

Abstract
Bidirectional (anterograde and retrograde) motor-based intraflagellar transport (IFT) governs cargo transport and delivery processes that are essential for primary cilia growth and maintenance and for hedgehog signaling functions. The IFT dynein-2 motor complex that regulates ciliary retrograde protein transport contains a heavy chain dynein ATPase/motor subunit, DYNC2H1, along with other less well functionally defined subunits. Deficiency of IFT proteins, including DYNC2H1, underlies a spectrum of skeletal ciliopathies. Here, by using exome sequencing and a targeted next-generation sequencing panel, we identified a total of 11 mutations in WDR34 in 9 families with the clinical diagnosis of Jeune syndrome (asphyxiating thoracic dystrophy). WDR34 encodes a WD40 repeat-containing protein orthologous to Chlamydomonas FAP133, a dynein intermediate chain associated with the retrograde intraflagellar transport motor. Three-dimensional protein modeling suggests that the identified mutations all affect residues critical for WDR34 protein-protein interactions. We find that WDR34 concentrates around the centrioles and basal bodies in mammalian cells, also showing axonemal staining. WDR34 coimmunoprecipitates with the dynein-1 light chain DYNLL1 in vitro, and mining of proteomics data suggests that WDR34 could represent a previously unrecognized link between the cytoplasmic dynein-1 and IFT dynein-2 motors. Together, these data show that WDR34 is critical for ciliary functions essential to normal development and survival, most probably as a previously unrecognized component of the mammalian dynein-IFT machinery.
Main Text
Primary or sensory cilia, hair-like organelles conserved as nonmotile monocilia on the surface of most cells of mammals, birds, amphibians, and fish, are implicated in diverse cellular signaling pathways, most prominently the hedgehog (Hh) pathway.1 Primary cilia extend from the cell surface, having a microtubule-based core axoneme that nucleates from, and is anchored by, a mother centriole-derived basal body, with their assembly and disassembly being tightly linked to the cell cycle.2 A microtubule- and ATP-dependent bidirectional intraflagellar transport (IFT) system uses motor proteins to move the molecules required for ciliogenesis and signaling from the cell body into cilia and back out again after use. Base-to-tip anterograde IFT is directed by kinesin-2 with cargos carried on IFT-B carrier complexes. Retrograde tip-to-base IFT is directed by the IFT dynein-2 complex, with cargos attached to IFT-A carrier complexes, although IFT-A has also been implicated in the regulation of anterograde IFT.3,4 The IFT dynein-2 motor complex contains an ATPase/motor heavy chain dynein subunit (DYNC2H1)5 and less well functionally defined subunits.6,7 The IFT-A and IFT-B complexes consist of at least 6 and 14 subunits, respectively.8
Ciliary defects arising from mutations in genes that encode ciliary proteins lead to complex developmental disorders in human and other vertebrates, termed ciliopathies. Human ciliopathies are genetically heterogeneous disorders characterized by variable expression of different disease features and clinical overlap between different disease entities. This is illustrated by the skeletal ciliopathies, which include the short-rib polydactyly syndromes (SRPS I–V [MIM 263530, MIM 263520, MIM 263510, MIM 269860, MIM 614091]), asphyxiating thoracic dystrophy or Jeune syndrome (JATD [MIM 208500, MIM 611263, MIM 613091, MIM 613819, MIM 614376]), Sensenbrenner syndrome or cranioectodermal dysplasia (CED [MIM 218330, MIM 613610, MIM 614099, MIM 614378]), Ellis-van Creveld syndrome (MIM 225500), and Mainzer-Saldino syndrome (MSS [MIM 266920]).9 Skeletal ciliopathies are frequently caused by mutations in intraflagellar transport (IFT) genes and molecular genetics has confirmed that these are at least partially allelic disorders, for example, SRPS III/Verma-Naumoff syndrome (MIM 263510) and JATD,10–12 Sensenbrenner syndrome and JATD,13 and MSS and JATD.14,15 Work in animal models such as knockout mice suggests that defective IFT leads to impaired hedgehog signaling, which disturbs chondrogenic and osteogenic cellular proliferation and differentiation, leading to chondrodysplasia phenotypes.4,16,17
