does not cause bleeding.
Integrin signaling involves the binding of several molecules to the cytoplasmic domain of integrin β subunits including talin6,7, kindlins10,11, c-Src12,13, and Gα138 (Fig. 1a). Co-immunoprecipitation of Gα13 with various β3 C-terminal truncation mutants suggests that Gα13 binding involves the β3 sequence between K729 and T741 (Fig. 1b, E.D. Fig 2a), but not the kindlin-/c-Src-binding sequences (Fig. 1a, b). Alignment of different β cytoplasmic domains reveals an ExE motif in this region, in which the first and third Glu residues are conserved among most β subunits, but not β8 (Fig. 1a). The ExE motif-containing β1, β2 and β3 all bound Gα13, but not β8 (Fig. 1c, E.D. Fig 2f). Wild-type (Wt) and E732A mutant β3 bound to Gα13, but not E731A, E733A, AAA (Fig. 1d, E.D. Fig 2b), DED or QSE mutants (E.D. Fig 2e), indicating that the first and third Glu within the ExE motif are important for Gα13 binding. Synthetic peptides containing the EEERA sequence inhibited Gα13-β3 interaction (see below), verifying this ExE motif-containing Gα13 binding site.
Fig. 1. Mutually exclusive binding of talin and Gα13 to β3.

(a) The sequence of human β3 cytoplasmic domain and its alignment with other β subunits showing conserved ExE motifs and binding sites for talin, kindlins and c-Src. (b) Co-immunoprecipitation of Wt and truncated mutant β3 with Gα13 and talin using anti-β3 or control pre-immune rabbit serum. Immunoprecipitates and CHO cell lysates (10% of that used in immunoprecipitation) were immunoblotted (IB) with indicated antibodies. (c) Binding of purified recombinant Gα13 to glutathione bead-bound glutathione S-transferase (GST), GST-β1 cytoplasmic domain fusion protein (GST-β1CD), GST-β2CD, GST-β3CD, or GST-β8CD. (d) Coimmunoprecipitation of CHO cell-expressed Wt or ExE motif mutated β3 with Gα13 and talin using anti-β3 or pre-immune rabbit serum. (e) Coimmunoprecipitation of CHO cell-expressed integrin αIIbβ3 with Gα13 and THD following transfection with cDNA encoding THD. (f, g) Inhibition of the binding of THD (20 nM) (f) or Gα13 (40 nM) (g) to immobilized GST-β3CD proteins (Wt and negative control mutants) by increasing concentrations of Gα13 (f) or THD (g). Bound Gα13 or THD was detected using anti-Gα13 or anti-talin.
The ExE motif is located in a talin-binding region (Fig. 1a)14,15. Over-expression of the integrin-binding talin head domain (THD) in αIIbβ3-expressing cells inhibited Gα13 co-immunoprecipitation with β3 (Fig. 1e). Purified recombinant THD and Gα13 directly competed for binding to purified GST-β3 cytoplasmic domain fusion protein (GST-β3CD) (Fig. 1f, g), indicating that Gα13 and talin are mutually exclusive in binding to β3. Interestingly, the binding of talin and Gα13 is regulated temporally during integrin signaling (Fig. 2). The first wave of talin association with αIIbβ3 occurred following thrombin-stimulated inside-out signaling (Fig. 2a, b) and before the onset of integrin ligation (as indicated by platelet aggregation (Fig. 2c)). Following integrin ligation, however, talin association with αIIbβ3 was diminished (Fig. 2a, b). The second wave of talin-β3 association occurred after full platelet aggregation (Fig. 2a-c), the timing of which correlates with clot retraction. Opposite to the waves of talin binding, the Gα13-β3 association was even lower than the basal level during inside-out signaling when the first talin binding wave occurred (Fig. 2a, b), but peaked after integrin ligation when the first talin binding wave subsided, and then decreased again during the second talin-binding wave (Fig. 2a, b). Thus, inside-out and various phases of outside-in signaling are associated with coordinated and opposing waves of Gα13 and talin binding to β3.
Fig. 2. Dynamics of talin and Gα13 binding to β3 and the role of talin in integrin signaling.
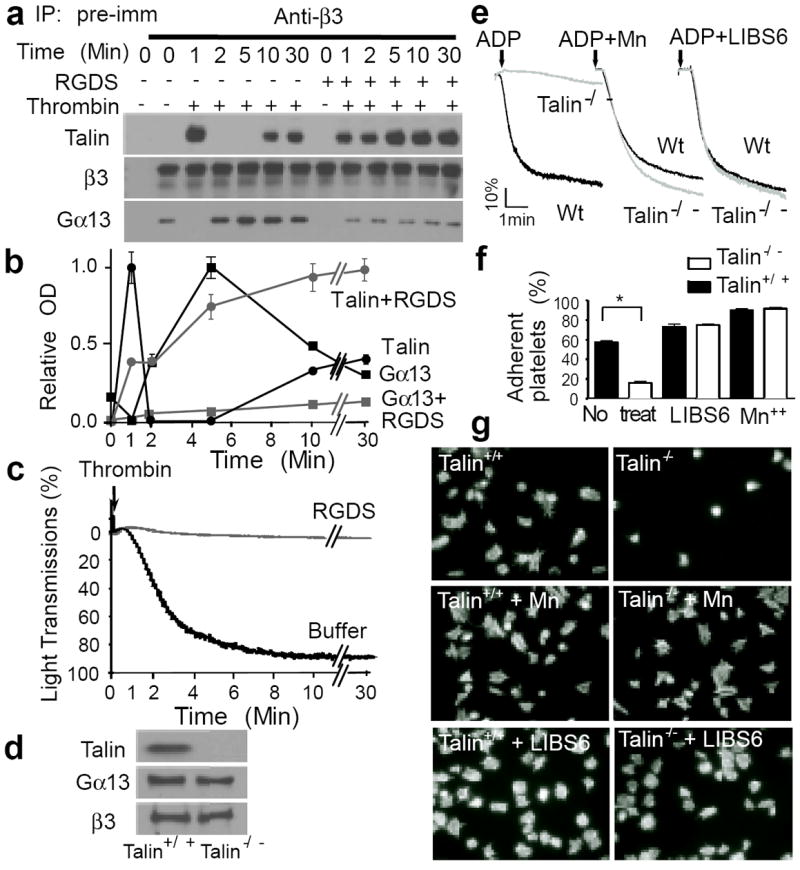
(a, b and c) Human platelets were stimulated with 0.025 U/ml α-thrombin (in an aggregometer) with or without 2 mM integrin inhibitor RGDS, solubilized at various time points, immunoprecipitated with anti-β3 or pre-immune rabbit serum, and immunoblotted for Gα13, talin, and β3 (additional controls in E.D. Fig 3d). (a) Typical immunoblots. (b) Quantification of immunoblots (mean ± SD, 3 experiments). (c) Turbidity changes indicating integrin-dependent platelet aggregation. (d) Immunoblotting of talin-1 in Wt and talin-1-/- mouse platelets. (e) Aggregation of Wt and talin-1-/- platelets stimulated with 5 μM ADP in the presence of 20μg/ml fibrinogen, with or without 1 mM MnCl2 or 0.3μg/ml LIBS6. (f) Adhesion of unstimulated mouse platelets to immobilized fibrinogen for 1 hour, with or without 1 mM MnCl2 or 0.18μg/ml LIBS6 (quantified as percentage of loaded platelets, mean ± SD, n=4, *p<0.001). (g) Images of phalloidin-stained mouse platelets spreading on fibrinogen for 1 hour, with or without 1 mM MnCl2 or 0.18μg/ml LIBS6 (quantification in E.D. Fig 4e).
Importantly, an increase in Gα13 binding to integrin can only be induced when integrin is activated in the presence of fibrinogen, but not by integrin activation alone (E.D. Fig 3a). Conversely, the integrin inhibitors RGDS (Fig. 2a, b) or EDTA (E.D. Fig 3b, c) prevented dissociation of talin from β3 and inhibited Gα13-β3 interaction in thrombin-stimulated platelets. Thus, the switch from a talin-bound to a Gα13-bound state of αIIbβ3 is initiated by the binding of macromolecular ligands.
