

Abstract
Short-rib polydactyly (SRP) syndrome type III, or Verma-Naumoff syndrome, is an autosomal-recessive chondrodysplasia characterized by short ribs, a narrow thorax, short long bones, an abnormal acetabulum, and numerous extraskeletal malformations and is lethal in the perinatal period. Presently, mutations in two genes, IFT80 and DYNC2H1, have been identified as being responsible for SRP type III. Via homozygosity mapping in three affected siblings, a locus for the disease was identified on chromosome 9q34.11, and homozygosity for three missense mutations in WDR34 were found in three independent families, as well as compound heterozygosity for mutations in one family. WDR34 encodes a member of the WD repeat protein family with five WD40 domains, which acts as a TAK1-associated suppressor of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway. We showed, through structural modeling, that two of the three mutations altered specific structural domains of WDR34. We found that primary cilia in WDR34 mutant fibroblasts were significantly shorter than normal and had a bulbous tip. This report expands on the pathogenesis of SRP type III and demonstrates that a regulator of the NF-κB activation pathway is involved in the pathogenesis of the skeletal ciliopathies.
Main Text
The short-rib polydactyly (SRP) group of skeletal dysplasias falls within the ciliopathy spectrum of disorders. This group includes four perinatal lethal conditions (SRP types I–IV) and two other similar skeletal disorders, Ellis-van Creveld (EVC [MIM 225500]) syndrome and asphyxiating thoracic dysplasia (ATD [MIM 208500, MIM 611263, MIM 613091, MIM 613819, MIM 614376]), all inherited as autosomal-recessive disorders. Ellis-van Creveld syndrome is usually a nonlethal skeletal disorder associated with a long narrow chest, short stature, atrial septal defects, oral frenulum, and polydactyly. ATD is characterized by respiratory insufficiency at birth and long-term survivors are affected by retinal degeneration and cystic renal and liver abnormalities. In ATD, increased lethality can result from pulmonary, renal, and liver failure. ATD is phenotypically related to short-rib polydactyly syndrome type III (SRP type III [MIM 263510]), which is also known as Verma-Naumoff syndrome. Indeed, both disorders share similar radiological features including short ribs and a trident-shaped acetabular roof of the pelvis. However, in SRP type III, the radiographic abnormalities are more profound with very shortened tubular bones with both round and laterally spiked metaphyseal ends as well as polydactyly.1,2 SRP type III is uniformly lethal as a result of pulmonary hypoplasia and other concomitant organ system abnormalities.
Mutations in a gene encoding a protein involved in intraflagellar transport (IFT), IFT80 (MIM 611177), have been identified in 3/39 individuals with ATD.3 We also identified IFT80 mutations in one SRP type III case, supporting the idea that ATD and SRP type III can be allelic with variable severity.4 More recently mutations in cytoplasmic dynein 2 heavy chain 1 (DYNC2H1 [MIM 603297]) have been identified in cases affected either with SRP type III or ATD.5,6 DYNC2H1 is a component of the cytoplasmic dynein complex and is directly involved in transport along microtubules via its large motor domain. These previous molecular and phenotypic data support the fact that SRP type III and ATD can be allelic and both demonstrate locus heterogeneity. The proposed functions of the identified genes confirm that the SRP skeletal dysplasia group belongs to the ciliopathy spectrum of disease.5
Under an approved human subjects protocol, DNA samples were collected from 15 SRP type III cases, in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and proper informed consent was obtained. We identified homozygosity for a mutation in IFT80 in one family4 and mutations in DYNC2H1 in three other families.5,6 Among the 11 remaining cases, we excluded mutations in IFT80 and DYNC2H1 and focused on a consanguineous Algerian family with three affected individuals. The first affected individual was diagnosed with SRP type III or severe asphyxiating thoracic dysplasia (ATD) by ultrasound at 23 weeks because of the findings of micromelia, curved femurs, and short thorax. The proband died on day 7 of life resulting from respiratory insufficiency and abnormal skeletal findings were confirmed by postnatal radiographs (Figure 1). The second case was a suspected recurrence and the pregnancy was terminated at 26 weeks of gestation. The third case was diagnosed at 20 weeks of gestation and was stillborn at 42 weeks of gestation.
Figure 1.