This study focused on the chondrodysplasia Jeune syndrome to further explore the role of IFT in skeletal development and disease. JATD is a rare autosomal-recessive disorder with a prevalence estimated at 1 per 100,000–130,000 live births. The predominant skeletal manifestations in affected individuals are shortened limbs and ribs causing a reduced thoracic capacity, with occasional polydactyly and brachydactyly.9,18 Although SRPS forms are inevitably lethal perinatally because of cardiorespiratory failure, survival rates are 40%–80% for JATD cases.7,19 However, affected individuals who survive may develop extraskeletal ciliopathy features affecting the kidney, eye, or more rarely the liver later in life.20,21 Defective rhodopsin transport via the connecting cilium in retinal photoreceptor cells is proposed as the basis for the development of retinal dystrophy.22 The disease mechanism underlying the renal phenotype, observed in approximately 20% of all JATD cases,15,19 has not been determined but might not depend primarily on hedgehog signaling.23 Mutations in genes encoding components of the IFT system have been implicated as causing JATD. Predominantly skeletal phenotypes are caused by mutations in DYNC2H1 (MIM 603297)7,11,19 encoding the IFT dynein-2 retrograde ATPase/motor heavy chain dynein,5 accounting for more than a third of cases, and also mutations in IFT80 (MIM 611177) encoding an IFT-B anterograde transport complex component.16,24 Mutations in IFT139 (TTC21B [MIM 612014]), IFT140 (MIM 614620), and WDR19 (IFT144 [MIM 608151]), encoding components of the IFT-A retrograde transport complex, are associated with skeletal phenotypes in addition to renal insufficiency.13–15,25 IFT140 mutations also appear to be frequently connected to retinal phenotypes.14,15 In addition, we have described mutations in both JATD and SRPS individuals in WDR60, which encodes a WD40 domain-containing protein that has recently been implicated in IFT in Chlamydomonas reinhardtii and planaria.12,26
We performed whole-exome sequencing (WES) on 61 unrelated individuals with a primary diagnosis of Jeune syndrome based on classical clinical and radiological findings, mainly short ribs with small/narrow thorax and small ilia with acetabular spurs, by using 3–5 μg of genomic DNA and Agilent V2 or Truseq whole-exome kits as previously described.7,12,15,27 All samples were obtained with the approval of the UCL-ICH/Great Ormond St. Hospital (08/H0713/82) (54 samples) and the University of Queensland (UQ; #2011000876) (7 samples) Research Ethics Committees, with informed consent. Analysis entailed sequencing of enriched libraries as 75 base paired-end reads (Illumina HiSeq), removing reads that failed quality filtering via the Illumina GA Pipeline, and mapping those reads that passed filtering to human genome reference (UCSC Genome Browser hg19) by using BWA v.0.5.9r16.28 Realignment around known indels from the 1000 Genomes Pilot study29 and recalibration of base quality scores was performed with GATK 1.1.5.30 Variant calling and independent filtering were performed with SAMtools mpileup v.0.1.1731 and GATK UnifiedGenotyper v.1.3.31,32 merging these callsets. Exome variant profiles were filtered to remove all variants occurring in dbSNP132, the 1000 Genomes, and NHLBI Exome project with a minor allele frequency >0.5% for UCL samples as described7,12,15,27 by means of EVAR software tool v.0.2.2 beta, and a quality score equivalent to a base error rate <0.1%. From the remaining variants, those affecting protein coding regions (nonsynonymous changes and indels) and known splice sites up to 15 base pairs away from the exon-intron boundary were then filtered against an in-house exome database to remove variants occurring with a frequency of >0.5%. Remaining biallelic variants were prioritized according to their presence in cilia proteome databases and mutation type. Coverage of the genes known to cause JATD when deficient was manually analyzed with Integrated Genome Viewer (IGV), and copy-number variants were also analyzed with the ExomeDepth program.33 WES of the Australian samples, quality control, and filtering against previously reported variants followed a very similar protocol12 with exclusion of previously reported variants from both external and internal databases with minor allele frequency (MAF) >0.1%; further filtering was performed according to the observed Mendelian inheritance pattern, functional annotation, genomic conservation, and presence in the cilia proteome databases.
This analysis revealed biallelic, probably damaging variants in WDR34 (MIM 613363; RefSeq accession number NM_052844.3) in five UCL cases and in a sixth case from Australia (SKDP-78.3), who had consanguineous parents. None of the total of ten affected individuals in these six families survived beyond infancy. We subsequently analyzed an additional 52 UCL-ascertained affected individuals with the clinical diagnosis of JATD, via a targeted next-generation sequencing (NGS) replication panel of selected JATD candidate genes. This revealed an additional four individuals with JATD from three families (UCL-120, UCL-123, and UCL-130) carrying biallelic variants in WDR34 predicted to be damaging.
