

Abstract
Acute and sustained hypoxic pulmonary vasoconstriction (HPV), as well as chronic pulmonary hypertension (PH), is modulated by nitric oxide (NO). NO synthesis can be decreased by asymmetric dimethylarginine (ADMA), which is degraded by dimethylarginine dimethylaminohydrolase–1 (DDAH1). We investigated the effects of DDAH1 overexpression (DDAH1tg) on HPV and chronic hypoxia–induced PH. HPV was measured during acute (10 min) and sustained (3 h) hypoxia in isolated mouse lungs. Chronic PH was induced by the exposure of mice to 4 weeks of hypoxia. ADMA and cyclic 3′,5′-guanosine monophosphate (cGMP) were determined by ELISA, and NO generation was determined by chemiluminescence. DDAH1 overexpression exerted no effects on acute HPV. However, DDAH1tg mice showed decreased sustained HPV compared with wild-type (WT) mice. Concomitantly, ADMA was decreased, and concentrations of NO and cGMP were significantly increased in DDAH1tg. The administration of either Nω-nitro-l-arginine or 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one potentiated sustained HPV and partly abolished the differences in sustained HPV between WT and DDAH1tg mice. The overexpression of DDAH1 exerted no effect on the development of chronic hypoxia–induced PH. DDAH1 overexpression selectively decreased the sustained phase of HPV, partly via activation of the NO–cGMP pathway. Thus, increased ADMA concentrations modulate sustained HPV, but not acute HPV or chronic hypoxia–induced PH.
Keywords: asymmetric dimethylarginine, cyclic guanosine monophosphate, hypoxia-induced pulmonary hypertension, hypoxic pulmonary vasoconstriction, nitric oxide
Exposure of the lung vasculature to hypoxia induces vasoconstriction of the precapillary vessels (hypoxic pulmonary vasoconstriction [HPV]) and, if chronic, vascular remodeling develops, fixing pulmonary hypertension (PH). HPV can be divided into two phases inducible by alveolar hypoxia, lasting seconds to minutes (acute HPV), or minutes to hours (sustained HPV). Such a biphasic response of the lung vasculature to hypoxia has been reported for isolated pulmonary arteries, as well as for isolated lungs (1, 2). Both phases of HPV play an important role in matching local perfusion to ventilation, thus optimizing pulmonary gas exchange. Acute HPV is finally triggered by an intracellular calcium increase, whereas sustained HPV additionally depends on calcium-sensitizing factors released by the endothelium, resulting in the search for an “endothelium-derived vasoconstrictive factor” (3, 4). In contrast to acute HPV, the increase of pulmonary arterial pressure (PAP) during sustained HPV is not spontaneously reversible upon reexposure to normoxia, and has been suggested to facilitate the development of PH. PH is characterized by vascular remodeling and vasoconstriction, resulting in an increased workload for the right heart. Vascular alterations in PH may also be triggered by endothelial dysfunction, resulting in smooth muscle cell proliferation, platelet aggregation, and fibroblast proliferation.
Nitric oxide (NO) is one of the major endothelium-derived vasoactive mediators controlling a diverse range of pulmonary functions, such as the regulation of pulmonary vascular resistance (2). It is synthesized from l-arginine and oxygen by a family of three NO synthases (NOSs), all of which are expressed in the lung. Endothelial NOS appears to play a predominant role in the regulation of pulmonary vascular tone in healthy animals (5). NO stimulates soluble guanylate cyclase (sGC) to synthesize cyclic 3′,5′-guanosine monophosphate (cGMP), which activates cGMP-dependent protein kinases, resulting in inhibition of platelet aggregation, vasodilatation, and immunomodulation (6). Moreover, NO interacts with other heme-containing molecules and proteins containing reactive thiol groups. The synthesis of NO depends on molecular oxygen, and hypoxia has been shown to reduce NO production (7). Hypoxia can also inhibit the activity of NOS by suppression of the uptake of l-arginine by pulmonary arterial endothelial cells (8). Although NO is the most important pulmonary vasodilator, the pharmacological inhibition of NOS exerts only a minor effect on normoxic tone, but results in the enhancement of acute HPV in isolated rabbit lungs (2, 7). With regard to chronic hypoxia, vascular responses have partly been attributed to decreased concentrations of NO (9). In line with this, the pharmacological increase of sGC activity reversed hypoxia-induced PH in mice (10).
All three isoforms of NOS can be inhibited by asymmetric ω-NG, NG-dimethylarginine (ADMA), which is generated by the lung (11). Chronic hypoxia–induced PH was demonstrated to be associated with increased pulmonary concentrations of ADMA (12). Plasma ADMA concentrations were also increased in patients with severe PH (13). These findings suggest that NOS inhibition by ADMA may contribute to hypoxia-induced PH.
ADMA is degraded by dimethylarginine dimethylaminohydrolase (DDAH) 1 and 2, both expressed in the lung (14). The activity of DDAH enzymes in the lung was reported to be decreased in chronic hypoxia–induced PH after 1 week of exposure to hypoxia. This finding correlated with decreased expression of the dimethylarginine dimethylaminohydrolase–1 (DDAH1) isoform, decreased NO concentrations, and increased ADMA concentrations (12). DDAH1 overexpression also decreased systemic blood pressure (15), whereas DDAH1 knockout caused increased systemic and pulmonary blood pressure (16).
Against this background, we hypothesized that the overexpression of DDAH1 may affect the responses of the lung vasculature to acute, sustained, and chronic hypoxia by interfering with the ADMA–NO–cGMP pathway. We thus investigated the effects of DDAH1 overexpression on HPV in isolated ventilated and perfused mouse lungs, as well as on the development of chronic hypoxia–induced PH in mice.
Materials and Methods
For detailed protocols, please refer to the online supplement
Animals
Adult wild-type (WT) mice (C57BL/6J) were purchased from Charles River (Sulzfeld, Germany). DDAH1-overexpressing mice were generated as previously described (15). All animal experiments were approved by the local authorities (Regierungspräsidium Giessen). Plasma ADMA concentrations of DDAH1-overexpressing mice were decreased, whereas systemic arterial pressure (SAP) was unchanged (see Figure E1 in the online supplement), in accordance with previous findings (17).