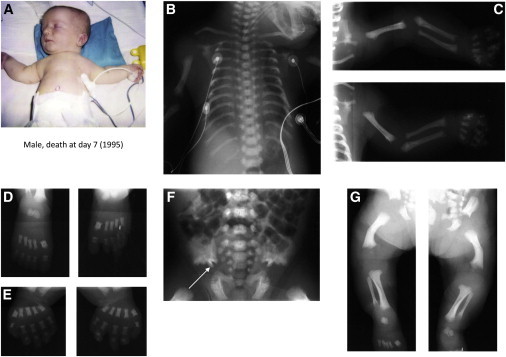
Clinical and Radiological Features of Individuals 1 and 2 in Family 1
Clinical and radiological features of the case 1. Male deceased at day 7 of respiratory distress.
(A) Note the short and narrow thorax, short limbs, and brachydactyly.
(B) Note the long and narrow thorax with short ribs.
(C–E, G) Upper and lower limbs: Note the shortened tubular bones, round metaphyseal ends with lateral spike, and the delayed bone age.
(D and E) Hands and feet: Note round metaphyseal ends with lateral spikes of square and thickset metatarsus and phalanx and the poor mineralization of phalanx.
(F) Note the trident aspect of the acetabular roof (arrow).
We performed genome-wide homozygosity mapping with (1) a 4 cM average marker density (1,000 markers, deCODE genetics) on DNA samples from the three affected individuals and (2) GeneChip Human Mapping 250K NspI arrays (Affymetrix) on DNA samples from two of the affected individuals. A single region of homozygosity shared by the three individuals was identified on chromosome 9. With the MERLIN program, the maximum LOD score was Z = 2.4 (θ = 0) for the region delimited by the markers at loci D9S1819 and D9S1847. Analysis of SNP genotypes delineated the critical region of homozygosity; the centromeric boundary defined by SNP_A-4229219 (rs2798429) and the telomeric boundary by SNP_A-2284253 (rs10114591) (9q34.11, 3.6 Mb) (Figure S1 available online). This region contains 94 genes, and among them, 33 genes referenced in the cilia database (cildb v.2) were considered as candidate genes. After excluding mutations in ODF2 (outer dense fiber protein 2) (MIM 602015) by direct sequencing, we considered WD repeat-containing protein 34 (WDR34 [MIM 613363], NCBI accession number NM_052844.3). We performed direct Sanger sequence analysis (oligonucleotide sequences provided on request) and identified homozygosity for a missense mutation in the three affected cases (ex7, c.1022C>T [p.Ala341Val]) (Table 1, Figure 2). The mutation was not identified among 242 control chromosomes. With the use of the PolyPhen web program, the consequence of this mutation was predicted as benign. However, the SIFT web program (Sorting Intolerant From Tolerant) predicted the substitution at position 341 from Ala to Val to affect protein function with a score of 0.02.
Table 1.
Clinical Features of the Three Families with WDR34 Mutations
| Family | Origin | CS | Individual | Age of Affected Individuals | Diagnosis | Polydactyly | Visceral Anomalies | Cleft Lip and/or Palate | Nucleotide Change | Amino Acid Change | Location | WD40 Repeat-Containing Domain |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Algerian | yes | 1 | death at day 7 | ATD/SRPIII | none | none | no | c.1022C>T | p.Ala341Val | exon 7 | no |
| 1 | Algerian | yes | 2 | TP at 26 wg | SRP III | none | hypotrophic lungs | no | c.1022C>T | p.Ala341Val | exon 7 | no |
| 1 | Algerian | yes | 3 | SB at 42 wg | SRP III | none | none | no | c.1022C>T | p.Ala341Val | exon 7 | no |
| 2 | Arabic | yes | 4 | carried to term | ATD/SRPIII | none | none | no | c.1061C>T | p.Thr345Met | exon 7 | no |
| 3 | Indian | yes | 5 | TP at 22 wg | SRPIII | yes | none | no | c.1339C>T | p.Arg447Trp | exon 8 | domain 4 |
| 4 | European descent | no | 6 | TP at 19 wg | ATD | none | none | no | c.1340G>A; c.982−2T>G | p.Arg447Gln | exon 8; intron 6 | domain 4 |
Abbreviations are as follows: CS, consanguinity; TP, terminated pregnancy; SB, stillbirth; wg, week of gestation.
Figure 2.
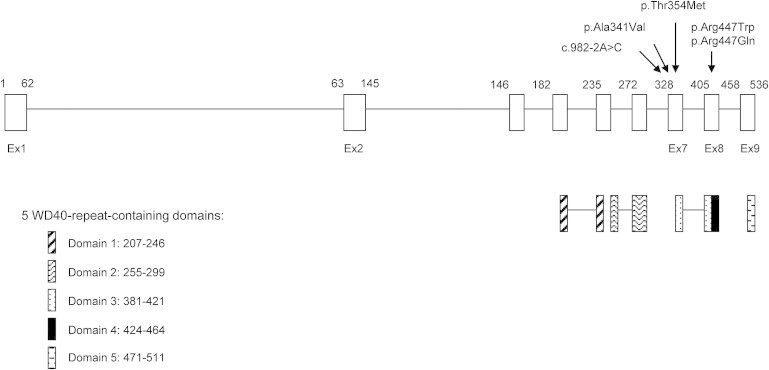
Location of the WDR34 Mutations
Location of the WDR34 mutation and the five WD40-repeat-containing domains.