The clinical details of all 14 individuals from the total of 9 JATD families harboring biallelic WDR34 mutations are summarized in Table 1, and representative examples of their JATD phenotypes are shown in Figure 1. Although most individuals did not survive past the neonatal period because of a severe respiratory phenotype, and therefore we cannot predict whether they would have developed renal, hepatic, or retinal involvement later in the disease course, individual UCL-123.1 is currently 8 years of age and has been diagnosed with rod-cone dystrophy, indicating that mutations in WDR34 may lead to retinal degeneration. A total of 11 different WDR34 variants were found in these families (Table 2) and were validated and confirmed to segregate appropriately within families by Sanger sequencing (Figure S1 available online) where DNA of other family members was available. The variants comprised eight predicted missense changes, one frameshift, one nonsense, and one essential donor splice site mutation. Prediction of the functional effects of these mutations was performed with PolyPhen-2 and Mutation Taster (Table 2). Most variants were predicted to be damaging and deleterious to protein function by these programs, respectively, but the homozygous c.616C>T (p.Arg206Cys) variant (rs148543026, MAF 0.123% in the NHLBI EVS control exome database) detected in both affected individuals from UCL family 120 was predicted to be benign by both programs, although it results in a substitution at the start of the first WD40 domain of WDR34 (Figure 2A). In keeping with previous reports that Jeune syndrome cases are presumed hypomorphs,7,15 no affected individuals were found to carry two predicted null alleles. The six homozygous variants were restricted to consanguineous unions and probands from an ethnic minority. Ten variants were private, and one missense change (c.1177G>A [p.Gly393Ser]) was shared between two families of Turkish origin. This variant was confirmed to be absent from 100 Turkish control chromosomes and thus is unlikely to be a polymorphism. A homozygous missense variant identified in one consanguineous family, UCL-101 (c.65C>T [p.Ala22Val]), occurs with a MAF of 0.6% in NHLBI EVS and is therefore unlikely to be deleterious to protein function by itself. We also identified a homozygous missense variant in this family in DYNC2H1 reported in NHLBI EVS with a similar MAF of 0.6% (rs103107198). Although highly speculative, it is possible to suggest that these mutations could act together in trans to cause Jeune syndrome. Digenic inheritance has previously been proposed for Jeune syndrome and the short-rib polydactyly syndrome associated with DYNC2H1 mutations.34,41 The affected individuals in families UCL-57 and UCL-101 were also heterozygous for predicted damaging missense changes in WDR19 and IFT140, respectively (WDR19: c.2720C>T [p.Ala907Val], PolyPhen-2 possibly damaging [0.614], Mutation Taster damaging [0.99]; and IFT140: c.4058C>G [p.Pro1353Arg], PolyPhen-2 possibly damaging [0.839], Mutation Taster disease causing [0.99]). However, the significance of these variants for the phenotype expression is again unknown.
Table 1.