Isolated Ventilated and Perfused Mouse Lungs in the Determination of HPV
For the determination of the strength of acute and sustained HPV, isolated ventilated and perfused lungs were used. This system offers the advantage of examining the effects of DDAH1 overexpression in HPV without disturbance by the systemic effects exerted by DDAH1. Preparation of the lungs was performed as described previously (18). Mice were deeply anesthetized with intraperitoneal pentobarbital sodium (100 mg/kg body weight), and were anticoagulated with heparin (1,000 U/kg body weight) by intravenous injection. After achieving deep anesthesia, mice were intubated via a tracheostoma and ventilated with room air at a tidal volume of 300 μl, a frequency of 90 breaths/minute, and a positive end-expiratory pressure of 3 cm H2O, using a piston pump (Minivent Type 845; Hugo Sachs Elektronik, March-Hugstetten, Germany). Midsternal thoracotomy was followed by the insertion of catheters into the pulmonary artery and left atrium. Using a peristaltic pump (REGLO Digital MS-4/12; Ismatech SA, Labortechnik-Analytik, Glattbrugg, Switzerland), perfusion with sterile Krebs-Henseleit buffer (Serag-Wiessner, Naila, Germany) via the pulmonary artery was initiated at 4°C and a flow of 0.2 ml/minute. In parallel with the onset of artificial perfusion, ventilation was changed from room air to a premixed normoxic normocapnic gas mixture with 21% O2 and 5.3% CO2, balanced with N2 (Air Liquide; Deutschland GmbH, Ludwigshafen, Germany). The lungs were removed from the thorax without the interruption of ventilation and perfusion, and were freely suspended from a force transducer for the monitoring of organ weight in a temperature-equilibrated, humidified chamber at physiologic body temperature. After rinsing the lungs with at least 20 ml buffer, the perfusion circuit was closed for recirculation. Meanwhile, the flow was slowly increased from 0.2 to 2.0 ml/minute (total system volume, 15 ml). Left atrial pressure was set at 2.0 mm Hg, and the whole perfusion system was equilibrated to physiologic body temperature. Pressures in the pulmonary artery and left atrium were registered with pressure transducers.
For hypoxic ventilation, a gas mixture containing 1% O2 and 5.3% CO2, balanced with N2, was used. After a normoxic ventilation period of 15 minutes, hypoxic ventilation was applied for 10 minutes (acute phase), followed by normoxic ventilation for 15 minutes Afterward, a hypoxic ventilation period of 3 hours (sustained phase) was applied, followed by 15 minutes of normoxic ventilation. Finally, a second acute hypoxic challenge was performed for 10 minutes, followed by 10 minutes of normoxic ventilation. We used 4.5 nM of the thromboxane mimetic U 46619 (Sigma-Aldrich, St. Louis, MO) as a hypoxia-independent vasoconstrictor. Nω-nitro-l-arginine (L-NNA) and 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one (ODQ) were purchased from Sigma-Aldrich (Steinheim, Germany) and applied to the buffer, resulting in final concentrations of 400 μM and 10 μM, respectively. The dose of ODQ was chosen to avoid the unspecific activation of NOS inhibition (19). L-NNA is a competitive NOS inhibitor that binds noncovalently to endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase, and neuronal nitric oxide synthase, with slow on/off kinetics at eNOS and nNOS. L-NNA was chosen instead of Nω-nitro-l-arginine methyl ester, because that compound has to be hydrolyzed to the active L-NNA (20). L-NNA is also preferred in comparison to the NOS inhibitor Nω-monomethyl-l-arginine, which can lead to the uncoupling of eNOS (21). ODQ is a highly selective, irreversible inhibitor of soluble guanylyl cyclase. The binding of ODQ oxidizes the prosthetic heme group and is competitive with NO. The inhibition of soluble guanylyl cyclase is time-dependent, with complete inactivation in 10 minutes (22). Both substances are stable in aqueous solutions for at least 1 day at room temperature.
In vivo hemodynamics, morphometry, and determination of right ventricular hypertrophy for the quantification of PH were undertaken after the exposure to chronic hypoxia.
For exposure to chronic hypoxia, mice were kept in normobaric hypoxia (inspiratory O2 10%) in a ventilated chamber for 28 days, as described elsewhere (18). Right ventricular systolic pressure (RVSP), the degree of muscularization of small pulmonary arteries, and right ventricular hypertrophy (ratio of the right ventricle (RV) and the left ventricle + septum [LV + S]) were determined as described previously (18). The mice were anesthetized with an intraperitoneal injection of a combination of ketamine and xylazine (100 mg/kg and 10 mg/kg body weight, respectively). Afterward, they were placed on a temperature-controlled table to maintain body temperature in the physiological range. They were tracheostomized and artificially ventilated with a gas volume of 10 ml/kg body weight (SAR830A/P; IITC, Woodland Hills, CA). A positive end-expiratory pressure of 1.0 cm H2O was used throughout. The SAP was monitored by cannulating the left carotid artery with a polyethylene cannula connected to a fluid-filled force transducer (B. Braun Melsungen AG, Melsungen, Germany). The right jugular vein was used for catheterization of the RV with a custom-made silicone catheter, and RVSP was measured. After the measurement of RVSP, total blood was collected directly from the RV for hematocrit measurement and plasma preparation. Hematocrit was measured immediately by a capillary centrifugation technique. Afterward, the lungs were flushed with saline, and fixation was performed by the perfusion of 3.5–3.7% formaldehyde (Otto Fischar GmbH und Co. KG, Saarbrücken, Germany) at a constant pressure of 22 cm H2O in the pulmonary artery and 11 cm H2O in the trachea. The lung and the heart were removed en bloc. Lung lobes were embedded in paraffin, and sections of 3 μm were cut from all lobes. The degree of muscularization of small peripheral pulmonary arteries was assessed by double staining with an anti–α–smooth muscle actin antibody (dilution, 1:900; clone 1A4; Sigma-Aldrich, St. Louis, MO) and anti-human von Willebrand factor antibody (dilution, 1:900; DAKO, Hamburg, Germany). Sections were counterstained with methyl green and examined by light microscopy through the use of a computerized morphometric system (Qwin; Leica, Wetzlar, Germany). At ×40 magnification, 100 intra-acinar vessels (diameter, 10–50 μm) accompanying either the alveolar ducts or alveoli were analyzed. Each vessel was categorized as nonmuscularized (no apparent smooth muscle), partly muscularized (with a partial smooth muscle layer), or fully muscularized (with a complete smooth muscle layer). The percentage of pulmonary vessels in each muscularization category was determined by dividing the number of vessels in that category by the total number counted.
