

Abstract
Microvascular injury and increased vascular leakage are prominent features of radiation-induced lung injury (RILI), and often follow cancer-associated thoracic irradiation. Our previous studies demonstrated that polymorphisms in the gene (MIF) encoding macrophage migratory inhibition factor (MIF), a multifunctional pleiotropic cytokine, confer susceptibility to acute inflammatory lung injury and increased vascular permeability, particularly in senescent mice. In this study, we exposed wild-type and genetically engineered mif−/− mice to 20 Gy single-fraction thoracic radiation to investigate the age-related role of MIF in murine RILI (mice were aged 8 wk, 8 mo, or 16 mo). Relative to 8-week-old mice, decreased MIF was observed in bronchoalveolar lavage fluid and lung tissue of 8- to 16-month-old wild-type mice. In addition, radiated 8- to 16-month-old mif−/− mice exhibited significantly decreased bronchoalveolar lavage fluid total antioxidant concentrations with progressive age-related decreases in the nuclear expression of NF-E2–related factor–2 (Nrf2), a transcription factor involved in antioxidant gene up-regulation in response to reactive oxygen species. This was accompanied by decreases in both protein concentrations (NQO1, GCLC, and heme oxygenase–1) and mRNA concentrations (Gpx1, Prdx1, and Txn1) of Nrf2-influenced antioxidant gene targets. In addition, MIF-silenced (short, interfering RNA) human lung endothelial cells failed to express Nrf2 after oxidative (H2O2) challenge, an effect reversed by recombinant MIF administration. However, treatment with an antioxidant (glutathione reduced ester), but not an Nrf2 substrate (N-acetyl cysteine), protected aged mif−/− mice from RILI. These findings implicate an important role for MIF in radiation-induced changes in lung-cell antioxidant concentrations via Nrf2, and suggest that MIF may contribute to age-related susceptibility to thoracic radiation.
Keywords: radiation pneumonitis, lung vascular permeability, macrophage migratory inhibition factor, Nrf2, antioxidant system, aging
Radiation-induced lung injury (RILI) is a dose-dependent adverse consequence of the total body or partial irradiation that is routinely used to treat a variety of malignancies (1). Although RILI develops in 20–30% of patients undergoing radiotherapy for thoracic-associated malignancies (1), factors influencing the likelihood of a patient to develop RILI remain largely unknown. The role of genetic susceptibility factors and single-nucleotide polymorphisms associated with the development of RILI is well documented (2, 3). However, few reliable predictor markers are available, as based on clinical and dosimetric parameters for the development of RILI (2, 3). Improved biomarkers and therapeutic targets are needed to tailor personalized therapeutic strategies successfully that decrease the morbidity and mortality associated with RILI. One potential target involves the gene encoding the macrophage migration inhibition factor (MIF), a protein we previously showed to be associated with susceptibility to acute inflammatory lung injury and increased vascular permeability (4). We previously identified polymorphisms in the MIF gene linked to acute lung injury and sepsis (5), and others have reported the association of MIF with a variety of other lung diseases, including sarcoidosis (6), asthma (7), and pulmonary fibrosis (8).
MIF is a multifunction pleiotropic cytokine with a variety of biological functions, including the regulation of cytokine secretion, the counterregulation of glucocorticoids in inflammation, the inhibition of oxidative stress–induced cell death, the migration and proliferation of fibroblasts in wound-healing and repair (9), and the activation of both the mitogen-activated protein kinase pathway and the Jun-activation domain–binding protein–1 (Jab-1) pathway (10–12). However, the ubiquitous expression of MIF implies that it has biological functions independent of those related to immune responses. Intracellular MIF concentrations are elevated in oxidative stress, which in turn modulates cellular glutathione reduced ester (GSH; an antioxidant) concentrations by altering the cellular GSH/GSSG balance. In this regard, MIF has been documented to protect cells from oxidative stress–induced cell death (13–15). Likewise, genetic variation in the expression of MIF, which is encoded in a functionally polymorphic locus (16, 17), is associated with variable responsiveness of the human heart to ischemia (18, 19).
Oxidative stress is a major factor in the initiation and perpetuation of RILI (20–22). Our previous expression-profiling data from radiated mouse lungs demonstrated significant deregulation of the redox-sensitive transcription factor NF-E2–related factor–2 (Nrf2) in RILI (23), a key protein in orchestrating cellular antioxidant defenses and maintaining redox homeostasis. Nrf2 is activated by the disruption of basal ubiquitin-dependent Nrf2 degradation by the 26S proteasome, leading to nuclear Nrf2 accumulation and gene induction (24). Activators of the Nrf2 pathway restore redox homeostasis by increasing the antioxidant response element (ARE)/electrophilic response element–mediated expression of Phase II antioxidant enzymes. Age-related susceptibility to disease may be attributed to stress-induced vascular reactive oxygen species (ROS) production and impaired antioxidant function. Aged animals not only demonstrate significantly elevated vascular ROS production in response to stress compared with younger animals, but also exhibit decreased Nrf2 concentrations (25–27). We have observed that MIF-deficient aged mice are more susceptible to ventilator-induced lung injury, a form of oxidative stress–induced injury, consistent with a role for MIF in oxidant/antioxidant lung homeostasis (unpublished data). Moreover, reduced endogenous MIF expression is evident in the hearts of aged mice, contributing to increased susceptibility to ischemic injury (28). In addition to its cytokine function, MIF regulates the antioxidant system in animals by binding and activating transcription factors such as Nrf2 and AP-1 (29).