Summary of Clinical Findings in WDR34 Individuals
| Consanguinity | Affected Person(s) | Mutation | Thorax | Polydactyly | Other Skeletal Features | Renal Phenotype | Liver Phenotype | Other Remarks |
|---|---|---|---|---|---|---|---|---|
| Family UCL-102 (Turkish) | ||||||||
| yes | 102.5 | c.1177G>A (p.Gly393Ser) homozygous | long and narrow thorax, short horizontal ribs | no | short long bones, trident acetabulum with spurs | no (infant) | no (infant) | died aged 4 months |
| 102.1a | died | |||||||
| Family UCL-104 (Turkish) | ||||||||
| no | 104.4 | c.1177G>A (p.Gly393Ser) heterozygous; c.1541_1542delCA (p.Thr514Argfs∗11); heterozygous | short horizontal ribs | no | short long bones, brachydactyly, mild bowing of femora and humeri, squared iliac wings and irregular sciatic notches, handlebar clavicles | no | no | fetus |
| 104.1 | fetus | |||||||
| Family UCL-57 (Turkish) | ||||||||
| yes | 57.1 | c.1169C>T (p.Pro390Leu) homozygous | short horizontal ribs | no | trident acetabulum with spurs | bilateral nephrocalcinosis but no cysts (infant) | no (infant) | died |
| Family UCL-60 (Australian) | ||||||||
| no | 60.1 | c.443G>T (p.Cys148Phe) heterozygous; c.1372+1G>A heterozygous | narrow bell-shaped thorax; short horizontal ribs with cupped ends | no | rhizomelic long bone shortening, brachydactyly, broad thumbs and toes, trident acetabulum with squared hypoplastic iliae and acetabular spurs | no (infant) | no (infant);protruding abdomen with ptosis of liver and spleen | polyhydramnios, died aged 46 days |
| Family SKDP-78 (Arab) | ||||||||
| yes | SKDP78.3 | c.1339C>T (p.Arg447Trp) homozygous | short horizontal ribs | no | short long bones (observed at 22 and 25 weeks gestation), trident acetabulum with spurs | no (neonate) | no (neonate) | born at 34 weeks gestation (polyhydramnios), elevated csf spaces, foot malformation, umbilical hernia, died aged 2 days due to respiratory insufficiency |
| SKDP78.1 | narrow thorax, short ribs | no | short extremities | no (neonate) | no (neonate) | died immediately after birth due to respiratory insufficiency | ||
| Family UCL-101 (Turkish) | ||||||||
| yes | 101.2 | c.65C>T (p.Ala22Val) homozygous; rs201715229, MAF 0.6% | short horizontal ribs | no | trident acetabulum with spurs | no (infant) | no (infant) | died in infancy |
| 101.1 | ||||||||
| Family UCL-120 (German, ethnic minority) | ||||||||
| ethnic minority | 120.1 | c.616C>T (p.Arg206Cys) homozygous | short ribs and narrow thorax | yes | trident acetabulum with spurs | no (8 years old) | no | |
| 120.4 | no | no (infant) | no (infant) | severe respiratory distress, recurrent infections, home ventilation | ||||
| Family UCL-123 (Russian) | ||||||||
| no | 123.1 | c.472C>T (p.Gln158∗) heterozygous; c.1307A>G (p. Lys436Arg) heterozygous | short ribs | no | trident acetabulum with spurs | no (8 years old) | no | rod-cone dystrophy, cryptorchidism, obesity, speech and language delay |
| Family UCL-130 (Pakistani) | ||||||||
| yes | 130.1 | c.1229G>T (p.Ser410Ile) homozygous | narrow thorax with short ribs | no | trident acetabulum with spurs | no (8.5 years old) | no | ERG at lower limit of normal |
UCL-102.1 was previously reported as affected individual 5 in Tüysüz et al.18
Figure 1.
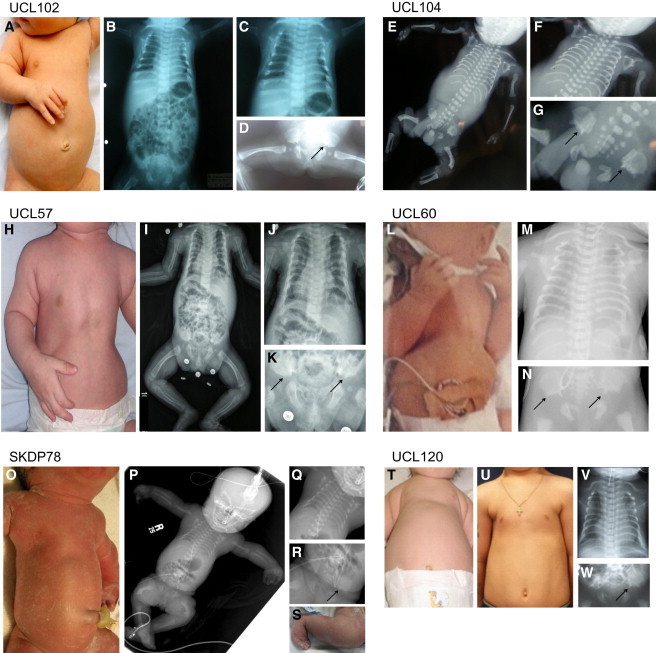
Clinical Features of WDR34 Jeune Syndrome Cases
UCL102.1 (A–D), fetus UCL104.1 (E–G), UCL57.1 (H–K), UCL60.1 (L–N), SKDP78.3 (O–S), and UCL120.1 (T–W) presented with a protruding abdomen and narrow thorax (A, E, H, L, O, T), short horizontal ribs (B, C, F, I, J, M, P, Q, V), and acetabular spurs (as indicated by arrows) (D, G, K, N, R, W). Handlebar clavicles were also observed (C, F, J, M, V). Fetuses UCL104.1 (E) and SKDP78.3 (P) also showed severely shortened femora. SKDP78.3 additionally presented with a foot malposition after birth (S). Individual UCL120.1 presented with a narrow thorax and short ribs at birth (T, V), with indications of improvement with age (U, at 6.5 years of age).