The RV was separated from the LV + S. After separation, they were placed on glass slides and dried for 1 week at room temperature. The right to left ventricle plus septum weight ratio (RV/LV + S) was calculated as an index of right ventricular hypertrophy.
Measurement of ADMA, cGMP, and NO Metabolites
For the determination of ADMA, an ADMA kit from DLD Diagnostika, GmbH (Hamburg, Germany) was used. The concentration of cGMP was measured by a cGMP kit (Cayman, Hamburg, Germany). The metabolic products of NO (nitrite, nitrate, and peroxynitrite), summarized as NOx, were determined in perfusate samples by using the chemiluminescence detector Nitric Oxide Analyzer, Sievers 280 (FMI GmbH, Seeheim, Germany) (7). For the determination of ADMA, cGMP, and NO metabolites in lung tissue, samples were prepared as described previously (12).
Western Blotting
Western blotting was performed as described previously, with slight modifications (18).
Statistical Analysis
All data are expressed as means ± SEMs. Comparisons of multiple groups were performed by ANOVA with the Student-Newman-Keuls post hoc test. For the comparison of two groups, a Student t test was performed. P < 0.05 was considered statistically significant for all analyses.
Results
Acute and Sustained HPV in DDAH1-Overexpressing Mice
Isolated perfused and ventilated lungs were exposed to normoxic ventilation (21% O2) for 15 minutes, followed by a period of 10-minute exposure to hypoxia (1% O2), with subsequent normoxic ventilation for 15 minutes. After that, a 3-hour period of sustained hypoxic ventilation was applied, followed by normoxic ventilation for 15 minutes and another acute hypoxic challenge (Figure 1A). No differences in ΔPAP were evident during the first acute hypoxic ventilation period between isolated lungs from WT and DDAH1-overexpressing (DDAH1tg) mice. However, during sustained hypoxic ventilation, the strength of HPV was decreased in isolated lungs from DDAH1tg mice compared with WT mice. This decrease became statistically significant at Minute 160, and lasted till the end of the experiments. Performing a second 10-minute hypoxic ventilation period after 15 minutes of normoxic ventilation at Minute 235 produced an acute HPV response of the same magnitude for both mouse strains (Figure 1A). In the course of 255 minutes of normoxic ventilation, no significant differences in PAP between isolated lungs from WT and DDAH1tg mice could be detected (Figure 1B).
Figure 1.

Effects of dimethylarginine dimethylaminohydrolase (DDAH) overexpression on pulmonary arterial pressure (PAP) during normoxic and hypoxic ventilation of isolated lungs. (A) Increase of PAP (ΔPAP) during normoxic and hypoxic ventilation was referenced to PAP values at time point 0 (normoxic ventilation). *Significant difference (P < 0.05) between lungs from wild-type mice (WT) and DDAH1-overexpressing mice (DDAH1tg). No significant difference in ΔPAP was evident during the second acute hypoxic ventilation, performed at Minutes 235–245, when related to PAP values at the onset of hypoxic ventilation (235 minutes). Each data point is derived from n = 8–10 lungs per group. (B) PAP during 255 minutes of normoxic ventilation. Each data point is derived from n = 3 lungs per group.
ADMA, NO, and cGMP Concentrations in Acute and Sustained HPV
ADMA concentrations in the perfusate of isolated lungs from DDAH1tg mice were significantly decreased at Minutes 10, 20, and 220, compared with WT mice (Figure 2A). In addition, a significant increase in ADMA concentrations was evident at 220 minutes during hypoxic ventilation in isolated lungs from WT and DDAH1tg mice, compared with the other time points (Figure 2A). ADMA concentrations in the lung homogenate of isolated lungs from WT mice were also significantly increased after 220 minutes of hypoxic ventilation, compared with 10 minutes of normoxic ventilation, whereas only a trend toward higher ADMA concentrations was observed in the lung homogenate of isolated lungs from DDAHtg animals (Figure E2). The concentration of NO metabolites was significantly increased at Minute 220 in isolated lungs from DDAH1tg mice compared with WT mice, and compared with the other time points in isolated lungs from DDAH1tg mice (Figure 2B). Correspondingly, cGMP concentrations in the buffer of isolated lungs were significantly increased at Minute 220 in DDAH1tg mice compared with WT mice, and compared with the other time points in DDAH1tg mice (Figure 2C). When comparing the average release of ADMA, NOx, and cGMP, ADMA release was evidently decreased, and NOx and cGMP release was increased, in isolated lungs from DDAH1tg mice compared with WT mice at time point 220 minutes (Figure E3). Investigation on the cellular level revealed that ADMA concentrations were increased in pulmonary arterial endothelial cells (PAECs) after 3 hours of hypoxic incubation (1% O2), but not in pulmonary arterial smooth muscle cells or in the presence of unspecific radical scavengers and thus also oxygen radical scavengers N-acetyl-l-cysteine (NAC) and 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) (Figure E4).
Figure 2.
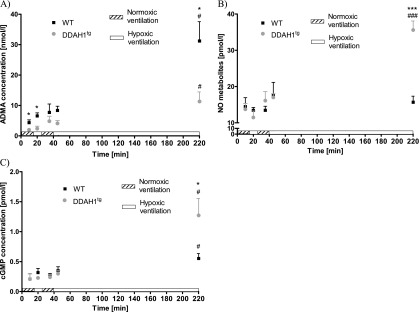
Concentrations of asymmetric dimethylarginine (ADMA), nitric oxide (NO) metabolites, and cyclic 3′,5′-guanosine monophosphate (cGMP) at different time points during hypoxic and normoxic ventilation in the perfusate of isolated lungs. Data are given for (A) ADMA, (B) NO metabolites (NOx), and (C) cGMP for lungs from wild-type (WT) and DDAH1 overexpressing mice (DDAHtg). Hypoxic ventilation, 1% O2; normoxic ventilation, 21% O2. *P < 0.05 and ***P < 0.001, significant difference between WT and DDAHtg mice during hypoxic ventilation. #P < 0.05 and ###P < 0.001, significant difference between hypoxic ventilation at 220 minutes and normoxic ventilation at 10 minutes (n = 4–8 per group).
An increase in smooth muscle tone is positively correlated with the inhibition of the myosin light chain phosphatase activity achieved by phosphorylation at the Thr-696 residue of its subunit myosin phosphatase target subunit–1 by Rho-kinase (23). Phosphorylation at this subunit was decreased in the lung homogenate of isolated lungs from DDAH1tg mice, compared with WT mice, after 220 minutes of hypoxic ventilation (Figure E5).