In this study, we hypothesized that MIF plays a key role in regulating RILI by modulating antioxidant defenses in the lungs of senescent mice. We speculated that age-related alterations in MIF expression contribute to impaired Nrf2-mediated antioxidant function and increased RILI susceptibility. To test our hypothesis, we used a previously characterized murine model of RILI (23), and assessed injury severity and MIF expression levels in lung tissues and in bronchoalveolar lavage (BAL) fluid obtained from wild-type (WT) mice of variable ages. To assess the involvement of MIF-regulated, Nrf2-driven antioxidant genes in the development of RILI in aged mif−/− mice, we measured antioxidant concentrations in BAL fluid as well as the expression levels of antioxidant proteins and genes from lung homogenates. Nrf2 protein expression was evaluated in the nuclear extract isolated from radiated lung homogenates of WT and mif−/− mice of increasing age. In addition, we silenced MIF in human pulmonary arterial endothelial cells (ECs) challenged with H2O2 to induce oxidative stress, and then evaluated Nrf2 expression levels relative to MIF expression levels. In separate experiments, MIF-silenced cells subjected to oxidative stress injury were subsequently treated with recombinant MIF (rMIF), and Nrf2 expression levels were assessed. Finally, we treated aged mif−/− mice with the antioxidants GSH and N-acetyl cysteine (NAC) before RILI challenge, and evaluated lung injuries. Our findings implicate an important role for MIF in radiation-induced changes in lung cell antioxidant concentrations, and suggest that MIF may contribute to age-related susceptibility to the untoward effects of thoracic radiation.
Materials and Methods
Animals and Reagents
All experiments and animal care procedures were approved by the Animal Care and Use Committee of the University of Illinois at Chicago. Female C57BL/6 (20–25 g) mice, aged 8–10 weeks, were purchased from Jackson Laboratory (Bar Harbor, ME), and mif−/− mice in the pure C57BL/6 background (30) were inbred according to approved University protocols.
Model of RILI and Antioxidant Administration
Female mif−/− mice, aged 8 weeks, 8 months, and 16 months, as well as age-typed and type-matched control mice (C57BL/6), were subjected to thoracic radiation (20 Gy), as described previously (23). A separate group of mif−/− and WT aged mice (16 mo old) were treated with 100 mg/kg NAC or 15 nM/kg GSH, both purchased from Sigma-Aldrich (St. Louis, MO), via intraperitoneal injection three times a week, beginning 1 hour after irradiation and continuing for 4 weeks after irradiation. Mice were then killed, and lung vascular leakage and inflammation were assessed via BAL fluid protein concentrations and cell counts at 4 to 6 weeks, as previously described (31). Lungs were harvested and stored at −80°C for protein and mRNA analysis.
Lung Histology
To characterize histological alterations, lungs from each experimental group were inflated to 30 cm H2O with 10% formalin for histological evaluation by hematoxylin-and-eosin staining.
Total Antioxidant Status
The total antioxidant capacity in freshly collected BAL fluid was performed using a commercial kit (Cayman’s Antioxidant Assay Kit; Cayman Chemicals, Ann Arbor, MI), using the protocol supplied by the manufacturer.
MIF Quantification in BAL and Plasma
The concentration of MIF in BAL fluid was measured by an ELISA (MIF ELISA Kit; Wuhan EIAab Science Co., Ltd., China), following the instructions from the manufacturer.
Western Blotting of MIF, Nrf2, and Antioxidant Proteins in Lung Homogenate
Lung tissue was homogenized using a tissue homogenizer (Tissue Ruptor; Qiagen, Valencia, CA) with RIPA buffer (Cell Signaling Technology, Inc., MA) containing protease and phosphatase inhibitors. Lysates were used for Western blotting, performed according to standard protocols.
Nuclear Extraction and Nrf2 Expression in Lung
Nuclei were isolated from lung cells using a Nuclear Extraction Kit (Pierce, Rockford, IL), following the manufacturer’s instructions. Nrf2 expression was then analyzed using a Western blotting technique.
Assessment of Gene Expression by Real-Time RT-PCR
Total RNA was extracted from frozen lungs with a combined protocol using TRIzol reagent (Invitrogen, Carlsbad, CA) and an RNeasy kit (Qiagen), and RT-PCR analysis was performed as we have previously described (32). Detailed methods of the RT-PCR analysis are included in the online supplement.
MIF Small, Interfering RNA Transfection and Induction of ROS in ECs
Human pulmonary-artery ECs were grown as previously described (33). Cells were transfected with MIF small, interfering RNA (siRNA), and silencing efficacy was assessed by Western blotting with subsequent experimentation 72 hours after siRNA transfection. For ROS generation, cells were treated with H2O2 (500 nM) for 2 hours. Total protein was extracted, and Western blotting was performed according to previously described protocols (34). Our protocol is detailed in the online supplement.
rMIF-Induced Nrf2 Salvage in MIF-Silenced ECs
MIF siRNA-transfected human pulmonary artery ECs were supplemented with 100 ng/ml rMIF, 72 hours after transfection. For ROS generation, ECs were treated with H2O2, 24 hours after rMIF supplementation, as described previously. Total protein was extracted, and Western blotting was performed according to previously described protocols (34).