The opposing waves of talin and Gα13 binding to β3 suggest that the interaction of these two proteins with β3 selectively mediates inside-out and outside-in signaling, respectively. This hypothesis was tested using talin-knockout16 and shRNA-induced talin-knockdown platelets, which are defective in ADP/fibrinogen-induced, integrin-dependent aggregation (Fig. 2d, e; E.D. Fig 4a, c). Their defective aggregation was fully corrected with manganese or an integrin-activating antibody (LIBS6) (Fig. 2e, E.D. Fig 4c), which activate integrins independently of inside-out signaling. These data confirm a role for talin in inside-out signaling6,15,17. It is established that inside-out signaling is not the only pathway of αIIbβ3 activation. Integrin-fibrinogen interaction may occur independently of inside-out signaling when fibrinogen changes conformation, either by immobilization or conversion to fibrin18,19. This is because the initial contact of the exposed ligand recognition sequence, RGD, with resting integrins triggers “ligand-induced integrin activation”20. Interestingly, adhesion of resting talin-knockout or -knockdown platelets to immobilized fibrinogen was defective (Fig. 2f, E.D. Fig 4b), indicating the importance of talin in platelet adhesion to immobilized fibrinogen in the absence of inside-out signaling. However, addition of manganese/integrin-activating antibody fully corrected talin-knockout/knockdown platelet adhesion and spreading (and also the spreading of talin-binding defective mutant β3-expressing CHO cells21) on immobilized fibrinogen (Fig. 2f, g, E.D. Fig 4b, d, e). Thus, the role of talin in resting platelet adhesion to fibrinogen is solely due to its importance in “ligand-induced integrin activation”. Because cell spreading requires the early phase of outside-in signaling, these data further demonstrate that talin is not required for the early phase of outside-in signaling leading to cell spreading once its role in integrin activation is bypassed.
To assess whether Gα13 binding to the ExE motif selectively mediates outside-in signaling without perturbing talin-dependent integrin function, Wt and AAA mutant β3-transfected bone marrow stem cells (from β3-/- mice) were transplanted into irradiated β3-/- mice. The platelets from the recipient mice expressed similar levels of Wt or AAA mutant β3 (Fig. 3a, E.D. Fig 5a). The AAA mutation inhibited β3 interaction with Gα13, but not talin (nor c-Src) (Fig. 3b, E.D. Fig 5b), during integrin signaling. The AAA mutation also had no effect on agonist-induced soluble fibrinogen binding (Fig. 3c). Thus, the ExE motif is not required for talin-dependent inside-out signaling. In contrast, the AAA mutant β3-expressing platelets were defective in spreading on immobilized fibrinogen (Fig. 3d, E.D. Fig 5c, d). Thus, Gα13 binding deficiency in β3 causes a selective defect in integrin outside-in signaling and platelet spreading. Similarly, αIIb/AAA and more conserved αIIb/DED or αIIb/QSE mutants expressed in CHO cells, all defective in Gα13 binding (E.D. Fig 2e), were also defective in spreading on fibrinogen (Fig. 3e, f, E.D. Fig 6a, b, c). However, AAA mutant β3 expressed in CHO cells had no negative effect on THD binding, in contrast to the Y747A mutant (E.D. Fig 6d, e). Additionally, AAA-expressing cells showed defects in integrin-dependent activation of c-Src (as shown by phosphorylation at Tyr416) and transient inhibition of RhoA during cell spreading (Fig. 3g, E.D. Fig 6f), both of which are important elements of outside-in signaling. Together with previous studies that identified β3 sequences mediating talin binding (Fig. 1a)6,15,17,22, our data suggest that talin and Gα13 dynamically interact with distinct recognition sequences in the same region of β3 to serve as a molecular switch controlling the direction of integrin signaling.
Fig. 3. The selective role of Gα13-ExE binding in integrin outside-in signaling.
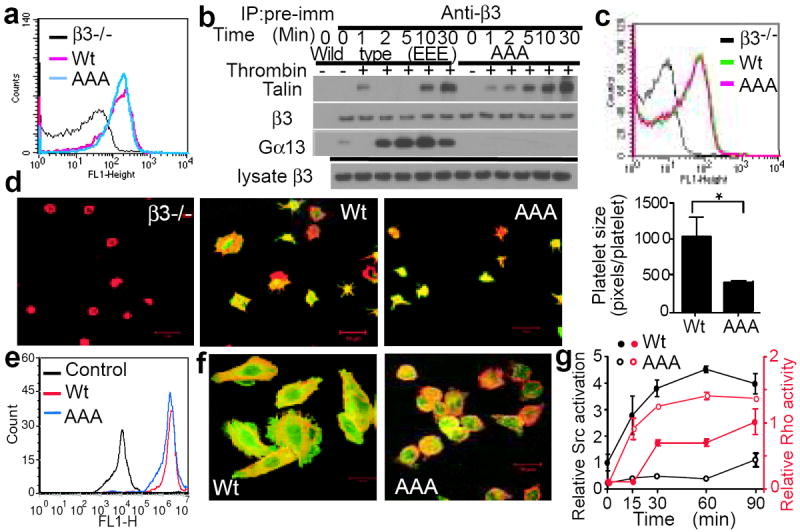
(a) Flow cytometric analysis of β3 expression in platelets from β3-/- mice transplanted with Wt or AAA-mutant β3-transfected bone marrow stem cells. β3-/- platelets served as negative control. (b) Mouse platelets expressing Wt or AAA mutant β3 were stimulated with 0.025 U/ml α-thrombin, solubilized at various time points, immunoprecipitated with anti-β3 or pre-immune rabbit serum, and immunoblotted for Gα13, talin, and β3. (c) PAR4AP-induced binding of Oregon-Green-labelled fibrinogen to Wt or AAA-mutant αIIbβ3-expressing platelets with β3-/- platelets as a negative control. (d) Confocal images of β3-/- platelets and β3-/- platelets expressing Wt or AAA-mutant β3 spreading on fibrinogen and surface area quantification (mean ± SE). Merged anti-β3 (green) and Alexa Fluor 546-conjugated phalloidin (red) fluorescence. (e) Flow cytometric analysis of Wt or β3 E731-733A (AAA) mutant αIIbβ3 expression in CHO-1b9 cells. (f) Confocal images of phalloidin (red)/ anti-β3 (green)-double stained Wt and AAA-mutant αIIbβ3-expressing CHO cell spreading on fibrinogen (45min). (g) RhoA activation and c-Src Tyr416 phosphorylation in Wt or AAA-mutant αIIbβ3-expressing CHO-1b9 cells adherent to fibrinogen (mean ± SD, n=3).
The specific role of the ExE motif in outside-in signaling prompted us to design selective inhibitors of outside-in signaling. We synthesized several myristoylated ExE motif-containing β3 peptides: mP5 (Myr-EEERA), mP6 (Myr-FEEERA) and mP13 (Myr-KFEEERARAKWDT). These peptides inhibited co-immunoprecipitation between Gα13 and β3 (Fig. 4a, E.D. Fig 7a-d), indicating that the minimal sequence of EEERA is sufficient to bind Gα13. In contrast, only mP13, but not mP6 (nor mP5), inhibited talin association with β3 (Fig. 4a), indicating that mP6 does not interact with talin. mP6 inhibited platelet spreading on fibrinogen (Fig. 4b, E.D. Fig 7e), but had no effect on either agonist-induced fibrinogen/PAC1 binding to platelets (Fig. 4c, E.D. Fig 7f, g) or platelet adhesion to immobilized fibrinogen (Fig. 4d). Interestingly, mP6 did not inhibit, but rather accelerated, platelet-dependent clot retraction (Fig. 4e). These data indicate that the ExE-based inhibitor, mP6, selectively inhibits the early phase of outside-in signaling without affecting talin-dependent inside-out signaling, ligand-induced integrin activation, or the late phase of outside-in signaling associated with the second wave of talin binding. In contrast, mP13 inhibited inside-out and outside-in signaling, as it inhibited fibrinogen binding (E.D. Fig 7h), platelet adhesion (Fig. 4d) and clot retraction (Fig. 4e) (not reversed by manganese, as previously shown using talin-/- platelets16). Thus, mP6 selectively interferes with the early phase of outside-in signaling, but mP13 affects all phases of integrin signaling. Importantly, mP6 inhibited the second wave of thrombin-induced platelet aggregation in vitro (Fig. 4f), and when injected into mice as micelles, was as potent as the currently used integrin antagonist Integrilin in inhibiting laser-induced arteriolar thrombosis (Fig. 4g, h, E.D. Fig 8a, b, videos in Supplementary Materials) and FeCl3-induced occlusive carotid artery thrombosis (Fig. 4i, E.D. Fig 8c). More strikingly, at the concentration in which both Integrilin and mP6 similarly inhibited occlusive thrombosis, Integrilin dramatically prolonged tail bleeding and increased blood loss, whereas mP6 had no such adverse effect (Fig. 4j, E.D. Fig 8d). Thus, we have discovered a novel anti-thrombotic that prevents thrombosis without causing bleeding.