Table 2.
Details of Identified WDR34 Mutation
| Consanguinity | Mutation | Exon | Polyphen2 | Mutation Taster | Protein Domain Affected | Mutations in Other Ciliopathy Genes (from WES Data) |
|---|---|---|---|---|---|---|
|
Family UCL-102 (Turkish) | ||||||
| yes | c.1177G>A (p.Gly393Ser) homozygous | 7 | probably damaging (0.993, HumVar/Div) | disease causing (0.9999) | mutation in WD3 | no |
| Family UCL-104 (Turkish) | ||||||
| no |
c.1177G>A (p.Gly393Ser) heterozygous | 7 | probably damaging (0.993, HumVar/Div) | disease causing (0.999) | mutation in WD3 | no |
| c.1541_1542delCA(p.Thr514Argfs∗11) heterozygous | 9 | - | disease causing (1.0) | mutation in WD5 | ||
|
Family UCL-57 (Turkish) | ||||||
| yes | c.1169C>T (p.Pro390Leu) homozygous | 7 | probably damaging (0.926, HumVar/Div) | disease causing (0.999) | mutation in WD3 | WDR19 c.2720C>T heterozygous |
| Family UCL-60 (Australian) | ||||||
| no |
c.443G>T (p.Cys148Phe) heterozygous | 1 | benign (0.114, HumVar/Div) | disease causing (0.999) | not in specific domain | no |
| c.1372+1G>A heterozygous | intron 8 | - | disease causing (1.0) | mutation in WD4 | ||
| Family SKDP-78 (Arab) | ||||||
| yes | c.1339C>T (p.Arg447Trp) homozygous | 8 | probably damaging (0.998, HumVar/Div) | disease causing (0.999) | mutation in WD4 | no |
| Family UCL-101 (Turkish) | ||||||
| yes | c.65C>T (p.Ala22Val) homozygous; rs201715229, MAF 0.6% | 1 | benign (0.3304, HumVar); probably damaging (0.969, HumDiv) | polymorphism (0.94); rs201715229 | not in specific domain | DYNC2H1 rs103107198 (MAF 0.6%) homozygous; IFT140 c.4058C>G heterozygous |
| Family UCL-120 (German, ethnic minority) | ||||||
| ethnic minority | c.616C>T (p.Arg206Cys) homozygous | 4 | benign (0.001, HumVar/Div) | polymorphism; rs148543026; MAF 0.123% in NHLBI EVS | beginning of WD1 | |
| Family UCL-123 (Russian) | ||||||
| no |
c.472C>T (p.Gln158∗) heterozygous | 3 | - | disease causing (1.0) | not in specific domain | |
| c.1307A>G (p. Lys436Arg) heterozygous |
8 |
benign (0.005, HumVar/Div) |
disease causing (0.973) |
mutation in WD4 |
||
| Family UCL-130 (Pakistani) | ||||||
| yes | c.1229G>T (p.Ser410Ile) homozygous | 8 | possibly damaging (0.685, HumVar/Div) | disease causing (0.999) | mutation in WD3 | |
Figure 2.
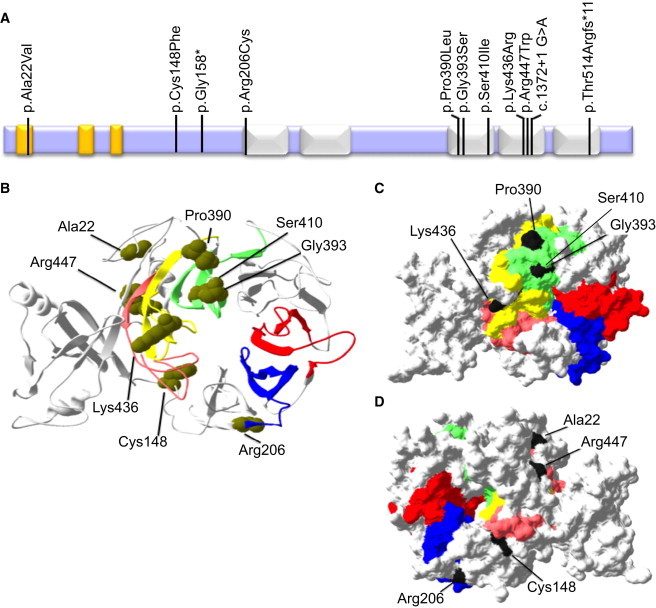
Modeling the Effect of Mutations on WDR34 Protein Structure
(A) Predicted structure of the WDR34 protein with five WD40 domains (white) and three low-complexity regions (yellow), showing the location of the 11 mutations identified in this study.