Inhibition of the NO–cGMP Pathway in Acute and Sustained HPV
To investigate the involvement of the NO–sGC–cGMP-pathway in the DDHA1-mediated inhibition of sustained HPV, L-NNA, an inhibitor of NOSs, and ODQ, an inhibitor of sGC, were applied in isolated lung experiments during acute and sustained HPV.
The administration of L-NNA in isolated lungs from WT mice 5 minutes before the first acute hypoxic challenge at a dose of 400 μM increased the hypoxia-induced pressure elevation (ΔPAP) during Minutes 55–60, compared with untreated WT mice (Figure 3A). The administration of ODQ in isolated lungs from WT mice 5 minutes before the first acute hypoxic challenge at a dose of 10 μM caused a significant increase of ΔPAP from Minutes 170–225 and at Minutes 245 and 255, compared with isolated lungs from untreated WT mice, and from Minutes 175–205 and 215–220, compared with L-NNA–treated isolated lungs from WT mice (Figure 3A). The administration of L-NNA in isolated lungs of DDAH1tg mice 5 minutes before the first acute hypoxic challenge caused a significant increase of ΔPAP during the time period from Minutes 55–70, compared with the isolated lungs of untreated DDAH1tg mice (Figure 3B). The administration of ODQ in isolated lungs of DDAH1tg mice 5 minutes before the first acute hypoxic challenge caused a significant increase of ΔPAP at the time periods starting from Minutes 55–80 and 95–225 and at Minute 245, compared with the untreated isolated lungs of DDAH1tg mice (Figure 3B).
Figure 3.

Effects of Nω-nitro-l-arginine (L-NNA) and 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one (ODQ) on pulmonary arterial pressure (PAP) during normoxic and hypoxic ventilation of isolated lungs. (A) Increase of PAP (ΔPAP) during normoxic and hypoxic ventilation was referenced to PAP values at time point 0 (normoxic ventilation) in the presence of L-NNA and ODQ in isolated lungs from wild-type (WT) mice. *Significant difference (P < 0.05) between untreated WT control lungs and L-NNA–treated lungs. #Significant difference (P < 0.05) between untreated WT control lungs and ODQ-treated lungs. §Significant difference (P < 0.05) between ODQ-treated and L-NNA–treated lungs. No significant difference in ΔPAP was evident during the second acute hypoxic ventilation, performed at Minutes 235–245, when related to PAP values at the onset of hypoxic ventilation (235 minutes). Each data point is derived from n = 4–10 lungs per group. (B) Increase of PAP (ΔPAP) during normoxic and hypoxic ventilation was referenced to PAP values at time point 0 (normoxic ventilation) in the presence of L-NNA and ODQ in isolated lungs from DDAH1-overexpressing (DDAHtg) mice. *Significant difference (P < 0.05) between untreated WT control lungs and L-NNA treated lungs. #Significant difference (P < 0.05) between untreated WT control lungs and ODQ-treated lungs. No significant difference in ΔPAP was evident during the second acute hypoxic ventilation, performed at Minutes 235–245, when related to PAP values at the onset of hypoxic ventilation (235 minutes). Each data point is derived from n = 5–8 lungs per group. (C) Increase of PAP (ΔPAP) during normoxic and hypoxic ventilation was referenced to PAP values at time point 0 (normoxic ventilation) in the presence of L-NNA in isolated lungs from WT and DDAHtg mice. *Significant difference (P < 0.05) between lungs of WT and DDAHtg mice. No significant difference of ΔPAP was evident during the second acute hypoxic ventilation performed at Minutes 235–245, when related to PAP values at the onset of hypoxic ventilation (235 minutes). Each data point is derived from n = 5–7 lungs per group. Data are derived from L-NNA–treated groups in A and B. (D) Increase of PAP (ΔPAP) during normoxic and hypoxic ventilation was referenced to PAP values at time point 0 (normoxic ventilation) in the presence of ODQ in isolated lungs from WT and DDAHtg mice. *Significant difference (P < 0.05) between lungs of WT and DDAH1tg mice. No significant difference in ΔPAP was evident during the second acute hypoxic ventilation, performed at Minutes 235–245, when related to PAP values at the onset of hypoxic ventilation (235 minutes). Each data point is derived from n = 4–5 lungs per group. Data are derived from the ODQ-treated groups in A and B. (E) PAP during 255 minutes of normoxic ventilation in the presence of L-NNA. Each data point is derived from n = 3 lungs per group. (F) PAP during 255 minutes of normoxic ventilation in the presence of ODQ. Each data point is derived from n = 3 lungs per group.
When directly comparing the effects of DDAH1 overexpression in the presence of L-NNA, as depicted in Figures 3A and 3B, ΔPAP was significantly lower in isolated lungs from DDAH1tg mice compared with WT mice from Minutes 210–255 (Figure 3C). When directly comparing the effects of DDAH1 overexpression in the presence of ODQ, as depicted in Figures 3A and 3B, ΔPAP was lower in the time periods from 195–225 minutes and 240–255 minutes in isolated lungs from DDAH1tg mice compared with WT mice (Figure 3D).
During 255 minutes of normoxic ventilation, no significant differences in PAP values were evident between isolated lungs from WT or DDAH1tg mice (Figures 3E and 3F) after the administration of L-NNA or ODQ (Figures 3E and 3F, compared with Figure 1B).
Effects of Chronic Hypoxia in DDAH1tg Mice
The exposure of mice to 4 weeks of chronic hypoxia induced PH in WT and DDAH1tg mice, as determined in vivo by increased RVSP (Figure 4A), increased ratios of right ventricular weight to the weight of the left ventricle plus septum (Figure 4B), and an increased portion of fully muscularized vessels at the expense of nonmuscularized vessels (Figure 4C). No differences in the development of PH could be detected when DDAH1tg mice were compared with WT mice (Figures 4A–4C). Correspondingly, no differences in ADMA concentrations (Figure 5A), concentrations of NO metabolites (Figure 5B), or cGMP concentrations (Figure 5C) were evident in the whole-lung homogenate of DDAH1tg mice compared with WT mice after exposure to chronic hypoxia. DDAH1 protein content was increased in lungs from DDAH1-overexpressing mice. Chronic hypoxia reduced the DDAH1 protein concentration by a similar percentage in DDAH1tg and WT mice, compared with normoxic mice (Figure 5D). The concentration of DDAH1 mRNA was increased in pulmonary arteries and lung homogenate from DDAH1tg mice compared with WT mice (Figure E6A). The DDAH2 protein concentration was not different in the lung homogenate of DDAH1tg and WT mice (Figure E6B). DDAH activity in the lung homogenate under basal normoxic conditions was unchanged (Figure E6C).