Fig. 4. A new anti-thrombotic that does not cause bleeding.
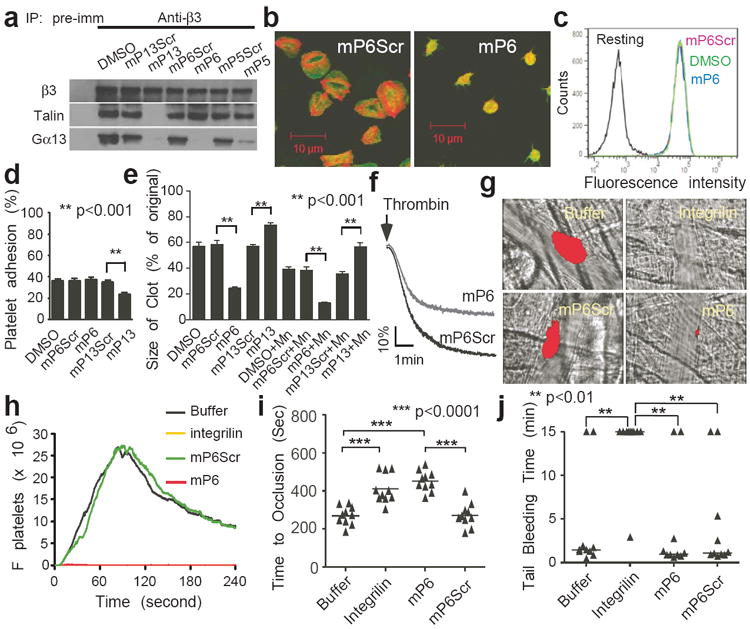
(a) The effects of 500 μM mP13, mP6, or mP5 on co-immunoprecipitation of β3 with Gα13 or talin in thrombin-stimulated platelets in comparison with scrambled controls (also see E.D. Fig 7). (b) Confocal images of phalloidin (red)/anti-β3 (green)-double-stained human platelets treated with 100 μM mP6 or mP6Scr spreading on immobilized fibrinogen (1 hour). (c) PAR4AP-induced Oregon-Green-labelled fibrinogen binding to human platelets pre-treated with DMSO or 100 μM mP6Scr or mP6. (d) Effect of mP6 and mP13 (250 μM) on resting platelet adhesion to immobilized fibrinogen as compared with scrambled peptides (mean ± SD, n=4). (e) Effect of mP6 or mP13 (250 μM) on clot retraction of human platelet-rich plasma, with or without 1 mM manganese (mean ± SD, n=3). (f) Effects of 10 μM mP6 or mP6Scr micelles on platelet aggregation induced by 0.03 U/ml thrombin. (g) Comparison of mP6 micelle (5 μmol/kg) with Integrilin (12 μmol/kg) and their respective controls in inhibiting laser-induced arteriolar thrombosis in mice. Representative images at 60 seconds after injury are shown. Platelet thrombi were indicated by DyLight 649-labelled nonblocking rat anti-mouse GPIbβ (red). (h) Quantification of (g). The median integrated platelet fluorescence (Fplatelets) during thrombosis at 30 injury sites in 3 mice. (i) Comparison of mP6 (5 μmol/kg) with Integrilin (5 μmol/kg) and their respective controls in inhibiting FeCl3-induced carotid artery thrombosis in mice. (j) Comparison of mP6 (5 μmol/kg) with Integrilin (5 μmol/kg) and controls in mouse tail bleeding time analysis.
Together, our study provides a conceptual advance by revealing a molecular switch controlling the directions and consequences of integrin signaling. We show that the switch between inside-out and outside-in signaling is mediated by coordinated but opposing waves of talin and Gα13 binding to distinct yet adjacent sequences within the β3 cytoplasmic domain. The discovery of this signaling switch forms a conceptual basis for selectively inhibiting outside-in signaling without perturbing the ligand binding function of integrins. Importantly, we translated this new concept into a potent novel anti-thrombotic, which, unlike currently available integrin antagonists or other anti-thrombotics, potently inhibits arterial thrombosis without the adverse effect of bleeding (Fig. 4g-j), a potentially life-threatening problem that limits the clinical use of current anti-integrin and anti-thrombotic therapies.
Online only methods
Animals and Reagents
Integrin β3-/- mice were obtained from the Jackson Laboratory. Talin-1(fl/fl), PF4-Cre mice were kindly provided by Dr. Brian Petrich and Dr. David Critchley16. Animal usage and protocol were approved by the institutional animal care committee of the University of Illinois at Chicago. For all animal experiments, mice with similar age, weight, and sex ratio (1:1, except for laser-induced thrombosis) were used for control and specific treatment. The individual mice chosen for specific treatment were decided randomly. Human integrin β3 cDNA was cloned into pcDNA3.1 vector following digestion with Hind III and Xho I, or pLenti6-V5/Dest vector following digestion with EcoR I, Mfe I, and Xho I. Truncation mutants and integrin E to A mutants were either previously reported23 or generated using PCR and cloned into pcDNA3.1 vector by Bam HI and Xho I. The primer sequences used are: (1): ITGB3-UP: 5’-GCGAAGCTTGCCGCCATGGACCGAGCGCGGCCGCGGCCCCGGCCGCTCT-3’; (2): ITGB3-728DN: 5’-GCGCTCGAGTCAAGCGAATTCTTTTCGGTCGTGGATGGTGATGAG-3’; (3): ITGB3-715DN: 5’-GCGCTCGAGTCACCAGATGAGCAGGGCGGCAAGGCCAATGAGCAG-3’; (4): Itgb3-E731A-up: 5’-AAGAATTCGCTAAATTTGCAGAAGAACGCGCCAGAGCAA-3’; (5): Itgb3-E732A-up: 5’-AAGAATTCGCTAAATTTGAGGCAGAACGCGCCAGAGCAA-3’; (6): Itgb3-E733A-up: 5’-AAGAATTCGCTAAATTTGAGGAAGCACGCGCCAGAGCAA-3’; (7): Itgb3-E731-733A-up: 5’-AAGAATTCGCTAAATTTGCAGCAGCACGCGCCAGAGCAA-3’; (8): Mfe-ITGB3-Up: 5’-CCGCAATTGGCCGCCATGGACCGAGCGCGGCCGCGGCCCCGGCCGCTCT-3’; (9): Xho I-ITGB3-DN: 5’- GCGCTCGAGTTAAGTGCCCCGGTACGTGATATTG-3’. Human integrin β8-CD cDNA was cloned into pGEX4T-1 vector following digestion with Bam HI and Xho I. Primer sequences used are: (1): ITGB8-UP: 5’- CGTGGATCC ATTAGACAGGTGATACTACAATGG -3’; (2): ITGB8-Dn: 5’- GCGCTCGAGTTAGA AGTTGCACCTGAAAGTTTC -3’. GST-β3CD and recombinant Gα13 purification was described previously8. Human talin head domain (THD) cDNA, corresponding to N-terminal talin amino acid residues 1-433, was cloned into pcDNA3.1 vector and pMal-C2 vector between EcoR I and Xho I sites. Anti-RhoA antibody was purchased from Cytoskeleton, Inc.; anti-Gα13(sc410), anti-c-Src (sc18), anti-talin (sc7534), and anti-integrin β3 (sc6627) antibodies were from Santa Cruz Biotechnology, Inc.; anti-Gα13(26004) was from NewEast; anti-phospho-Src Y416 antibody was obtained from Cell Signaling; anti-talin (TA205) was from Millipore; anti-talin antibody 8d4 (T3287) was obtained from Sigma; PAC1 antibody (340507) and anti-mouse αIIb antibody MWReg3 (14-0411) were obtained from BD Biosciences; anti-human integrin β3 antibody MAb15, LIBS6 and 8053 rabbit serum were kindly provided by Dr. Mark Ginsberg (University of California, San Diego, La Jolla, CA); Lipofectamine 2000, viraPower lentivirus expression system, Alexa Fluor 546-conjugated phalloidin, Fluor 488-conjugated anti-mouse secondary antibody, talin-1 shRNA plasmids (NM-011602), and non-specific shRNA control vector were from Invitrogen; Y-27632 is from Calbiochem; Fibrinogen from Enzyme Research Laboratories.