Statistical Analysis
Two-way ANOVA was used to compare the means of data from two or more experimental groups. If significant differences were present previously ANOVA (P < 0.05), a least significant differences test was performed post hoc. Subsequently, differences between groups were considered statistically significant when P values were less than 0.05. Results are expressed as means ± SEs.
Results
mif−/− Mice Demonstrate Age-Dependent Increases in RILI Susceptibility
Using a well-characterized preclinical model of RILI (22), female C57BL/6 (WT) and mif−/− mice of three different age groups (8 wk of age, young adults; 8 and 16 mo of age, aged adults) were exposed to a single dose of whole thoracic radiation (20 Gy) to assess age-dependent RILI susceptibility in terms of BAL fluid protein (vascular leakage) and inflammatory cell influx (inflammation). Radiation induced significant increases in the concentrations of protein and total cells in the BAL fluid of WT and mif−/− mice (aged 8 wk) compared with respective nonradiated control mice. Notably, no difference was discernible in the response to RILI between young (8-wk-old) WT and young (8-wk-old) mif−/− mice when assessed 6 weeks after radiation (Figures 1A–1D). However, aged (16-mo-old) WT mice did exhibit significant changes in injury levels compared with young (8-wk-old) WT mice after radiation, with significant increase in BAL cell count, implicating age as a potential contributing factor in RILI susceptibility. In contrast, aged (8 and 16 mo of age) mif−/− mice demonstrated significant RILI susceptibility, proportional to age, characterized by increased lung vascular leakage, inflammation, and percent body weight loss, compared with age-matched, irradiated WT mice (Figures 1E–1H). Moreover, BAL fluid from irradiated aged mif−/− mice demonstrated increased neutrophils compared with age-matched, radiated WT mice (Figure 1G). These observations were confirmed by a histologic evaluation of lung sections, which demonstrated significant age-dependent increases in injury with a marked inflammatory cell influx in radiated mif−/− mice 6 weeks after radiation, compared with age-matched WT radiated control mice (Figure 2).
Figure 1.

Migration inhibition factor knockout (mif−/−) mice demonstrate age-dependent increased susceptibility to radiation-induced lung injury (RILI). mif−/− and wild-type (WT) control (Ctrl) mice, aged 8 weeks, 8 months, and 16 months, received single-dose 20 Gy radiation to the thorax, and bronchoalveolar lavage (BAL) fluid was collected 6 weeks afterward. Compared with control mice, RILI-challenged 8-week-old WT and mif−/− mice exhibited a significant increase in BAL protein and total cell counts. However, no difference was evident between irradiated (IR) WT and mif−/− mice (A and B). Notably, 8-month-old and 16-month-old mif−/− mice demonstrated significant increases in BAL fluid protein and cell counts, compared with irradiated WT mice (C–G, n = 8/group). *P < 0.05, compared with respective uninjured control mice. #P < 0.05, compared with respective irradiated control mice. Notably, macrophages represented the dominant cell type (> 75%) in both groups of aged (16-mo-old) mice, although a significant increase was evident in the percentage of BAL neutrophils (15–20%), and lymphocytes (Lympho) (10%) in aged mif−/− mice after radiation, compared with irradiated, aged WT animals. (H) Weight change during the period of 6 weeks after radiation. Weight gain in both control, nonirradiated groups were comparable, whereas aged mif−/− mice exhibited significant weight loss after radiation, compared with aged WT mice. W, weeks.
Figure 2.

Histological evaluation of irradiated lung tissue from 16-month-old WT and mif−/− mice. Radiation significantly induced increased inflammatory cell influx in hematoxylin-stained lungs from aged mif−/− mice, compared with their irradiated age-matched WT control mice, 6 weeks after radiation.
Reductions in MIF Expression Contribute to Age-Related RILI Susceptibility
These data indicate RILI susceptibility in mif−/− mice, suggestive of a protective regulatory role of MIF in response to ionizing radiation. To link MIF expression with age-dependent susceptibility, we next determined whether MIF expression is down-regulated in senescent mice by examining the expression levels of MIF in 8-week-old and 16-month-old mouse lungs. Western blot analysis of MIF protein expression in the lungs of nonradiated WT mice revealed markedly decreased concentrations of protein in 16-month-old compared with 8-week-old mice (Figure 3A), supporting our hypothesis that an age-associated reduction in MIF expression is an important factor in RILI susceptibility and pathogenesis. However, in radiated WT mice, MIF protein expression was significantly elevated across all age groups, consistent with the protective role of MIF in RILI. We further quantified secreted MIF in BAL fluid in all age groups of WT control and irradiated mice (Figure 3B). Our data indicate an age-dependent decrease in MIF release in mouse lungs, even under control conditions, whereas exposure to ionizing radiation did not change MIF concentrations in the BAL fluid from the 16-month-old group, compared with 16-month-old WT control mice.
Figure 3.