Absence of any tissue to further study the consequences of the predicted mutation prompted us to perform whole-exome analysis on one of the affected individuals to determine whether other mutations in the region of homozygosity could be responsible for the disorder. Across the exome, there were 22,165 variants in 10,359 genes. Excluding variants listed in dbSNP and UTR, ncRNA, synonymous, and heterozygote variants narrowed the number of variants to 22 in 15 genes present in the homozygous state. Among those, four homozygous variants in four genes encoded ciliary proteins: VPS10 (vacuolar protein sorting 10 [MIM 602005]), DCLK2 (doublecortin like kinase 2 [MIM 613166]), SPTBN5 (spectrin beta non erythrocytic 5 [MIM 605916]), and WDR34. Finally, in the region of homozygosity on chromosome 9, there was only one homozygous variant, the c.1022C>T change previously identified by direct sequencing of WDR34. This homozygous change was absent from data sets including dbSNP129, the 1000 Genomes Project, and in-house exome data.
We then combined candidate gene sequencing of WDR34 and whole-exome analysis in 30 independent cases with the ATD/SRP type III spectrum phenotype. In two individuals (both of whom had SRP type III) from consanguineous families, homozygosity for mutations in exons 7 and 8 (one in each individual) were identified (ex7 c.1061C>T [p.Thr354Met]; ex8 c.1339C>T [p.Arg447Trp]) (Table 1, Figure 2) The mutations cosegregated with the disease in each family and were not identified among 220 control chromosomes. In an independent family with a proband (R00-326) affected by ATD, compound heterozygosity for the following mutations were identified: ex8 c.1340G>T, p.Arg447Gln, and c.982−2T>G. The mutations segregated within the family. A source of RNA was not available from the affected individual to confirm the predicted consequence of the intron 6 splice site mutation. Interestingly, the same arginine, p.447, was substituted in the homozygous and heterozygous state to methionine and glutamine, respectively. These changes were predicted as possibly and probably damaging by the PolyPhen and SIFT web programs, respectively.
WDR34 encodes a protein of 536 amino acids that is highly conserved in evolution, with human and mouse WDR34 sharing 83% identity (Ensembl). The expression pattern of mouse Wdr34 mRNA shows that the gene is ubiquitously expressed in all examined tissues, including brain, thymus, heart, lung, liver, spleen, kidney, testis, and intestine.7 It is primarily localized within the cytoplasm in 293-Flag-WDR34-transfected HEK293 cells.7 It is a member of the WD repeat protein family with five WD40 domains (details in Figure 2). WD40 repeats are minimally conserved regions of approximately 40 amino acids, typically bracketed by Gly-His and Trp-Asp (GH-WD) and facilitate formation of heterotrimeric or multiprotein complexes. These proteins are involved in intracellular trafficking, cargo recognition, and protein binding.8,9 In coat proteins (such as COPI, COPII, and clathrin), they are essential for vesicular cage formation.10
Many WD40 domain-containing proteins have been previously implicated in the ciliopathies and specifically in disorders with skeletal abnormalities. Indeed, mutations in IFT80 (also called WDR56) are responsible for the ATD/SRP type III spectrum; mutations in WDR35 (MIM 613602) and WDR10 (also called IFT122) (MIM 606045) produce Sensenbrenner syndrome (MIM 218330), which is characterized by craniosynostosis and facial, ectodermal, and skeletal anomalies,11–15 and WDR35 mutations have been found in an unclassified form of SRP.16 Finally, mutations in WDR19 (also called IFT144) (MIM 608151) have been identified in Sensenbrenner syndrome, ATD, and nephronophthisis (NPHP [MIM 256100]).17
WD40 repeats are configured into β strands and turns, referred to as a bladed beta-propeller folded structure. To determine the effect of the mutations (p.Ala341Val, p.Thr354Met, p.Arg447Trp) on this structure, we undertook homology modeling of the WDR34 mutations. Although a three-dimensional structure has not been determined for WDR34, the 2.50 Å coordinate set for the yeast α/β’-COP subcomplex of the COPI vesicular coat (PDB accession number 3mkq),18 a known beta-propeller folded structure, was used as a template for modeling the human WDR34 protein. Using the Swiss-Model program18 in the automated mode, the three-dimensional structure of human WDR34 protein (residues 210–515) was compared with residues 1–301 of the yeast β’-COP N-terminal β-propeller. The predicted structures (Figure S2) were visualized with the Swiss-Pdb Viewer 3.7. The yeast β’-COP N-terminal β-propeller is characterized by a regular and compact structure involving short connecting loops between the β strands at both axial ends of the β-propeller. The effect is to create relatively flat axial ends, which are important for formation of interactions at the triskelion (three arms) center. At the triskelion center, the β’-COP subunits associate through pairwise interactions involving a small, circumscribed area of the axial end of one N-terminal β-propeller and the sides of the adjacent β-propeller. The p.Thr354Met missense substitution is located in an area that is predicted to be important for interactions at the triskelion center, suggesting that the mutation might alter the configuration of the propellers. The precise function of WDR34 in vesicle formation is unknown, so it is not possible to predict the functional impact of the mutation. The p.Arg447Trp substitution is predicted to disrupt the fourth WD40 repeat, which may influence cargo binding, but the functional impact for p.Ala341Val substitution was not clear from this analysis.