Purified Gα13 and THD binding to integrin cytoplasmic domains
GST-tagged integrin cytoplasmic domain proteins were coated onto Pierce Glutathione-coated plates (15140) overnight at 4°C. After washing twice with NP40 buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, 1 mM NaF) with complete protease inhibitor cocktail tablets (1 tablet/5 ml buffer, Roche), purified THD or Gα13 proteins were added onto the plate in NP40 buffer (for Gα13 binding, buffer contained 30 μM AlF4-). Bound THD or Gα13 was estimated with anti-talin or anti-Gα13 antibody, horse radish peroxide (HRP)-conjugated secondary antibody, and 3,3’,5,5’-Tetramethylbenzidine Substrates (Pierce, 34021). The wells were washed three times with NP40 buffer between each of these steps. The reactions were terminated with 1 M sulphuric acid and measured for OD450. For the competitive inhibition assay, increasing concentrations of THD or Gα13 was added to the reactions.
Platelet preparation
Studies using human blood were approved by the institutional review board at the University of Illinois at Chicago, and informed consent was obtained from all donors. Washed human platelets from healthy donors who have not taken medication within 2 weeks prior to donation and platelets from 8-12 week-old mice were prepared as previously described and resuspended in modified Tyrode’s buffer13.
Platelet aggregation assay
Platelet aggregation and secretion were measured in a turbidometric platelet aggregometer (Chronolog) at 37°C with stirring (1000 rpm). Washed platelets (3 × 108/ml) in modified Tyrode’s buffer were stimulated with thrombin (Enzyme Research Laboratories). Aggregation traces shown are representative of at least three independent experiments.
Fibrinogen and PAC1 binding assay
For the fibrinogen binding assay, washed human or mouse platelets resuspended in modified Tyrode’s buffer were incubated with 10 μg/ml Oregon Green-conjugated fibrinogen (Molecular Probes) and PAR4AP as described previously23. The reaction was diluted with PBS and analyzed by flow cytometry using an Accuri C6 flow cytometry (BD). PAC1 binding was measured with FITC-labelled PAC1 antibody (Molecular Probe).
Co-immunoprecipitation
As previously described8, platelets or CHO-1b9 cells expressing recombinant integrin αIIbβ323 were solubilized in NP40 lysis buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, 1 mM NaF), with complete protease inhibitor cocktail tablets (1 tablet/5 ml buffer, Roche). Lysis debris was cleared after centrifugation at 14,000g for 10 min. Lysates were then immunoprecipitated with rabbit anti-Gα13 IgG, anti-integrin β3 rabbit serum or an equal amount of rabbit IgG or pre-immune serum for 2 hours before Protein A/G sepharose beads were added. After incubation of Protein A/G sepharose beads for 45min at 4 °C, beads were centrifuged down and washed for six times with NP40 lysis buffer. Immunoprecipitates were analyzed by immunoblotting.
RhoA activity assay
Platelets or αIIbβ3-expressing CHO cells in modified Tyrode’s buffer or adherent on immobilized fibrinogen were solubilized in cold NP40 lysis buffer at 4°C, and debris-cleared lysates were incubated for 1 hour with purified GST-RBD beads, washed, and then immunoblotted with an anti-RhoA monoclonal antibody, as previously described8.
Bone marrow transplantation
As previously described8, bone marrow stem cells were isolated from femur and tibias of 6-8 week old integrin β3-/-, or C57/BL6 mice using the MACS lineage cell depletion kit (Miltenyi Biotec). Stem cells were subsequently infected twice with concentrated lenti-virus containing shRNA or cDNA constructs, as described in Animals and Reagents section, using a Lenti-X concentrator (Clontech). The cells were then retro-orbitally injected into irradiated recipient mice (5Gy for integrin β3-/- mice and 9.6Gy for C57/BL6 mice, one million cells per recipient mice) one day after irradiation.
Platelet Adhesion Assay
As previously described23, washed platelets were pre-incubated with vehicle or peptides, or with either 1 mM MnCl2 or 0.18 μg/ml LIBS6 prior to plating. After 1 hour incubation at 37°C, adherent platelets were estimated by measuring platelet phosphatase activity with 0.3% p-nitrophenyl phosphate in 1% Triton X-100, 50 mM sodium acetate, pH 5.0 for 1 hour at 37°C. The reaction was stopped with 1 M NaOH. Results were determined by reading OD405. Statistic significance was determined using t-test (n=3).
Cell spreading, immunofluorescence and confocal microscopy
Washed platelets or αIIbβ3-expressing CHO cells suspended in modified Tyrode’s buffer were added to 100 μg/ml fibrinogen (Enzyme Research Laboratories)-coated cover slides and incubated at 37°C for various lengths of time. Cells were fixed, permeabilized, blocked with 0.5% bovine serum albumin in modified Tyrode’s buffer, stained with mAb15 (followed by Fluor 488-conjugated anti-mouse secondary antibody) and/or Alexa Fluor-546 conjugated phalloidin, and viewed with a Zeiss LSM510 META confocal microscope, as previously described8 or with Leica DM IRB fluorescence microscope, Photometrics CoolSNAP HQ camera and μManager software. Cell surface area was measured by NIH ImageJ analysis of 5-10 random images. Statistical significance was determined using t-test.
Clot Retraction Assay
As previously described8, human PRP was incubated with vehicle or peptides for 5 minutes at room temperature prior to stimulation with thrombin. The two-dimensional size of retracted clots was quantified using Image J software, and statistical significance was determined using t-test (n=3).
Peptide inhibitors
Myristoylated peptides were synthesized and purified at the Research Resource Center at the University of Illinois at Chicago. These peptides include: mP13 (Myr-KFEEERARAKWDT), mP5 (Myr-EEERA), mP6 (Myr-FEEERA), and the corresponding control peptides mP13Scr (Myr-EEARERKDWAKFT), mP5Scr (Myr-EEARE), and mP6Scr (Myr-ERAFEE). The peptides were prepared in DMSO for use in vitro, and in micellar formulation for in vivo (and in vitro) use. For micellar formulation, PEG2000-DSPE, L-α-phosphatidylcholine, and peptides were mixed at a molar ratio of 45:5:2. The micelles were suspended to form micelle colloid in HEPES-saline buffer (10 mM HEPES, 150 mM NaCl, pH 7.4), peptide concentration 1 mM) as previously described24. mP6 is similar to mP6Scr in uptake by platelets (E.D. Fig 9a) and does not cause significant changes in hemogram in vivo (E.D. Fig 9b).
Estimation of peptide concentration in platelets
mP6 and mP6Scr peptides were dissolved and conjugated with 1-Pyrenyldiazomethane (PDAM) overnight in the dark in DMSO, or conjugated in methanol and incorporated into the micelle as described above. Platelets were incubated with the PDAM-conjugated peptides for 5 minutes at room temperature, pelleted via centrifugation, washed and lysed with NP40 lysis buffer, and the concentration of PDAM-conjugated peptide was estimated by measuring fluorescence intensity (absorption 340 nm/emission 395 nm) as previously described30. Platelet lysates (without peptide incubation) were used as a blank control. Standard curve was obtained using known concentrations of peptides added to platelet lysates.