MIF deficiency in senescence contributes to age-related RILI susceptibility. (A) Representative Western blotting and densitometry graph from lung homogenates of RILI-challenged mice (20 Gy) demonstrates decreased MIF expression in aged WT mice under control conditions. Densitometry analysis of the Western blotting confirmed decreased MIF expression in aged lungs. (B) Age-dependent decrease in MIF concentrations in BAL fluid of WT 8-month-old and 16-month-old mice, compared with 8-week-old mice (n = 5/group). *P < 0.05, compared with control mice. #P < 0.05, compared with 8-wk-old mif−/− mice. mos, months; wks, weeks.
Age-Dependent Alterations in Cellular Antioxidant Activity Levels in Lungs of WT and mif−/− Mice
Our previous genome-wide expression profiling study of RILI-challenged lung tissue identified the Nrf2 signaling pathway as one of five canonical pathways significantly dysregulated in RILI (23), with the up-regulated gene expression of both Nrf2 and MIF, suggesting the involvement of MIF in oxidative stress–mediated Nrf2 signaling in the pathogenesis of RILI. To validate the involvement of MIF in oxidant–antioxidant function in the pathogenesis of RILI, we next evaluated total cellular antioxidant activity levels in the BAL fluid of WT and mif−/− mice across all age groups at 6 weeks after radiation, along with control mice. Total antioxidant activity levels followed an age-dependent pattern of decrease in WT and mif−/− mice. However, MIF deletion drastically decreased antioxidant activity in all age groups, with radiation significantly down-regulating antioxidant activity levels in the BAL fluid of WT and mif−/− mice in an age-dependent manner (Figure 4). Moreover, the radiation-induced reduction in antioxidant activity in BAL fluid was significantly attenuated in 8-month-old and 16-month-old mif−/− mice, compared with their radiated WT control mice.
Figure 4.
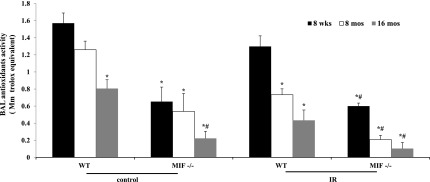
Age-dependent alterations in cellular antioxidant concentrations in WT and mif−/− mice. Nonradiated mif−/− mice demonstrated an age-dependent decrease of antioxidant concentrations in BAL fluid compared with WT age-matched control mice, whereas nonradiated 16-month-old mif−/− mice exhibited significantly reduced antioxidant concentrations in BAL fluid compared with age-matched WT mice. Radiation resulted in significantly reduced concentrations of antioxidants in the BAL fluid of both WT and mif−/− mice after 6 weeks, compared with nonradiated mice (n = 10/group). *P < 0.05, compared with 8-week-old WT uninjured control mice. #P < 0.05, compared with respective 8-week-old mif−/− control mice. **P < 0.05, compared with irradiated 8-week-old WT mice.
Nrf2 Expression Is Regulated by MIF
The transcription factor Nrf2 is activated by increased ROS concentrations that lead to Nrf2 translocation to the nucleus, the binding of AREs, and the transcription of antioxidant defense enzymes. Thus, to determine whether changes in antioxidant activity in mif−/− aged mice are attributable to impaired Nrf2 activation and nuclear translocation, we analyzed Nrf2 nuclear protein expression in the lung homogenates of WT and mif−/− mice at baseline and 6 weeks after exposure to radiation. Figure 5A illustrates an age-related decrease in lung nuclear Nrf2 expression in 16-month-old WT mice and mif−/− mice. MIF deletion contributes to further reductions in Nrf2 after exposure to radiation.
Figure 5.

NF-E2–related factor–2 (Nrf2) regulation by MIF in vitro and in vivo. (A) Representative Western blots and densitometric analyses of Nrf2 expression in the nuclear extracts of WT and mif−/− mouse lung homogenates are shown. mif−/− mice demonstrated significantly reduced Nrf2 expression levels compared with WT control mice, independent of age or irradiation. In addition, 16-month-old mif−/− mice demonstrated significantly reduced Nrf2 expression compared with 8-week-old mif−/− mice, independent of irradiation (n = 4/group). *P < 0.05, compared with respective WT control mice. #P < 0.05, compared with 8-week-old mif−/− control mice. (B and C) MIF-silenced endothelial cells (ECs) (short, interfering RNA; siRNA) were treated with H2O2 for 2 hours, and cell lysates were subjected to Western blotting for Nrf2. H2O2 induced increased Nrf2 expression in ECs, whereas MIF silencing attenuated both basal and H2O2-induced Nrf2 expression. Supplementing MIF-silenced cells with recombinant MIF (rMIF) partly restored Nrf2 protein expression in these cells (n = 4/group). *P < 0.05, compared with untreated control cells. #P < 0.05, compared with respective untransfected control cells. †P < 0.05, rMIF-treated compared with respective MIF-silenced cells.