(B) 3D-structural wild-type WDR34 model predicted by I-TASSER showing the predicted protein has a beta propeller structure formed by the five WD40 repeat sequences. The domains are colored as follows: WD40 aa 207–246, blue; WD40 aa 255–299, red; WD40 aa 381–421, green; WD40 aa 424–464, pink; and WD40 aa 471–511 yellow. Six missense substitutions affect residues (Arg206, Pro390, Gly393, Ser410, Leu436, and Arg447) in the WD40 domains and one alters a residue (Cys148) that lies in close proximity within the 3D structure.
(C) WD40 domains colored as in (B), showing the location of the Pro390, Gly393, Ser410, and Leu436 residues on the molecular surface of the beta propeller faces of the WDR34 I-TASSER model, suggesting a possible role in protein-protein interaction.
(D) Seen on the opposite side of the molecule, amino acid Arg206 is positioned directly adjacent to WD40 repeat 1 (aa positions 207–246, blue) and Cys148 lies outside of the WD40 domains but is predicted to be close by based on the three-dimensional structure, suggesting a possible role for these mutations in protein-protein interaction. WD40 domains colored as in (B).
WDR34 is a predicted multiple WD40 domain-containing protein and six of the eight putative missense substitutions identified occur within its WD40-repeat domains, although the other two, p.Ala22Val and p.Cys148Phe, are in regions without domain prediction (Figure 2A). The predicted effect of the putative mutations on WDR34 protein structure was modeled by I-TASSER,35 which showed that the “faces” of the five WD40 domains form a beta propeller site for protein-protein interactions (Figure 2B). 3D-protein modeling predicts that four missense substitutions (p.Pro390Leu, p.Gly393Ser, p.Ser410Ile, and Leu436Arg) are located in close proximity on the molecular surface of one of the beta propeller faces, suggesting a possible role in protein-protein interaction (Figures 2B and 2C). The missense substitutions p.Ala22Val, p.Cys148Phe, p.Arg206Cys, and p.Arg447Trp are close by within the 3D structure (Figures 2B and 2D), and all missense substitutions occur in this central portion of WDR34, close to or at the surface of the protein (Figures 2C and 2D). Furthermore, the missense substitutions affect highly conserved residues, the only exception being p.Arg206Cys, which is not well conserved (Figure S2).
WDR34 has previously been described to inhibit the NF-κB pathway component TAK1 in cell culture.36 Its function during mammalian development has not been investigated, but in zebrafish, knockdown of the wdr34 homolog dync2i1 leads to a typical ciliopathy phenotype.37 Furthermore, WDR34 is the human homolog of Chlamydomonas reinhardtii FAP133, an intermediate dynein chain that forms part of the retrograde IFT-dynein complex.6 We proceeded to investigate whether it is similarly part of the IFT dynein-2 complex in mammals, because this has not previously been determined. We first studied its behavior in murine cells (mouse embryonic fibroblasts, ATDC5 chondrocyte precursor cells, and inner medullary collecting duct [IMCD3] cells), in addition to normal human fibroblasts (Life Technologies, Sigma). These were all cultured under standard conditions at 37°C and 5% CO2 in DMEM-F12 Glutamax medium (Life Technologies, Invitrogen) with 10% fetal bovine serum FBS (Life Technologies, Invitrogen), followed by a 24 hr period of serum starvation (0.2% FBS) to induce ciliogenesis. High-resolution immunofluorescence analysis of WDR34 costained for markers of the axoneme (acetylated tubulin) or basal bodies/centrioles (gamma tubulin or pericentrin) revealed prominent staining of WDR34 around the basal bodies in ciliated human fibroblasts (Figure 3A), with occasional axonemal staining (Figure 3B). In non-serum-starved mouse embryonic fibroblasts (Figure 3C) and IMCD3 cells (Figure S3), WDR34 colocalized with the centrioles, and its staining also extended around the centrioles in a punctate vesicular pattern as shown by costaining with gamma tubulin during different cell cycle stages (Figure S3). In ciliated IMCD3 cells, WDR34 was localized to both centrioles, often in an asymmetric pattern with more intense staining around one centriole (Figure S3). In some cells, especially human fibroblasts, WDR34 staining was additionally observed at the nuclear membrane (Figure 3D). To confirm that the antibody recognizes WDR34, we preincubated the antibody with an excess of purified recombinant myc-tagged WDR34 protein prior to immunofluorescence analysis. This abrogated the staining pattern described above, with no observed colocalization around the basal body marker pericentrin (Figure S4). In addition, we were able to detect recombinant protein by immunoblotting (Figure S5), although this antibody was not capable of detecting specific bands of the predicted size for endogenous WDR34 in cell lysates (data not shown).