In vivo FeCl3-induced thrombosis and tail bleeding time
7-8 week-old C57/BL6 mice were anesthetized by isoflurane inhalation. Retro-orbital injection of peptide micelle or integrilin (5 μmol/kg mouse weight) were performed 15 minutes prior to experimentation. Carotid arterial thrombosis was induced with a filter paper disc (d = 2 mm) soaked with 1.2 μl of 7.5% FeCl3 28. Blood flow was monitored with a TS420 flow meter using a MA-0.5SB dopler probe (Transonic Systems, Ithaca, NY). Data were analyzed using one-way ANOVA. Tail bleeding time analysis were performed as previously described29. Time to stable cessation of bleeding was defined as no evidence of rebleeding for 60 seconds. Bleeding exceeding 15 minutes was immediately stopped by applying pressure. Statistical significance was determined using the Mann-Whitney test. Similar results were also obtained with a nonparametric ANOVA. For bleeding assays measuring total blood loss, cut mouse tails were immersed in Microcentrifuge tubes with 1.5 ml of 0.15 M NaCl at 37 °C for 15 minutes. The hemoglobin concentration in the tube was determined using a HemoCue photometer. Data were analyzed using one-way ANOVA. The experiments were performed in double-blinded fashion.
Intravital Microscopy and Laser-induced thrombosis
Similar to the methods described previously27, Wt male mice (6-8 weeks old) were anesthetized via IP injection of ketamine and xylazine and placed on a thermo-controlled blanket (37°C). The cremaster muscle was exteriorized and superfused with thermo-controlled (37°C) bicarbonate-buffered saline for the duration of experiments. Fluorescence and brightfield images were recorded using an Olympus BX61W microscope with a 60 x/1.0 NA water immersion objective and a high speed camera (Hamamatsu C9300) through an intensifier (Video Scope International). Fluorescence images were captured at 20 frames per second, and data were analyzed using Slidebook v5.5 (Intelligent Imaging Innovations). Arteriolar wall injury was induced with a micropoint laser ablation system (Photonics Instruments). Platelet accumulation was visualized by infusion of Dylight 649-labeled anti-mouse CD42c (Emfret, 0.05 μg/g body weight body weight) into mice. Vehicle control, Integrilin, scrambled peptide, or mP6 were infused 3 minutes prior to laser injury. Laser-induced thrombi were generated at different sites in the blood vessel, with new sites upstream of earlier thrombi. Data were collected for 5 minutes following laser injury. The kinetics of platelet accumulation was analyzed by median fluorescence values of the antibodies as a function of time in approximately 30 thrombi in 3 mice per group. Statistical difference of fluorescence intensity (mean ± SD) at selected time points was also determined using Welch t-test. The experiments were performed in double-blinded fashion.
Statistics
For parametric data, statistical significance was analysed using Student’s t-test (or Welch t-test for samples with nonequal variances) or ANOVA following determination of normal distribution and equal variances. For nonparametric data (bleeding time analysis), Mann-Whitney test was applied. Analyses were performed with GraphPad Prism 4 software. Sample size estimation was performed with Fisher’s exact test using GraphPad InStat 3.
Supplementary Material
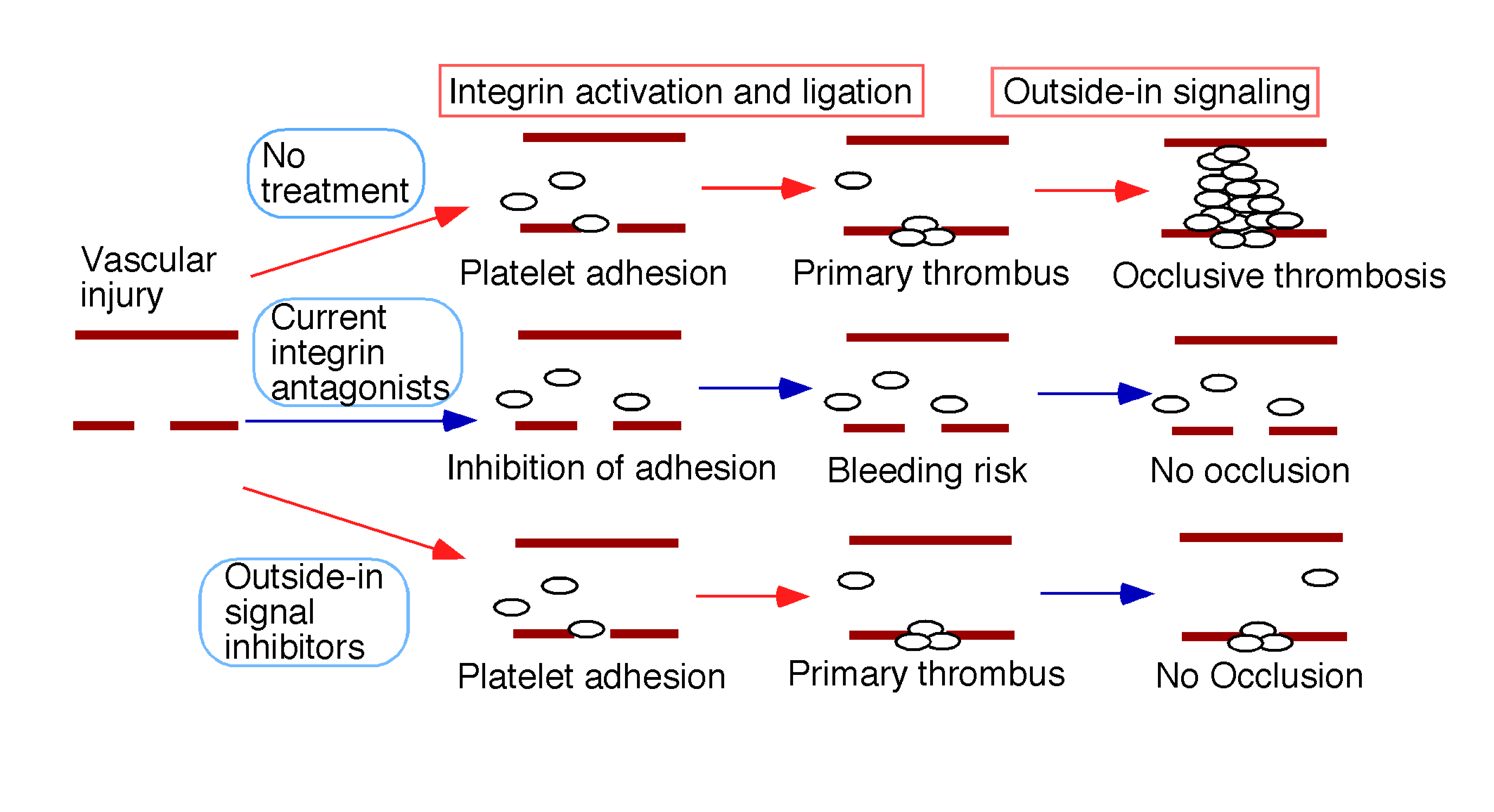
Blue arrows indicate steps that are inhibited.