MIF Silencing Reduces Nuclear Nrf2 Expression in Human Pulmonary Arterial Endothelial Cells
Under conditions of oxidative stress, Nrf2 activators disrupt the basal ubiquitin-dependent degradation of Nrf2 by the 26S proteasome, leading to nuclear Nrf2 accumulation and the induction of antioxidant genes. To evaluate the involvement of MIF in regulating Nrf2 translocation and accumulation in the nucleus, human lung ECs were challenged with H2O2, which resulted in the effective up-regulation of Nrf2 and MIF protein expression at 2 hours after challenge, compared with unchallenged cells. In contrast, MIF-silenced ECs (siRNA) produced a complete attenuation of H2O2-mediated Nrf2 up-regulated expression. However, Nrf2 expression levels were partly restored when rMIF was added to MIF-silenced ECs before challenge with H2O2 (Figures 5B and 5C).
Antioxidants Protect Aged mif−/− Mice from RILI
To confirm the role of MIF in the pathogenesis of RILI via Nrf2-mediated antioxidant regulation, we treated aged (16-mo-old) mif−/− and WT mice with the antioxidants GSH or NAC. Both NAC and GSH protected WT mice from RILI, as evidenced by decreased BAL fluid protein concentrations and total cell counts compared with RILI-challenged control mice. Interestingly, GSH supplementation also attenuated RILI in aged mif−/− mice, but NAC, a precursor of GSH and an Nrf2 substrate, was not protective in these animals (Figures 6A–6C).
Figure 6.

Effect of antioxidant treatment on RILI in mif−/− mice. Irradiated (20 Gy), aged (16-mo-old) mif−/− and WT mice were treated with the antioxidants glutathione ester (GSH, 15 nM/kg) or N-acetyl cysteine (NAC, 100 mg/kg), starting 1 hour after irradiation via intraperitoneal injection, three times per week, for 4 weeks. RILI was attenuated in mif−/− mice pretreated with GSH, although NAC pretreatment was not protective, as assessed by BAL fluid cell counts (A) and total protein concentrations (B), as well as inflammatory changes evident on lung histology (C; n = 5/group). *P < 0.05, compared with respective uninjured control mice. #P < 0.05, compared with respective irradiated control mice.
MIF Regulated Nrf2-Dependent Antioxidant Genes and Proteins in Aged Mouse Lungs
To determine whether age-related and MIF-regulated Nrf2 dysregulation impairs the ability of lungs to mount an effective antioxidant response to oxidative stress, aged (16-mo-old) WT as well as mif−/− mice were analyzed for the expression of Nrf2-induced antioxidant proteins, such as NQO1, HO-1, and GCLC (Figures 7A and 7B). Aged mif−/− mice lungs displayed significantly reduced antioxidant protein expression at baseline, compared with respective age-matched WT mouse lungs, confirming the interaction between MIF and the Nrf2 antioxidant system. Similarly, quantitative real-time RT-PCR was used to analyze the mRNA expression of additional known Nrf2 targets in aged mouse lungs, and revealed a significantly increased lung gene expression of Gpxn1, Prdx1, and Trxn1 (Figures 7C–7E) in irradiated WT mice compared with WT control mice. In contrast, aged mif−/− mice demonstrated a decreased mRNA expression of these genes, confirming the dysregulation of antioxidant defense genes in mice expressing MIF.
Figure 7.

Validation of MIF influence on Nrf2-regulated antioxidant gene expression. (A and B) Western blots and densitometric analyses are shown of Nrf2-mediated antioxidant protein (NQO1, HO-1, and GCLC) expression levels in lungs of aged (16-mo-old) WT mice and mif−/− mice (representative blots, n = 3/group). *P < 0.05, compared with WT control mice. (C–E) Quantitative real time RT-PCR was used to measure mRNA expression of Gpxn1, Prdx1, and Txn1 in aged (16-mo-old) WT and mif−/− mice at baseline and after irradiation (n = 3/group). *P < 0.05, compared with respective uninjured control mice. #P < 0.05, mif−/− compared with WT mice.
Discussion
RILI is a dose-limiting and potentially fatal toxicity of thoracic radiotherapy. Although research efforts have identified several plausible mechanisms responsible for the pathogenesis of RILI, few viable therapeutic options exist, and none have proven to be consistently effective. Aging is one of the major factors associated with RILI susceptibility, and an elucidation of the mechanisms associated with the age-related deterioration of protective responses to RILI may serve as potential preventive and therapeutic options. In the present study, we demonstrated for the first time that MIF, a cytokine which we previously identified as important in acute inflammatory lung injury (4), is markedly reduced in aged mouse lungs, and that the differential expression of MIF in lung tissue modulates RILI via Nrf2 activation and regulation of the antioxidant pathway.
Our data support the conclusion that in aged, wild-type mice, MIF expression is significantly reduced as measured in lung tissue, BAL fluid, and plasma. This decreased MIF expression in aged mice lungs is associated with dysregulated Nrf2 expression and the resultant antioxidant activities and production, as confirmed in mif−/− mice. However, we presume that radiation-induced cellular oxidative stress up-regulates MIF expression in lungs as a compensatory mechanism to protect lungs from RILI. Despite the presence of significant oxidative stress from radiation, mif−/− mice failed to demonstrate Nrf2 activation, validating the hypothesis that RILI susceptibility in aged mif−/− mice is attributable in part to decreased Nrf2 activation and an associated decrease in Nrf2-mediated gene expression. Moreover, the treatment of aged mif−/− mice with NAC, an antioxidant and a substrate for Nrf2, did not affect RILI susceptibility, whereas treatment with GSH, another antioxidant, was protective against RILI. These data strongly implicate a key role for MIF in age-related susceptibility to RILI.