Figure 4.
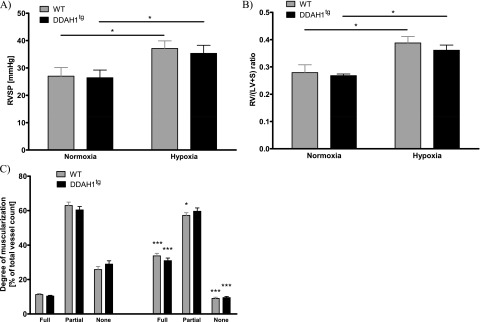
Effects of DDAH1 overexpression on the development of hypoxic-induced pulmonary hypertension. (A) Right ventricular systolic pressure (RVSP) in normoxia and after 4-week exposure to hypoxia in wild-type (WT) and DDAH1-overexpressing (DDAH1tg) mice. *Significant difference (P < 0.05) between normoxia and hypoxia (n = 10 per group). (B) Ratio of right ventricular weight (RV) and left ventricular plus septal weight (LV + S) during normoxia and after 4 weeks of exposure to hypoxia in WT and DDAH1tg mice. *Significant difference (P < 0.05) between normoxia and hypoxia (n = 10 per group). (C) Degree of vascular muscularization is given as portions of fully, partly, and not muscularized vessels in percentages [%] of total vessels during normoxia and after 4 weeks of exposure to hypoxia in WT and DDAH1tg mice. *P < 0.05 and ***P < 0.001, significant difference compared with the respective normoxic group and hypoxic group (n = 10 per group).
Figure 5.
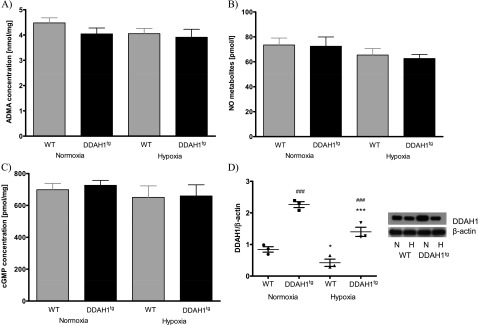
Concentrations of ADMA, NO metabolites, cGMP, and the DDAH1 protein quantified from homogenized lungs of normoxic or chronically hypoxic (4 weeks of hypoxia) wild-type (WT) and DDAH1-overexpressing (DDAH1tg) mice. Data are given for (A) ADMA, (B) NO metabolites, and (C) cGMP concentrations. (D) DDAH1 protein concentrations were normalized to β-actin. *P < 0.05 and ***P < 0.001, significant difference between normoxic (N) and hypoxic (H) lungs of WT and DDAH1tg mice. ###Significant difference (P < 0.001) between lungs of WT and DDAH1tg mice (n = 3 for DDAH1 protein, n = 4 for NO, n = 7 for ADMA and cGMP in each group, respectively).
Discussion
This study revealed that (1) DDAH1 gene overexpression resulted in the attenuation of sustained HPV in isolated mouse lungs, (2) ADMA concentrations were increased after 3 hours of hypoxic ventilation in isolated lungs from WT mice, but much less so in those from DDAH1tg mice, (3) the NO–cGMP pathway was enhanced in isolated lungs from DDAH1tg mice compared with WT mice, but this pathway was only partly responsible for the attenuation of sustained HPV, and (4) chronic hypoxia–induced PH in the intact animal was not affected by DDAH1 gene overexpression.
Our study showed that DDAH1 overexpression did not affect acute HPV or normoxic PAP in isolated lungs, but specifically decreased sustained HPV, partly by activation of the NO–cGMP pathway. In accordance with these findings, vasodilative NO (as reflected by NO metabolites) and the downstream effector cGMP were increased in the perfusate of isolated lungs from DDAHtg mice compared with WT mice during sustained hypoxia, but not during acute hypoxia or normoxia. Sustained HPV has been suggested to differ from acute HPV insofar as sustained HPV requires an intact endothelium for full expression, whereas acute HPV is less dependent on the endothelium (24). Although DDAH1 overexpression in the DDAH1tg mice used in this study occurred in a tissue-unspecific and cell type–unspecific manner (15), the main production of NO in the lung vasculature generally occurs in the endothelium (5, 25). Thus our results support the hypothesis that sustained HPV is specifically decreased in DDAH1tg mice via the activation of the endothelial NO–cGMP pathway, which is thought to be more important in sustained HPV.
Regulation of the NO–cGMP pathway may have occurred via ADMA, which was decreased in the perfusate of isolated lungs from DDAHtg mice compared with WT mice because of greater degradation. However, the decreased ADMA concentration only correlated with an increase of NOx and cGMP in the perfusate of isolated lungs of DDAHtg mice during sustained hypoxia, but not during acute hypoxia and normoxia. In this regard, ADMA plasma concentrations were found to be significantly lower in DDAH1tg mice, and NOS activity in skeletal or cardiac tissues and urinary nitrogen oxides was increased (15). The discrepancy within these results may be explained by the low concentration of ADMA at early time points in our isolated lung experiments, which may have been below the concentration required to inhibit NOS (26). ADMA concentrations were increased in the perfusate and homogenate of isolated lungs, as well as in isolated PAECs, but not in pulmonary arterial smooth muscle cells, incubated in hypoxia for 3 hours. The concentrations in the perfusate increased during the time course of the experiment, suggesting a continuous release during each time point of the experiment. However, the kinetics of intracellular ADMA production and intracellular concentration may be different, because the transport of ADMA across the cellular membrane is thought to be insufficient to equilibrate ADMA between the intracellular and extracellular space (26). In contrast, NO freely diffuses across cellular membranes, and thus the decreasing rate of NOx accumulation in the perfusate of isolated lungs from WT mice indicates decreased intravascular NO release during sustained hypoxia. In this regard, the activation of NOS and the resultant NO concentrations in hypoxia are subject to debate (27). The majority of studies suggest decreased NOS production in hypoxic lungs as a net result of altered NOS expression and activity. NOS activity is regulated, among other factors, by calcium, ADMA, and the presence of substrates such as oxygen and l-arginine. Thus, NOS stimulation during hypoxia (e.g., by calcium) may be counteracted by increased ADMA concentrations in WT mice. The hypoxia-induced stimulation of NOS is in accordance with the finding that NO concentrations were greatly increased during sustained hypoxia in DDAH1tg mice, despite even slightly increased ADMA concentrations. The concentrations of cGMP in the perfusate of isolated lungs showed a continuous increase during the time course of the experiment, indicating, in view of the lower NO release, a contribution of NO-independent GC activation. The NO-independent activation of sGC may occur via hydrogen peroxide (28) or carbon monoxide (29). In this regard, ADMA was recently suggested to induce cellular oxidative stress by the uncoupling of eNOS (30). Interestingly, Rho-kinase activity was decreased in isolated lungs from DDAH1tg mice compared with WT mice after 220 minutes of hypoxic ventilation. This finding is of particular interest because Rho-kinase activity had been suggested to be specifically important in the regulation of sustained HPV, but not acute HPV (31), thus possibly explaining the specific effect of DDAH1 overexpression on sustained HPV. Along this line, ADMA was previously shown to exert its effect on cell motility via Rho-kinase (32). In summary, our data on perfusate concentrations of ADMA, NOx, and cGMP support the hypothesis that DDAH1 overexpression results in the specific inhibition of the second phase of HPV because of the greater degradation of ADMA, which inhibits the NO–cGMP pathway during sustained HPV in isolated lungs from DDAH1tg mice to a lesser extent than in WT mice.