{kind=link}
(a and b) Lysates from CHO cells expressing similar levels of wild-type (Wt) αIIbβ3, β3 C-terminal truncation mutants (Δ759, Δ741, Δ728, and Δ715) complexed with Wt αIIb (a), and β3 ExE motif mutants (E731A, E732A, E733A and EEE to AAA) complexed with Wt αIIb (b) were immunoprecipitated with anti-Gα13 antibody or equal amount of control rabbit IgG. Immunoprecipitates and lysates (equivalent of 10% used for immunoprecipitation) were immunoblotted with anti-Gα13 and anti-β3 antibodies. (c and d) GST-β3CD (Wt) or GST-β3AAACD (AAA mutant) proteins immobilized in Glutathione-coated microtiter wells were incubated with increasing concentrations of Gα13 (c), or increasing concentrations of talin head domain (THD) (d). After washing, bound Gα13 and THD were respectively detected using anti-Gα13 or anti-talin (mouse IgG was used as a specificity control) followed by secondary HRP-labeled anti-IgG antibody. (e) In addition to the AAA mutation, conserved mutations of EEE to DED and EEE to QSE (as found in β5) were introduced to the β3 cytoplasmic domain. These mutants were co-transfected with Wt αIIb into CHO cells, which were sorted to achieve comparable expression levels with Wt αIIbβ3-expressing cells (as shown in E.D. Fig. 4e). Lysates from these cells were immunoprecipitated with anti-β3 or equal amount of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13 or anti-β3. (f) Lysates from human platelets (with or without stimulation with 0.025 u/ml thrombin) were immunoprecipitated with anti-Gα13 antibody or equal amount of control rabbit IgG. Immunoprecipitates were immunoblotted with anti-Gα13 and anti-β1 antibodies. Gα13 is associated with β1, which is significantly increased following thrombin stimulation.
{kind=link}
(a) To determine the effect of integrin activation and ligand occupancy on Gα13-β3 association, human platelets were incubated with or without 1 mM MnCl2 and 30 μg/ml fibrinogen for 5 minutes at 22°C. Platelet lysates were then immunoprecipitated with anti-β3 or pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-β3 or anti-Gα13. (b, c) Washed human platelets were stimulated with 0.025 U/ml α-thrombin with or without adding 2 mM EDTA (an inhibitor of the ligand binding function of integrins), stirred (1000 rpm) at 37°C, solubilized at various time points, and immunoprecipitated with anti-β3 or equal amounts of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13, anti-talin, or anti-β3 antibodies. (b) Western blot results. (c) Turbidity changes in platelet suspension indicating integrin-dependent platelet aggregation. Note the inhibitory effect of EDTA on talin dissociation and Gα13 binding to β3. (d) As additional controls for Fig. 2a to exclude the possibility of significant loss of talin and β3 in platelet lysates to insoluble fraction during integrin signaling, washed human platelets were stimulated with 0.025 U/ml α-thrombin in the absence or presence of 2 mM integrin inhibitor RGDS, stirred (1000 rpm) at 37°C, and then solubilized at various time points as in Fig. 2a. Solubilized platelets were centrifuged at 14,000g for 10 minutes to separate lysates from insoluble pellets. Pellets were dissolved in SDS sample buffer to the same volume as the lysates after diluting them 1:1 with 2x SDS sample buffer, and both were immunoblotted with anti-β3 and anti-talin antibodies. Note that the levels of talin and β3 in platelet lysates kept essentially constant during the course of platelet aggregation and, with low concentrations of thrombin used to stimulate platelets, very little insoluble β3 and talin were present in the pellet, which were detectable only after prolonged exposure (5 min exposure compared to 10 sec of normal exposure time) and with no obvious variation during the course of platelet aggregation.
{kind=link}
(a) Western blot comparison of talin-1 expression levels in mouse platelets derived from control shRNA- or talin-shRNA-transfected bone marrow stem cells. Western blots of Gα13, and integrin β1 and β3 are also shown. (b) Adhesion of un-stimulated mouse platelets to immobilized fibrinogen for 1 hour. Adherent platelets were quantified as percentage of total platelets loaded (mean ± SD, n=4). (c) Turbidity changes in mouse platelet suspension stimulated with 5 μM ADP in the presence of 20 μg/ml fibrinogen, with or without 1 mM MnCl2, as detected using an aggregometer. (d) Fluorescence microscopy images of phalloidin-stained mouse platelet spreading on fibrinogen for 1 hour, with or without 1 mM MnCl2. (e) Quantification of surface areas of individual adherent platelets as shown in Fig. 2g (mean ± SE).
{kind=link}
(a) Flow cytometric analysis of integrin αIIb/β3 expression levels in β3-/- mouse platelets transfected with Wt or AAA mutant β3 using bone marrow stem cell transplantation technology in comparison with C57BL/6 mouse platelets. β3-/- platelets were used as a negative control. αIIbβ3 complex was detected using an anti-mouse αIIb antibody. (b) Mouse platelets expressing recombinant Wt or AAA mutant β3 as in (a) were lysed and immunoprecipitated with anti-β3 or equal amounts of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Src or anti-β3 antibodies. (c and d) Spreading of phalloidin-stained Wt platelets (EEE), AAA mutant platelets and AAA mutant platelets incubated with mP6Scr or mP6 on immobilized fibrinogen for 1 hour. (c) Typical fluorescence microscopy images. (d) Quantification of surface areas of individual platelets (mean ± SE).
{kind=link}
(a) Expression levels of Wt or the ExE motif (QSE, DED, or AAA) mutants of β3 in complex with Wt αIIb in CHO cells, as determined by flow cytometry. Mouse IgG was used as a negative control. (b, c) Spreading of CHO-1b9 cells expressing Wt αIIbβ3, and QSE, DED, or AAA mutant αIIbβ3 on fibrinogen for 1 hour. (b) Quantification of surface areas of individual cells (mean ± SE). (c) Typical microscopy images. (d) Flow cytometric analysis of Wt αIIbβ3, AAA or Y747A mutant αIIbβ3 expression in CHO cells. Mouse IgG was used as a control. (e) CHO cells expressing Wt, AAA, or Y747A β3 without (upper panels) or with (lower panels) co-expression of recombinant THD were solubilized and immunoprecipitated with anti-β3 or pre-immune serum. 10% lysates and immunoprecipitates were immunoblotted with anti-talin, anti-Gα13, or anti-β3 antibodies. (f) Typical Western blots for Fig. 3g. Wt or AAA-mutant αIIbβ3-expressing CHO-1b9 cells were allowed to adhere to immobilized fibrinogen, solubilized at various time points, and analyzed for RhoA activation and c-Src Tyr416 phosphorylation.
{kind=link}
(a, b, c, and d) Washed human platelets were stimulated with 0.025 U/ml α-thrombin in the absence or presence of 250 μM myristoylated peptides, mP13 (a, b) and mP6 (c, d) with stirring (1000 rpm) at 37°C, and then solubilized at various time points. Lysates were immunoprecipitated with anti-β3 rabbit serum or equal amounts of pre-immune serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13, anti-talin, or anti-β3 antibodies. (a, c) Typical Western blot results. (b, d) Typical turbidity changes in platelet suspension indicating integrin-dependent platelet aggregation. (e) Quantification of human platelet spreading on immobilized fibrinogen for 1 hour, without or with treatment with DMSO, mP6Scr, or mP6 as shown in Fig. 4b (mean surface area ± SE). (f) Flow cytometric analysis of PAR4AP-induced Oregon-Green labeled soluble fibrinogen binding to human platelets pre-treated with 100 μM mP6Scr or 100 μM mP6 stimulated with increasing concentrations of PAR4 agonist peptide (PAR4AP). Integrilin-treated platelets were used as a negative control. (g) Flow cytometric analysis of 100 μM PAR4AP-induced PAC1 binding to human platelets pre-treated with 100 μM mP6Scr or mP6. Integrilin-treated platelets were used as negative control. (h) Flow cytometric analysis of PAR4AP-induced Oregon-Green labeled soluble fibrinogen binding to human platelets pre-treated with solvent DMSO, mP13Scr or mP13. Resting platelets were used as a negative control.
{kind=link}
(a) Representative images of laser-induced mouse cremaster arteriolar thrombosis (red) in the context of the bright-field microvascular histology, visualized by infusion of nonblocking rat anti-mouse GPIbβ antibody conjugated to DyLight 649. The C57BL/6 mice were injected with 5 μmol/kg micellar formulated mP6 or mP6Src (negative control), 12 μmol/kg Integrillin or buffer, 3 minutes before laser-induced arteriolar wall injury. White arrows indicate the directions of the blood flow. (b) The mean platelet fluorescence intensity for 30 thrombi (performed in 3 mice) for each treatment at selected time points (mean ± SE, n=30, t-test). Fluorescence in mP6- and Integrilin-treated mice is minimal. (c) Comparison of mP6 (5 μmol per kg) with the same dose of Integrilin and their respective controls in occlusion time of FeCl3-induced carotid artery thrombosis in mice. Typical arterial blood flow charts of FeCl3-induced occlusive thrombosis are shown. (d) Comparison of mP6 (5 μmol per kg) with the same dose of Integrilin and controls in mouse tail bleeding analysis. Released hemoglobin levels were used as a parameter to assess blood loss (mean ± SD, n=10).