MIF is a multifunction pleiotropic cytokine with variety of biological functions, and is a contributor to the symptoms of sepsis-induced acute lung injury, inflammatory bowel disease, asthma, fibrosis, and other diseases (35). MIF inhibition offers a potential therapeutic strategy in treating inflammatory disease. However, in our experiments, MIF deletion not only failed to protect mice from RILI, but dramatically exacerbated injury while pointing to altered MIF function unique to oxidative stress management in senescence. Because disease severity involves a complex interplay between environmental and genetic factors, the polymorphisms in MIF that regulate MIF expression may be relevant to the susceptibility of specific populations, including the elderly, in ROS-intensive disorders such as RILI and ischemic heart injury (18).
Epithelial and endothelial injury and barrier dysfunction may be precipitated by increased levels of the oxidative and nitrosative stress induced by direct ionizing radiation (36, 37). Nrf2 is a cellular sensor of radiation-induced oxidative stress (38). Consistent with the increased ROS and RNS observed in our preclinical model, the Nrf2 pathways were prominently deregulated in microarray analyses of lungs from RILI-challenged mice (23). Stress-activated transcription factors, such as Nrf2, play an important role in regulating the aging process by orchestrating the transcriptional response of cells to oxidative stress (25, 39). Previous studies suggest that the genetic depletion of Nrf2 also affects the aging process in mice, increasing age-related cancer morbidity and abrogating the anticancer effects of caloric restriction (40). Recent studies have demonstrated that Nrf2-driven pathways are functional in endothelial cells and confer important antioxidative, anti-inflammatory, and antiapoptotic effects (22, 26, 41, 42)
In 8-week-old mice, activated Nrf2 translocates to the nucleus in response to ROS, where binding to ARE occurs with the activation of transcription of Phase II and antioxidant defense enzymes, including NQO1 (a key plasma membrane redox component), HO-1, and γ-glutamylcysteine synthetase (the rate-limiting enzyme for glutathione synthesis). Aged animals demonstrate significantly increased vascular ROS production upon stress compared with younger animals, contributing to the majority of age-related increases in morbidity and mortality. Although the functioning of the antioxidant system is vital in attenuating increased vascular oxidative stress, accumulating evidence demonstrates an age-related decline in cellular glutathione (GSH) concentrations, attributable to diminished Nrf2 activation (43) and an impaired (Nrf2)–driven antioxidant defense mechanism (25–27). Conversely, endogenous MIF expression is reduced in aged hearts, and this cardiac MIF down-regulation results in an impairment of ischemic AMPK activation, leading to intolerance of the aged heart to ischemic injury (28). Our Western blotting analysis of nuclear extracts from the lungs of aged mice supports the translocation of impaired Nrf2 to the nucleus, especially in aged mif−/− mice compared with their WT control mice. Our findings suggest that aging is associated with a down-regulation of MIF expression and vascular Nrf2 expression at the protein level, contributing to the decline in antioxidant concentrations detected in BAL fluid from aged mice. Previous studies indicate that age-related oxidative stress promotes vascular inflammation in aged animals by activating the redox-sensitive transcription factor NF-κB (44). Our present study provides evidence that the loss of MIF during aging impairs the ability to mount an effective Nrf2-dependent antioxidant defense in response to lung oxidative stress in vivo. As a result, radiation significantly induces more oxidative stress in the lungs of aged mif−/− mice than in those of WT or 8-week-old knockout mice. MIF treatment rescued cardiomyocytes from nitric oxide–induced cell death while inducing ARE-regulated glutathione S-transferase Ya subunit and HO-1 gene expression in vitro (45), suggesting that MIF, apart from other pleotropic functions, acts as a regulator of the Nrf2-mediated antioxidant system. Consistent with this notion, the screening of cardioprotective drugs revealed that BTZO-1, an activator of the ARE, bound selectively to MIF, and that a reduction in cellular MIF concentrations suppressed the BITZO-1–mediated antioxidant protection of cardiomyocytes (46).
Our data suggest that the MIF regulation of Nrf2 occurs via direct binding to Nrf2 or via a binding partner, which together would activate Nrf2-mediated antioxidant responses under conditions of oxidative stress (45). MIF interacts with the JAB1/CSN5 signalosome and regulates cellular activities (29), and CSN5 in turn has been implicated in the functions of MIF (29). CSN5 is required for the down-regulation of genes regulated by Nrf2, in which context it is associated with the neddylation of Cul3, which is essential for the Cul3/Keap1-mediated degradation of Nrf2. CSN5 functions to maintain the stability of Cul3/Keap1, which serves as an E3 complex to degrade NRF2 (47, 48). Consistent with this, CSN5-deficient mice demonstrate an increased expression of antioxidant genes (HO-1) and lower mortality. Moreover, MIF also binds to JAB1/CSN5 intracellularly, implicating this as a potential mechanism of Nrf2 activation via the inhibition of CSN5.