To further understand the effect of WDR34 substitutions on cilia morphology, we showed that the structure of the cilia in cultured fibroblasts from affected individuals in two of the families (R97-375, p.Thr354Met and R07-106, p.Arg447Trp) appeared subjectively shorter with a bulbous tip (Figure 3), as observed in DYNC2H1 mutant cultured chondrocytes, suggesting a defect in cytoskeletal microtubule architecture with an abnormal elongation and/or maintenance of the axoneme by IFT to form the mature primary cilium.6 These findings may support a role of WDR34 in retrograde intraflagellar transport. The mean cilia length was 2.63 ± 0.44 μm for R97-375, 1.28 ± 0.23 μm for R07-106, and 3.74 μm ± 0.63 for control cells. Student’s t test analysis showed that cilia from both SRP cell lines were statistically significantly shorter than control (p < 0.05). As previously shown, WDR34 is a cytoplasmic protein and our observation of similar localization of the protein between control and diseased fibroblasts may suggest that the mutations do not lead to mislocalization or absence of the protein (Figure 4).
Figure 3.
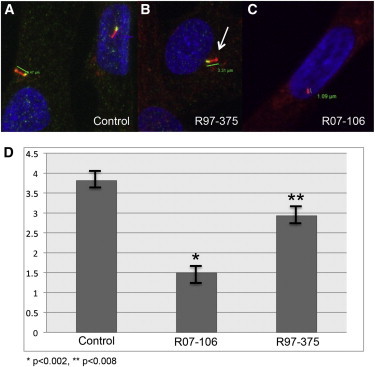
The p.Thr354Met and p.Arg447Trp Substitutions Produce Abnormal Primary Cilia in Fibroblasts
(A) Control fibroblast showing the primary cilium. Blue (DAPI) labels the nucleus, red (acetylated tubulin) labels the cilium, and yellow labels the basal body.
(B and C) Fibroblasts from affected individuals R97-375 (B) and R07-106 (C), showing statistically significantly shorter cilia. The arrow in (B) identifies the bulbous tip of the cilium.
(D) Bar graph showing variation in cilia length resulting from WDR34 mutations. n = 10. SD for control 0.43, R97-375 SD, 0.48, p < 0.0008 relative to control, SD for R07-106, 0.52, p < 0.0002 relative to control.
Figure 4.
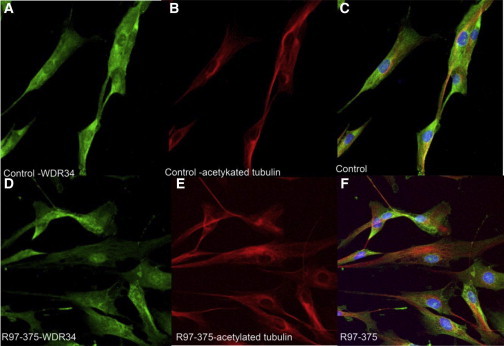
Localization of WDR34 Protein in Control and R97-375 Cultured Fibroblasts
Control fibroblasts (A–C) and R97-375 fibroblasts (D–F) showing localization of WDR34 (A, D), acetylated tubulin (B, E), and overlapping expression (C, F) (blue, DAPI, nucleus).