{kind=link}
(a) Estimation of intracellular levels of PDAM-conjugated mP6 and mP6Scr following incubation with platelets for 5 minutes. Platelets were pelleted by centrifugation, and the amounts of PDAM-conjugated peptides in platelet lysates were estimated (mean ± SD, n=3). (b) Hemogram of mouse whole blood before or 1 hour after injection of mP6 or mP6Scr (5 μmol per kg), showing no significant differences.
{kind=link}
Acknowledgments
We thank Drs. Tohru Kozasa, Barry Kreutz and Christina Chow for providing purified recombinant Gα13 protein; Drs. Brian Petrich and David Critchley for providing talin-/- mice. We acknowledge that Haixia Gong performed experiments for this project. This work is supported by grants from National Heart, Lung and Blood Institute (HL080264, HL062350 (X.D.), and HL109439 (J.C)).
Footnotes
Author contributions:
BS performed most experiments, participated in experimental design, data analysis and manuscript writing. XZ, KAO, AS, and MKD each performed a part of experiments and participated in aspects of data analyses and manuscript writing. KK and JC performed laser-induced thrombosis experiments and data analysis, SC-TL provided talin constructs and purified proteins, participated in discussions and data analyses; XD designed and directed the research, analyzed data and wrote the paper.
Competing financial interests
Patent pending. The authors declare no other competing financial interests.
References
- 1.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 2.Coller BS. Anti-GPIIb/IIIa drugs: current strategies and future directions. Thromb Haemost. 2001;86:427–443. [PubMed] [Google Scholar]
- 3.Serebruany VL, Malinin AI, Eisert RM, Sane DC. Risk of bleeding complications with antiplatelet agents: meta-analysis of 338,191 patients enrolled in 50 randomized controlled trials. American journal of hematology. 2004;75:40–47. doi: 10.1002/ajh.10451. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Moissoglu K, Schwartz MA. Integrin signalling in directed cell migration. Biol Cell. 2006;98:547–555. doi: 10.1042/BC20060025. [DOI] [PubMed] [Google Scholar]
- 6.Tadokoro S, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 7.Ye F, Kim C, Ginsberg MH. Molecular mechanism of inside-out integrin regulation. J Thromb Haemost. 2011;9(Suppl 1):20–25. doi: 10.1111/j.1538-7836.2011.04355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong H, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–343. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Current opinion in cell biology. 2012;24:600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 11.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181:439–446. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obergfell A, et al. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157:265–275. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flevaris P, et al. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–565. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patil S, et al. Identification of a talin-binding site in the integrin beta(3) subunit distinct from the NPLY regulatory motif of post-ligand binding functions. The talin n-terminal head domain interacts with the membrane-proximal region of the beta(3) cytoplasmic tail. J Biol Chem. 1999;274:28575–28583. doi: 10.1074/jbc.274.40.28575. [DOI] [PubMed] [Google Scholar]
- 15.Wegener KL, et al. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 16.Haling JR, Monkley SJ, Critchley DR, Petrich BG. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood. 2011;117:1719–1722. doi: 10.1182/blood-2010-09-305433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrich BG, et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. Journal of Experimental Medicine. 2007;204:3103–3111. doi: 10.1084/jem.20071800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coller BS. Interaction of normal, thrombasthenic, and Bernard-Soulier platelets with immobilized fibrinogen: Defective platelet-fibrinogen interaction in thrombasthenia. Blood. 1980;55:169–178. [PubMed] [Google Scholar]
- 19.Ugarova TP, et al. Conformational changes in fibrinogen elicited by its interaction with platelet membrane glycoprotein GPIIb-IIIa. J Biol Chem. 1993;268:21080–21087. [PubMed] [Google Scholar]
- 20.Du X, et al. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 21.Arias-Salgado EG, Lizano S, Shattil SJ, Ginsberg MH. Specification of the direction of adhesive signaling by the integrin beta cytoplasmic domain. J Biol Chem. 2005;280:29699–29707. doi: 10.1074/jbc.M503508200. [DOI] [PubMed] [Google Scholar]
- 22.Goksoy E, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Molecular Cell. 2008;31:124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xi XD, Bodnar RJ, Li ZY, Lam SCT, Du XP. Critical roles for the COOH-terminal NITY and RGT sequences of the integrin beta(3) cytoplasmic domain in inside-out and outside-in signaling. Journal of Cell Biology. 2003;162:329–339. doi: 10.1083/jcb.200303120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnadas A, Rubinstein I, Onyuksel H. Sterically stabilized phospholipid mixed micelles: in vitro evaluation as a novel carrier for water-insoluble drugs. Pharm Res. 2003;20:297–302. doi: 10.1023/a:1022243709003. [DOI] [PubMed] [Google Scholar]
- 25.O’Brien KA, Gartner TK, Hay N, Du X. ADP-stimulated activation of Akt during integrin outside-in signaling promotes platelet spreading by inhibiting glycogen synthase kinase-3beta. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2232–2240. doi: 10.1161/ATVBAHA.112.254680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delaney MK, Liu J, Zheng Y, Berndt MC, Du X. The role of Rac1 in glycoprotein Ib-IX-mediated signal transduction and integrin activation. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2761–2768. doi: 10.1161/ATVBAHA.112.254920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho J, et al. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of beta3 integrins. Blood. 2012;120:647–655. doi: 10.1182/blood-2011-08-372532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118:4215–4223. doi: 10.1182/blood-2010-12-323204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marjanovic JA, Li Z, Stojanovic A, Du X. Stimulatory roles of nitric-oxide synthase 3 and guanylyl cyclase in platelet activation. J Biol Chem. 2005;280:37430–37438. doi: 10.1074/jbc.M506518200. [DOI] [PubMed] [Google Scholar]
- 30.Nimura N, Kinoshita T, Yoshida T, Uetake A, Nakai C. 1-Pyrenyldiazomethane as a fluorescent labeling reagent for liquid chromatographic determination of carboxylic acids. Analytical chemistry. 1988;60:2067–2070. doi: 10.1021/ac00170a017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Blue arrows indicate steps that are inhibited.
(a and b) Lysates from CHO cells expressing similar levels of wild-type (Wt) αIIbβ3, β3 C-terminal truncation mutants (Δ759, Δ741, Δ728, and Δ715) complexed with Wt αIIb (a), and β3 ExE motif mutants (E731A, E732A, E733A and EEE to AAA) complexed with Wt αIIb (b) were immunoprecipitated with anti-Gα13 antibody or equal amount of control rabbit IgG. Immunoprecipitates and lysates (equivalent of 10% used for immunoprecipitation) were immunoblotted with anti-Gα13 and anti-β3 antibodies. (c and d) GST-β3CD (Wt) or GST-β3AAACD (AAA mutant) proteins immobilized in Glutathione-coated microtiter wells were incubated with increasing concentrations of Gα13 (c), or increasing concentrations of talin head domain (THD) (d). After washing, bound Gα13 and THD were respectively detected using anti-Gα13 or anti-talin (mouse IgG was used as a specificity control) followed by secondary HRP-labeled anti-IgG antibody. (e) In addition to the AAA mutation, conserved mutations of EEE to DED and EEE to QSE (as found in β5) were introduced to the β3 cytoplasmic domain. These mutants were co-transfected with Wt αIIb into CHO cells, which were sorted to achieve comparable expression levels with Wt αIIbβ3-expressing cells (as shown in E.D. Fig. 4e). Lysates from these cells were immunoprecipitated with anti-β3 or equal amount of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13 or anti-β3. (f) Lysates from human platelets (with or without stimulation with 0.025 u/ml thrombin) were immunoprecipitated with anti-Gα13 antibody or equal amount of control rabbit IgG. Immunoprecipitates were immunoblotted with anti-Gα13 and anti-β1 antibodies. Gα13 is associated with β1, which is significantly increased following thrombin stimulation.