In conclusion, our data implicate reduced macrophage migratory inhibition factor gene (MIF) and protein expression in conferring the susceptibility of aging mice to radiation lung injury. The mechanism of the involvement of this multifunctional pleiotropic cytokine and oxidoreductase appears to involve the dysregulation of Nrf2 and antioxidant activities and concentrations. These findings suggest that reduced MIF may contribute to age-related susceptibility to inflammatory disorders, and may offer a potential therapeutic strategy to reduce the untoward effects of thoracic radiation.
Acknowledgments
Acknowledgments
The authors are grateful to Ms. Lakshmi Natarajan for excellent technical assistance.
Footnotes
This study was supported by National Institutes of Health grants AI 42310 and P01 HL098050 (to V.N., J.G.N.G., S.M.D., J.R.J., and R.R.W.) and HL 58094 (to J.G.N.G.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0291OC on March 22, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Movsas B, Raffin TA, Epstein AH, Link CJ., Jr Pulmonary radiation injury. Chest. 1997;111:1061–1076. doi: 10.1378/chest.111.4.1061. [DOI] [PubMed] [Google Scholar]
- 2.Tucker SL, Zhang M, Dong L, Mohan R, Kuban D, Thames HD. Cluster model analysis of late rectal bleeding after IMRT of prostate cancer: a case-control study. Int J Radiat Oncol Biol Phys. 2006;64:1255–1264. doi: 10.1016/j.ijrobp.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Robnett TJ, Machtay M, Vines EF, McKenna MG, Algazy KM, McKenna WG. Factors predicting severe radiation pneumonitis in patients receiving definitive chemoradiation for lung cancer. Int J Radiat Oncol Biol Phys. 2000;48:89–94. doi: 10.1016/s0360-3016(00)00648-9. [DOI] [PubMed] [Google Scholar]
- 4.Wadgaonkar R, Dudek SM, Zaiman AL, Linz-McGillem L, Verin AD, Nurmukhambetova S, Romer LH, Garcia JG. Intracellular interaction of myosin light chain kinase with macrophage migration inhibition factor (MIF) in endothelium. J Cell Biochem. 2005;95:849–858. doi: 10.1002/jcb.20472. [DOI] [PubMed] [Google Scholar]
- 5.Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, Finegold D, Carter M, Leng L, Peng ZY, et al. The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. Faseb J. 2009;23:2403–2411. doi: 10.1096/fj.09-129445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amoli MM, Donn RP, Thomson W, Hajeer AH, Garcia-Porrua C, Lueiro M, Ollier WE, Gonzalez-Gay MA. Macrophage migration inhibitory factor gene polymorphism is associated with sarcoidosis in biopsy proven erythema nodosum. J Rheumatol. 2002;29:1671–1673. [PubMed] [Google Scholar]
- 7.Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, Lane SJ, Craft J, Nishihira J, Donnelly SC, et al. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci USA. 2005;102:14410–14415. doi: 10.1073/pnas.0507189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bargagli E, Prasse A, Olivieri C, Muller-Quernheim J, Rottoli P. Macrophage-derived biomarkers of idiopathic pulmonary fibrosis. Pulm Med. 2011;2011:717130. doi: 10.1155/2011/717130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dewor M, Steffens G, Krohn R, Weber C, Baron J, Bernhagen J. Macrophage migration inhibitory factor (MIF) promotes fibroblast migration in scratch-wounded monolayers in vitro. FEBS Lett. 2007;581:4734–4742. doi: 10.1016/j.febslet.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 10.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lolis E, Bucala R. Macrophage migration inhibitory factor. Expert Opin Ther Targets. 2003;7:153–164. doi: 10.1517/14728222.7.2.153. [DOI] [PubMed] [Google Scholar]
- 12.Donn RP, Ray DW. Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. J Endocrinol. 2004;182:1–9. doi: 10.1677/joe.0.1820001. [DOI] [PubMed] [Google Scholar]
- 13.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen MT, Lue H, Kleemann R, Thiele M, Tolle G, Finkelmeier D, Wagner E, Braun A, Bernhagen J. The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress–induced apoptosis. J Immunol. 2003;170:3337–3347. doi: 10.4049/jimmunol.170.6.3337. [DOI] [PubMed] [Google Scholar]
- 16.Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem. 1991;266:8162–8170. [PubMed] [Google Scholar]
- 17.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R. A functional promoter polymorphism in the macrophage migration inhibitory factor (mif) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3:170–176. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 18.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates amp-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 19.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, et al. Impaired macrophage migration inhibitory factor-amp-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–292. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 21.Eroglu C, Yildiz OG, Saraymen R, Soyuer S, Kilic E, Ozcan S. Aminoguanidine ameliorates radiation-induced oxidative lung damage in rats. Clin Invest Med. 2008;31:E182–E188. doi: 10.25011/cim.v31i4.4778. [DOI] [PubMed] [Google Scholar]
- 22.Fledderus JO, Boon RA, Volger OL, Hurttila H, Yla-Herttuala S, Pannekoek H, Levonen AL, Horrevoets AJ. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1339–1346. doi: 10.1161/ATVBAHA.108.165811. [DOI] [PubMed] [Google Scholar]