WDR34 has been implicated in the immune response as a negative regulator of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway, functioning by sequestering TAK1, TAB2, and TRAF6 by binding to its WD domains.7 We further hypothesize that the mutations identified in SRP type III individuals may disturb the stability or interaction of WDR34 with these proteins. This is based on findings that overexpression of WDR34 leads to diminished TAK1 phosphorylation7 and suggests that enhanced TAK1 activity may contribute to the cellular phenotype in the affected individuals with WDR34 substitutions resulting from partial loss of function. It has also been recently demonstrated that the cilium and the regulation of its structure and function are of fundamental importance in inflammation.19 In bovine primary chondrocytes, interleukin-1 (IL-1β and IL-1α) exposure increases pre-existing primary cilia length. This elongation occurred via protein kinases: cAMP-activated protein kinase A (PKA), protein kinase C (PKC), and MAP mitogen-activated protein kinases MEK-ERK. Moreover, adenylate cyclase (cAMP) levels and G protein subunits Gαi also regulate chondrocyte cilia length via PKA, but independent of IL-1, implicating complex immune regulation of cilia length.19
The identification of WDR34 mutations in four distinct families with SRP type III/severe ATD demonstrates that a negative regulator of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway is involved in the pathogenesis of the skeletal ciliopathies. Our findings of abnormal cilia with bulbous distal tip further support the role of WDR34 in retrograde intraflagellar transport.
Acknowledgments
Part of this work has been supported by a national grant from Programme Hospitalier de Recherche Clinique (PHRC AOM06031) DRCD, Assistance Publique-Hôpitaux de Paris. D.K., D.H.C., and D.L.R. are supported by National Institutes of Health grant HD22657 and D.H.C. and D.K. are supported by NIH grant R01 DE019567.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Alamut Interpretation Software 2.0 (gateway for PolyPhen-2, SIFT, SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and Human Splicing Finder), http://www.interactive-biosoftware.com
Ensembl Genome Browser, http://www.ensembl.org/index.html
GeneCards, http://www.genecards.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Swiss-Pdb Viewer 3.7, http://spdbv.vital-it.ch/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The NCBI accession number for WDR34 reported in this paper is NM_052844.3.
References
- 1.Ho N.C., Francomano C.A., van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am. J. Med. Genet. 2000;90:310–314. doi: 10.1002/(sici)1096-8628(20000214)90:4<310::aid-ajmg9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 2.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 4.Cavalcanti D.P., Huber C., Sang K.H., Baujat G., Collins F., Delezoide A.L., Dagoneau N., Le Merrer M., Martinovic J., Mello M.F. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J. Med. Genet. 2011;48:88–92. doi: 10.1136/jmg.2009.069468. [DOI] [PubMed] [Google Scholar]
- 5.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merrill A.E., Merriman B., Farrington-Rock C., Camacho N., Sebald E.T., Funari V.A., Schibler M.J., Firestein M.H., Cohn Z.A., Priore M.A. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am. J. Hum. Genet. 2009;84:542–549. doi: 10.1016/j.ajhg.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao D., Wang R., Li B., Yang Y., Zhai Z., Chen D.Y. WDR34 is a novel TAK1-associated suppressor of the IL-1R/TLR3/TLR4-induced NF-kappaB activation pathway. Cell. Mol. Life Sci. 2009;66:2573–2584. doi: 10.1007/s00018-009-0059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole D.G. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic. 2003;4:435–442. doi: 10.1034/j.1600-0854.2003.t01-1-00103.x. [DOI] [PubMed] [Google Scholar]
- 9.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 10.Lee C., Goldberg J. Structure of coatomer cage proteins and the relationship among COPI, COPII, and clathrin vesicle coats. Cell. 2010;142:123–132. doi: 10.1016/j.cell.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sensenbrenner J.A., Dorst J.P., Owens R.P. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig. Artic. Ser. 1975;11:372–379. [PubMed] [Google Scholar]
- 12.Levin L.S., Perrin J.C., Ose L., Dorst J.P., Miller J.D., McKusick V.A. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J. Pediatr. 1977;90:55–61. doi: 10.1016/s0022-3476(77)80764-6. [DOI] [PubMed] [Google Scholar]
- 13.Amar M.J., Sutphen R., Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome) Am. J. Med. Genet. 1997;70:349–352. doi: 10.1002/(sici)1096-8628(19970627)70:4<349::aid-ajmg3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 14.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A. Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A., Keighren M., Bahlo M., Bromhead C.J., Budd P. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 2011;88:508–515. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 19.Wann A.K., Knight M.M. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell. Mol. Life Sci. 2012;69:2967–2977. doi: 10.1007/s00018-012-0980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.