(a) To determine the effect of integrin activation and ligand occupancy on Gα13-β3 association, human platelets were incubated with or without 1 mM MnCl2 and 30 μg/ml fibrinogen for 5 minutes at 22°C. Platelet lysates were then immunoprecipitated with anti-β3 or pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-β3 or anti-Gα13. (b, c) Washed human platelets were stimulated with 0.025 U/ml α-thrombin with or without adding 2 mM EDTA (an inhibitor of the ligand binding function of integrins), stirred (1000 rpm) at 37°C, solubilized at various time points, and immunoprecipitated with anti-β3 or equal amounts of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13, anti-talin, or anti-β3 antibodies. (b) Western blot results. (c) Turbidity changes in platelet suspension indicating integrin-dependent platelet aggregation. Note the inhibitory effect of EDTA on talin dissociation and Gα13 binding to β3. (d) As additional controls for Fig. 2a to exclude the possibility of significant loss of talin and β3 in platelet lysates to insoluble fraction during integrin signaling, washed human platelets were stimulated with 0.025 U/ml α-thrombin in the absence or presence of 2 mM integrin inhibitor RGDS, stirred (1000 rpm) at 37°C, and then solubilized at various time points as in Fig. 2a. Solubilized platelets were centrifuged at 14,000g for 10 minutes to separate lysates from insoluble pellets. Pellets were dissolved in SDS sample buffer to the same volume as the lysates after diluting them 1:1 with 2x SDS sample buffer, and both were immunoblotted with anti-β3 and anti-talin antibodies. Note that the levels of talin and β3 in platelet lysates kept essentially constant during the course of platelet aggregation and, with low concentrations of thrombin used to stimulate platelets, very little insoluble β3 and talin were present in the pellet, which were detectable only after prolonged exposure (5 min exposure compared to 10 sec of normal exposure time) and with no obvious variation during the course of platelet aggregation.
(a) Western blot comparison of talin-1 expression levels in mouse platelets derived from control shRNA- or talin-shRNA-transfected bone marrow stem cells. Western blots of Gα13, and integrin β1 and β3 are also shown. (b) Adhesion of un-stimulated mouse platelets to immobilized fibrinogen for 1 hour. Adherent platelets were quantified as percentage of total platelets loaded (mean ± SD, n=4). (c) Turbidity changes in mouse platelet suspension stimulated with 5 μM ADP in the presence of 20 μg/ml fibrinogen, with or without 1 mM MnCl2, as detected using an aggregometer. (d) Fluorescence microscopy images of phalloidin-stained mouse platelet spreading on fibrinogen for 1 hour, with or without 1 mM MnCl2. (e) Quantification of surface areas of individual adherent platelets as shown in Fig. 2g (mean ± SE).
(a) Flow cytometric analysis of integrin αIIb/β3 expression levels in β3-/- mouse platelets transfected with Wt or AAA mutant β3 using bone marrow stem cell transplantation technology in comparison with C57BL/6 mouse platelets. β3-/- platelets were used as a negative control. αIIbβ3 complex was detected using an anti-mouse αIIb antibody. (b) Mouse platelets expressing recombinant Wt or AAA mutant β3 as in (a) were lysed and immunoprecipitated with anti-β3 or equal amounts of pre-immune rabbit serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Src or anti-β3 antibodies. (c and d) Spreading of phalloidin-stained Wt platelets (EEE), AAA mutant platelets and AAA mutant platelets incubated with mP6Scr or mP6 on immobilized fibrinogen for 1 hour. (c) Typical fluorescence microscopy images. (d) Quantification of surface areas of individual platelets (mean ± SE).
(a) Expression levels of Wt or the ExE motif (QSE, DED, or AAA) mutants of β3 in complex with Wt αIIb in CHO cells, as determined by flow cytometry. Mouse IgG was used as a negative control. (b, c) Spreading of CHO-1b9 cells expressing Wt αIIbβ3, and QSE, DED, or AAA mutant αIIbβ3 on fibrinogen for 1 hour. (b) Quantification of surface areas of individual cells (mean ± SE). (c) Typical microscopy images. (d) Flow cytometric analysis of Wt αIIbβ3, AAA or Y747A mutant αIIbβ3 expression in CHO cells. Mouse IgG was used as a control. (e) CHO cells expressing Wt, AAA, or Y747A β3 without (upper panels) or with (lower panels) co-expression of recombinant THD were solubilized and immunoprecipitated with anti-β3 or pre-immune serum. 10% lysates and immunoprecipitates were immunoblotted with anti-talin, anti-Gα13, or anti-β3 antibodies. (f) Typical Western blots for Fig. 3g. Wt or AAA-mutant αIIbβ3-expressing CHO-1b9 cells were allowed to adhere to immobilized fibrinogen, solubilized at various time points, and analyzed for RhoA activation and c-Src Tyr416 phosphorylation.
(a, b, c, and d) Washed human platelets were stimulated with 0.025 U/ml α-thrombin in the absence or presence of 250 μM myristoylated peptides, mP13 (a, b) and mP6 (c, d) with stirring (1000 rpm) at 37°C, and then solubilized at various time points. Lysates were immunoprecipitated with anti-β3 rabbit serum or equal amounts of pre-immune serum. Lysates (10%) and immunoprecipitates were immunoblotted with anti-Gα13, anti-talin, or anti-β3 antibodies. (a, c) Typical Western blot results. (b, d) Typical turbidity changes in platelet suspension indicating integrin-dependent platelet aggregation. (e) Quantification of human platelet spreading on immobilized fibrinogen for 1 hour, without or with treatment with DMSO, mP6Scr, or mP6 as shown in Fig. 4b (mean surface area ± SE). (f) Flow cytometric analysis of PAR4AP-induced Oregon-Green labeled soluble fibrinogen binding to human platelets pre-treated with 100 μM mP6Scr or 100 μM mP6 stimulated with increasing concentrations of PAR4 agonist peptide (PAR4AP). Integrilin-treated platelets were used as a negative control. (g) Flow cytometric analysis of 100 μM PAR4AP-induced PAC1 binding to human platelets pre-treated with 100 μM mP6Scr or mP6. Integrilin-treated platelets were used as negative control. (h) Flow cytometric analysis of PAR4AP-induced Oregon-Green labeled soluble fibrinogen binding to human platelets pre-treated with solvent DMSO, mP13Scr or mP13. Resting platelets were used as a negative control.
(a) Representative images of laser-induced mouse cremaster arteriolar thrombosis (red) in the context of the bright-field microvascular histology, visualized by infusion of nonblocking rat anti-mouse GPIbβ antibody conjugated to DyLight 649. The C57BL/6 mice were injected with 5 μmol/kg micellar formulated mP6 or mP6Src (negative control), 12 μmol/kg Integrillin or buffer, 3 minutes before laser-induced arteriolar wall injury. White arrows indicate the directions of the blood flow. (b) The mean platelet fluorescence intensity for 30 thrombi (performed in 3 mice) for each treatment at selected time points (mean ± SE, n=30, t-test). Fluorescence in mP6- and Integrilin-treated mice is minimal. (c) Comparison of mP6 (5 μmol per kg) with the same dose of Integrilin and their respective controls in occlusion time of FeCl3-induced carotid artery thrombosis in mice. Typical arterial blood flow charts of FeCl3-induced occlusive thrombosis are shown. (d) Comparison of mP6 (5 μmol per kg) with the same dose of Integrilin and controls in mouse tail bleeding analysis. Released hemoglobin levels were used as a parameter to assess blood loss (mean ± SD, n=10).
(a) Estimation of intracellular levels of PDAM-conjugated mP6 and mP6Scr following incubation with platelets for 5 minutes. Platelets were pelleted by centrifugation, and the amounts of PDAM-conjugated peptides in platelet lysates were estimated (mean ± SD, n=3). (b) Hemogram of mouse whole blood before or 1 hour after injection of mP6 or mP6Scr (5 μmol per kg), showing no significant differences.