- 23.Mathew B, Huang Y, Jacobson JR, Berdyshev E, Gerhold LM, Wang T, Moreno-Vinasco L, Lang G, Zhao Y, Chen CT, et al. Simvastatin attenuates radiation-induced murine lung injury and dysregulated lung gene expression. Am J Respir Cell Mol Biol. 2010;44:415–422. doi: 10.1165/rcmb.2010-0122OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapple SJ, Siow RC, Mann GE. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol. 2012;44:1315–1320. doi: 10.1016/j.biocel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 25.Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, Ballabh P, de Cabo R, Sonntag WE, Csiszar A. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of Nrf2-mediated antioxidant response. Am J Physiol. 2011;301:H363–H372. doi: 10.1152/ajpheart.01134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zakkar M, Van der Heiden K, Luong Le A, Chaudhury H, Cuhlmann S, Hamdulay SS, Krams R, Edirisinghe I, Rahman I, Carlsen H, et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. 2009;29:1851–1857. doi: 10.1161/ATVBAHA.109.193375. [DOI] [PubMed] [Google Scholar]
- 27.Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2–related factor–2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes. 2008;57:2809–2817. doi: 10.2337/db06-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, et al. Impaired macrophage migration inhibitory factor–AMP–activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–292. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, et al. Intracellular action of the cytokine mif to modulate ap-1 activity and the cell cycle through jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 30.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Muller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nonas SA, Moreno-Vinasco L, Ma SF, Jacobson JR, Desai AA, Dudek SM, Flores C, Hassoun PM, Sam L, Ye SQ, et al. Use of consomic rats for genomic insights into ventilator-associated lung injury. Am J Physiol. 2007;293:L292–L302. doi: 10.1152/ajplung.00481.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jayaraman S, Patel A, Jayaraman A, Holterman M, Prabhakar B. Transcriptome analysis of epigenetically modulated genome indicates signature genes in manifestation of Type 1 diabetes and its prevention in nod mice. PLoS ONE. 2013;8:e55074. doi: 10.1371/journal.pone.0055074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, et al. ABL tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21:4042–4056. doi: 10.1091/mbc.E09-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitra S, Sammani S, Wang T, Boone DL, Meyer NJ, Dudek SM, Moreno-Vinasco L, Garcia JG, Jacobson JR. Role of growth arrest and DNA damage–inducible alpha in AKT phosphorylation and ubiquitination after mechanical stress–induced vascular injury. Am J Respir Crit Care Med. 2011;184:1030–1040. doi: 10.1164/rccm.201103-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao L, Flores C, Fan-Ma S, Miller EJ, Moitra J, Moreno L, Wadgaonkar R, Simon B, Brower R, Sevransky J, et al. Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl Res. 2007;150:18–29. doi: 10.1016/j.trsl.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hallahan DE, Virudachalam S, Kuchibhotla J. Nuclear factor kappaB dominant negative genetic constructs inhibit X-ray induction of cell adhesion molecules in the vascular endothelium. Cancer Res. 1998;58:5484–5488. [PubMed] [Google Scholar]
- 37.Giaid A, Lehnert SM, Chehayeb B, Chehayeb D, Kaplan I, Shenouda G. Inducible nitric oxide synthase and nitrotyrosine in mice with radiation-induced lung damage. Am J Clin Oncol. 2003;26:e67–e72. doi: 10.1097/01.COC.0000077940.05196.86. [DOI] [PubMed] [Google Scholar]
- 38.Lee OH, Jain AK, Papusha V, Jaiswal AK. An auto-regulatory loop between stress sensors Inrf2 and Nrf2 controls their cellular abundance. J Biol Chem. 2007;282:36412–36420. doi: 10.1074/jbc.M706517200. [DOI] [PubMed] [Google Scholar]
- 39.Lewis KN, Mele J, Hayes JD, Buffenstein R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr Comp Biol. 2010;50:829–843. doi: 10.1093/icb/icq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, Cascajo MV, Tamashiro KL, Poosala S, Csiszar A, Ungvari Z, et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci USA. 2008;105:2325–2330. doi: 10.1073/pnas.0712162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C. Laminar flow induction of antioxidant response element–mediated genes in endothelial cells: a novel anti-inflammatory mechanism. J Biol Chem. 2003;278:703–711. doi: 10.1074/jbc.M203161200. [DOI] [PubMed] [Google Scholar]
- 42.Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, et al. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008;103:e1–e9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 43.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol. 2007;293:H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 45.Kimura H, Sato Y, Tajima Y, Suzuki H, Yukitake H, Imaeda T, Kajino M, Oki H, Takizawa M, Tanida S. BTZO-1, a cardioprotective agent, reveals that macrophage migration inhibitory factor regulates ARE-mediated gene expression. Chem Biol. 2010;17:1282–1294. doi: 10.1016/j.chembiol.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 46.Yukitake H, Kimura H, Suzuki H, Tajima Y, Sato Y, Imaeda T, Kajino M, Takizawa M. BTZO-15, an ARE-activator, ameliorates DSS- and TNBS-induced colitis in rats. PLoS ONE. 2011;6:e23256. doi: 10.1371/journal.pone.0023256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. 2008;118:239–247. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seldon MP, Silva G, Pejanovic N, Larsen R, Gregoire IP, Filipe J, Anrather J, Soares MP. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of nf-kappab rela phosphorylation at serine 276. J Immunol. 2007;179:7840–7851. doi: 10.4049/jimmunol.179.11.7840. [DOI] [PubMed] [Google Scholar]