However, our data also showed that activation of the NO–cGMP–pathway was not the exclusive mechanism by which sustained HPV was decreased in isolated lungs from DDAH1tg mice. When applying inhibitors of the NO–cGMP pathway, sustained HPV was enhanced in isolated lungs from both mouse strains. However, although the differences between both mouse strains were attenuated in the presence of L-NNA and ODQ, sustained HPV was still significantly lower in isolated lungs from DDAH1tg mice compared with WT mice. Thus, DDAH1 may also exert its effects through an ADMA-independent mechanism, as recently suggested (33, 34). In addition, the NOS-independent and sGC-independent effects of ADMA may exist, for instance, because of the induction of mitochondrial dysfunction (35) or the activation of Rho-kinase, as already discussed. Interestingly, L-NNA and ODQ enhanced HPV at different time points of hypoxia. This finding supports the notion that a NO-independent activation of sGC occurs during sustained hypoxia, as already discussed. Our finding that the inhibition of the NO–cGMP pathway in isolated lungs only tended to increase acute HPV is consistent with a study showing that endothelial NO modulated the second phase of HPV in particular (36), and that sGC-α1 deficiency did not change the response to acute hypoxia in the lungs of anesthetized mice (37). In contrast to our findings in mice, L-NNA was shown to increase acute HPV in isolated rabbit lungs (2, 38), and when comparing studies in mice, the gene deletion of eNOS resulted in an augmentation of acute HPV in isolated lungs (5). Differences in the applied setup and experimental protocol with the specific activation of NOS by specific experimental procedures may account for this discrepancy. In summary, the results of the inhibitor experiments again emphasized the finding that the endothelial NO–cGMP pathway is of higher importance for sustained HPV than for acute HPV in isolated lungs, and showed that DDAH1 overexpression at least partly exerted its effects via the NO–cGMP pathway.
In the intact animal model, DDAH1tg mice did not differ from WT mice in regard to RVSP, right heart hypertrophy, or the degree of muscularization under baseline conditions. Moreover, the overexpression of DDAH1 did not affect the development of hypoxia-induced PH compared with WT mice. Four weeks of exposure in mice to 10% hypoxia resulted in increased RVSP, right heart ratio, and vascular remodeling to a similar degree in both mouse strains. In line with these results, concentrations of neither ADMA nor NOx or cGMP were different in the lung homogenate of both mouse strains during normoxia or after exposure to chronic hypoxia, although DDAH1 protein expression in the lung homogenate and DDAH1 mRNA in pulmonary arteries were increased in DDAHtg mice compared with WT mice. Unchanged ADMA concentrations are unlikely to be related to a compensatory down-regulation of DDAH2, because the protein expression of DDAH2 was unaltered in the lung homogenate of DDAH1tg mice compared with WT mice. These unchanged ADMA concentrations are more likely related to our inability to detect differences in DDAH activity in the lung homogenate of the mouse strains under basal normoxic conditions, which has been previously reported in liver and kidney tissue. This observation was explained by a “ceiling effect of activity which was unsurpassed by hDDAH1 overexpression” (39). Thus differences in ADMA concentrations may only be detectable under conditions of decreased DDAH1 activity in WT mice, as occurred during sustained hypoxia. DDAH1 activity has been reported to be under redox control (34), which is in accordance with our finding that increased ADMA concentrations during hypoxia in WT PAECs cannot be found in the presence of radical scavengers. In addition, the similar concentrations of ADMA in the lung homogenate from the in vivo experiments may be attributed to the fact that measurements in lung homogenate do not represent the intravascular release from distinct cell types and mask site-specific ADMA release. This suggestion is supported insofar as we found differences in ADMA concentrations in the lung perfusate between DDAH1tg mice and WT mice in the isolated lung experiments, but not in the homogenate of isolated lungs. In line with the measurements of the perfusate, we did not find differences in NOx and cGMP from the lung homogenate of both mouse strains during normoxia, which may be attributed to low ADMA concentrations at baseline. In hypoxia, the DDAH1 protein concentration was decreased by a similar percentage in both mouse strains, which may partly explain the similar degree of PH in both WT and DDAH1tg mice. In this regard the down-regulation of DDAH1 in DDAH1 knockout mice caused an accumulation of ADMA, a decrease of NO signaling, and the development of PH (16). However, we could not find any differences in ADMA, NOx, or cGMP concentrations after chronic hypoxic exposure compared with normoxia in the lung homogenate of WT or DDAH1tg mice. In contrast, differences in ADMA concentrations could be detected not only in the perfusate, but also in the lung homogenate, of isolated lungs that were ventilated with hypoxia for 3 hours. Although ADMA concentrations were previously shown to be increased after 1 week of hypoxia (12), a detailed analysis of the time pattern revealed that ADMA was increased after 2 weeks, but not after 3 weeks, of hypoxic exposure (40). Thus in chronic hypoxia, the regulatory mechanisms that counteract decreased DDAH1 protein concentrations may result in unchanged ADMA concentrations. In this regard, we speculate that DDAH1 overexpression will effectively regulate pulmonary vascular responses to hypoxia with a time pattern similar to that of the ADMA increase during hypoxia. In further studies, time points in addition to 3 hours and 4 weeks of hypoxic exposure might be examined to answer this question. Regarding NO and cGMP concentrations, it remains debatable whether these concentrations are decreased or increased during chronic hypoxia. However, evidence indicates that the NO–cGMP pathway is activated by chronic hypoxia, but that NO concentrations are decreased because of substrate limitation, at least when the measurements were performed during persistent hypoxia (27). Our measurements were performed in the lung homogenate during normoxia after 4 weeks of chronic hypoxic incubation, which may explain the unchanged NOx and cGMP concentrations. In line with this, our group showed previously that 3 weeks of hypoxia did not alter the bioavailability of NO or the activity of sGC in homogenized lungs (41).
In summary, in the isolated lungs of mice overexpressing DDAH1, we found a decreased response of the pulmonary vasculature to sustained hypoxia, as well as decreased ADMA concentrations and increased NO–cGMP signaling, but no alterations as a response to acute hypoxia in isolated lungs and chronic hypoxia in intact animals. In this regard, the different phases of pulmonary vascular response to hypoxia were possibly shown to be regulated by different mechanisms (42). Acute HPV lasts for minutes, is completely reversible, and is thought to depend on an increase in intracellular calcium, whereas sustained HPV occurring after more than 30 minutes to hours of hypoxic exposure may be related to calcium sensitization, and is not completely reversible. In chronic hypoxic PH, additional structural alterations of the vasculature occur (31, 43). Chronic hypoxia–induced PH can be detected, at the earliest, 2–3 weeks after exposure to hypoxia (44). Accordingly, this time frame was chosen for our study of chronic hypoxic effects in contrast to sustained effects. Because ADMA concentrations are increased after 1 and 2 weeks of hypoxic exposure, but not after 3 weeks (as already described), we speculate that the suppressive effects of DDAH1 overexpression will be effective in the same time frame. Interestingly, NO production was suggested to be regulated differentially during short-term and chronic hypoxia, namely, during the short term by a hypoxia-specific inhibitor (which could be ADMA) and during chronic hypoxia by the general availability of a cofactor (which could be oxygen) (45). Thus a hitherto unknown “endothelium-derived contracting factor,” in analogy to NO as the “endothelium-derived relaxing factor,” has been sought to regulate sustained HPV (46). In this regard, we found that ADMA was increased during in vitro hypoxia in PAECs. Thus our study supports the speculation that ADMA could serve as an “endothelium-derived contracting factor.” However, further studies are necessary to prove that ADMA originates in pulmonary endothelial cells during sustained hypoxia in vivo.
In conclusion, DDAH1 overexpression specifically decreased sustained HPV, but did not alter the response of the pulmonary vasculature to acute or chronic hypoxia, and did not alter baseline parameters, reflecting the specific importance of increased ADMA concentrations during sustained HPV. The NO–cGMP pathway was partly responsible for the effects of DDAH1 overexpression on sustained HPV. It remains to be investigated how ADMA production is specifically regulated during sustained hypoxia, and by which additional NO–cGMP–independent pathway DDAH1 and/or ADMA exert their effects.
Footnotes
The work was supported by National Institutes of Health grants U01HL100397 and UM1HL113456 (J.P.C.), and by German Research Foundation grants WE 1978/4-1 and SFB/TR 84, Project C6 (N.W. and M.W.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0330OC on May 3, 2013.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Bennie RE, Packer CS, Powell DR, Jin N, Rhoades RA. Biphasic contractile response of pulmonary artery to hypoxia. Am J Physiol. 1991;261:L156–L163. doi: 10.1152/ajplung.1991.261.2.L156. [DOI] [PubMed] [Google Scholar]
- 2.Ketabchi F, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Egemnazarov B, Shid-Moosavi SM, Dehghani GA, Weissmann N, Sommer N. Effects of hypercapnia and NO synthase inhibition in sustained hypoxic pulmonary vasoconstriction. Respir Res. 2012;13:7. doi: 10.1186/1465-9921-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson TP, Ward JP, Aaronson PI. Hypoxia induces the release of a pulmonary-selective, Ca(2+)-sensitising, vasoconstrictor from the perfused rat lung. Cardiovasc Res. 2001;50:145–150. doi: 10.1016/s0008-6363(01)00192-4. [DOI] [PubMed] [Google Scholar]
- 4.Robertson TP, Hague D, Aaronson PI, Ward JP. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol. 2000;525:669–680. doi: 10.1111/j.1469-7793.2000.t01-1-00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fagan KA, Tyler RC, Sato K, Fouty BW, Morris KG, Jr, Huang PL, McMurtry IF, Rodman DM. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol. 1999;277:L472–L478. doi: 10.1152/ajplung.1999.277.3.L472. [DOI] [PubMed] [Google Scholar]
- 6.Gkaliagkousi E, Ferro A. Nitric oxide signalling in the regulation of cardiovascular and platelet function. Front Biosci. 2011;16:1873–1897. doi: 10.2741/3828. [DOI] [PubMed] [Google Scholar]
- 7.Grimminger F, Spriestersbach R, Weissmann N, Walmrath D, Seeger W. Nitric oxide generation and hypoxic vasoconstriction in buffer-perfused rabbit lungs. J Appl Physiol. 1995;78:1509–1515. doi: 10.1152/jappl.1995.78.4.1509. [DOI] [PubMed] [Google Scholar]
- 8.Durmowicz AG, Stenmark KR. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- 9.Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- 10.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113:286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 11.Bulau P, Zakrzewicz D, Kitowska K, Leiper J, Gunther A, Grimminger F, Eickelberg O. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am J Physiol Lung Cell Mol Physiol. 2007;292:L18–L24. doi: 10.1152/ajplung.00076.2006. [DOI] [PubMed] [Google Scholar]
- 12.Millatt LJ, Whitley GS, Li D, Leiper JM, Siragy HM, Carey RM, Johns RA. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia–induced pulmonary hypertension. Circulation. 2003;108:1493–1498. doi: 10.1161/01.CIR.0000089087.25930.FF. [DOI] [PubMed] [Google Scholar]
- 13.Gorenflo M, Zheng C, Werle E, Fiehn W, Ulmer HE. Plasma levels of asymmetrical dimethyl-l-arginine in patients with congenital heart disease and pulmonary hypertension. J Cardiovasc Pharmacol. 2001;37:489–492. doi: 10.1097/00005344-200104000-00016. [DOI] [PubMed] [Google Scholar]
- 14.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol. 2004;24:1023–1030. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 15.Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson AJ, et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation. 2003;108:3042–3047. doi: 10.1161/01.CIR.0000101924.04515.2E. [DOI] [PubMed] [Google Scholar]
- 16.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 17.Jacobi J, Maas R, Cardounel AJ, Arend M, Pope AJ, Cordasic N, Heusinger-Ribeiro J, Atzler D, Strobel J, Schwedhelm E, et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E–deficient mice by lowering asymmetric dimethylarginine. Am J Pathol. 2010;176:2559–2570. doi: 10.2353/ajpath.2010.090614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roth M, Rupp M, Hofmann S, Mittal M, Fuchs B, Sommer N, Parajuli N, Quanz K, Schubert D, Dony E, et al. Heme oxygenase–2 and large-conductance Ca2+-activated K+ channels: lung vascular effects of hypoxia. Am J Respir Crit Care Med. 2009;180:353–364. doi: 10.1164/rccm.200806-848OC. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DB. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1056–L1065. doi: 10.1152/ajplung.90329.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;2012:318087. doi: 10.1155/2012/318087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Druhan LJ, Forbes SP, Pope AJ, Chen CA, Zweier JL, Cardounel AJ. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry. 2008;47:7256–7263. doi: 10.1021/bi702377a. [DOI] [PubMed] [Google Scholar]
- 22.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 23.Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- 24.Aaronson PI, Robertson TP, Ward JP. Endothelium-derived mediators and hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol. 2002;132:107–120. doi: 10.1016/s1569-9048(02)00053-8. [DOI] [PubMed] [Google Scholar]
- 25.Sherman TS, Chen Z, Yuhanna IS, Lau KS, Margraf LR, Shaul PW. Nitric oxide synthase isoform expression in the developing lung epithelium. Am J Physiol. 1999;276:L383–L390. doi: 10.1152/ajplung.1999.276.2.L383. [DOI] [PubMed] [Google Scholar]
- 26.Teerlink T, Luo Z, Palm F, Wilcox CS. Cellular ADMA: regulation and action. Pharmacol Res. 2009;60:448–460. doi: 10.1016/j.phrs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L575–L582. doi: 10.1152/ajplung.2001.280.4.L575. [DOI] [PubMed] [Google Scholar]
- 28.Neo BH, Kandhi S, Wolin MS. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol. 2010;299:H1235–H1241. doi: 10.1152/ajpheart.00513.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182. doi: 10.1165/rcmb.2006-0333TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohan S, Fung HL. Mechanism of cellular oxidation stress induced by asymmetric dimethylarginine. Int J Mol Sci. 2012;13:7521–7531. doi: 10.3390/ijms13067521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wojciak-Stothard B, Torondel B, Tsang LY, Fleming I, Fisslthaler B, Leiper JM, Vallance P. The ADMA/DDAH pathway is a critical regulator of endothelial cell motility. J Cell Sci. 2007;120:929–942. doi: 10.1242/jcs.002212. [DOI] [PubMed] [Google Scholar]
- 33.Leiper J, Vallance P. New tricks from an old dog: nitric oxide–independent effects of dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol. 2006;26:1419–1420. doi: 10.1161/01.ATV.0000229598.55602.17. [DOI] [PubMed] [Google Scholar]
- 34.Pope AJ, Karrupiah K, Kearns PN, Xia Y, Cardounel AJ. Role of dimethylarginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J Biol Chem. 2009;284:35338–35347. doi: 10.1074/jbc.M109.037036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol. 2008;294:C1407–C1418. doi: 10.1152/ajpcell.00384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang F, Woodmansey PA, Morice AH. Acute hypoxic vasoconstriction in isolated rat small and large pulmonary arteries. Physiol Res. 1995;44:7–18. [PubMed] [Google Scholar]
- 37.Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, Pellens M, Gillijns H, Swinnen M, Graveline A, et al. Soluble guanylate cyclase–alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation. 2007;116:936–943. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- 38.Ketabchi F, Egemnazarov B, Schermuly RT, Ghofrani HA, Seeger W, Grimminger F, Shid-Moosavi M, Dehghani GA, Weissmann N, Sommer N. Effects of hypercapnia with and without acidosis on hypoxic pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol. 2009;297:L977–L983. doi: 10.1152/ajplung.00074.2009. [DOI] [PubMed] [Google Scholar]
- 39.Schwedhelm E, von Leitner EC, Atzler D, Schmitz C, Jacobi J, Meinertz T, Münzel T, Baldus S, Cooke JP, Böger RH, et al. Extensive characterization of the human DDAH1 transgenic mice. Pharmacol Res. 2009;60:494–502. doi: 10.1016/j.phrs.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Yildirim AO, Bulau P, Zakrzewicz D, Kitowska KE, Weissmann N, Grimminger F, Morty RE, Eickelberg O. Increased protein arginine methylation in chronic hypoxia: role of protein arginine methyltransferases. Am J Respir Cell Mol Biol. 2006;35:436–443. doi: 10.1165/rcmb.2006-0097OC. [DOI] [PubMed] [Google Scholar]
- 41.Kirsch M, Kemp-Harper B, Weissmann N, Grimminger F, Schmidt HH. Sildenafil in hypoxic pulmonary hypertension potentiates a compensatory up-regulation of NO–cGMP signaling. FASEB J. 2008;22:30–40. doi: 10.1096/fj.06-7526com. [DOI] [PubMed] [Google Scholar]
- 42.Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci USA. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pak O, Aldashev A, Welsh D, Peacock A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J. 2007;30:364–372. doi: 10.1183/09031936.00128706. [DOI] [PubMed] [Google Scholar]
- 44.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death–dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 45.Shaul PW, Wells LB, Horning KM. Acute and prolonged hypoxia attenuate endothelial nitric oxide production in rat pulmonary arteries by different mechanisms. J Cardiovasc Pharmacol. 1993;22:819–827. doi: 10.1097/00005344-199312000-00007. [DOI] [PubMed] [Google Scholar]
- 46.Aaronson PI, Robertson TP, Knock GA, Becker S, Lewis TH, Snetkov V, Ward JP. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol. 2006;570:53–58. doi: 10.1113/jphysiol.2005.098855. [DOI] [PMC free article] [PubMed] [Google Scholar]