Figure 3.
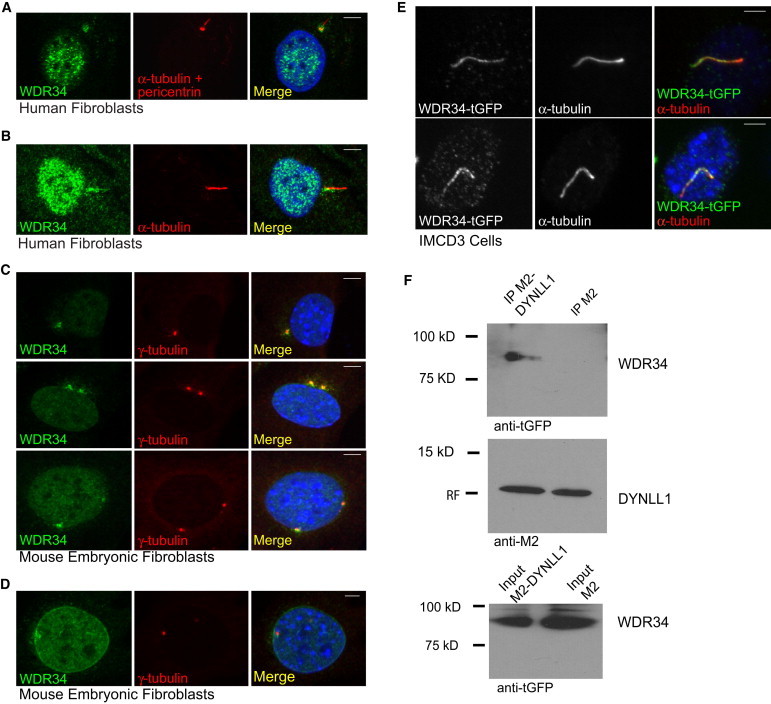
Centrosomal and Ciliary Localization of WDR34 Protein
(A and B) Serum-starved control human fibroblasts stained with polyclonal rabbit anti-WDR34 antibody (1:100, HPA0407641, Sigma) (green), compared to mouse monoclonal antibodies for anti-acetylated tubulin (1:1000, IgG2b, clone 6-11B-1, Sigma) and anti-pericentrin (IgG, mAmbcam28144, Abcam) antibodies to stain cilia and basal bodies/centrioles (red), respectively, counterstained with DAPI (blue). WDR34 localizes around the basal body (A) and is occasionally also detected at the ciliary axoneme (B).
(C) Centriolar colocalization of WDR34 (green) with gamma tubulin (1:200 IgG1 clone GTU-88, Sigma, red) in mouse embryonic fibroblast cells (MEFs) counterstained with DAPI (blue).
(D) In mouse embryonic fibroblasts, WDR34 staining (green) could also be detected at the nuclear membrane during interphase compared to gamma tubulin (red) and DAPI (blue), whereas general nuclear staining was observed throughout all cell cycle phases (A–D).
(E) Murine IMCD3 cells with low levels of exogenous human WDR34-tGFP expression (TrueORF WDR34 clone from Origene) show localization of the protein to the axoneme. For overexpression studies, cells were serum starved for 24 hr beginning 24 hr after transfection and stained with anti-turboGFP (1:2,000, #AB513, Evrogen). For immunofluorescence analysis, secondary antibodies used were goat anti-mouse IgG1 Alexa Fluor 647, goat anti-mouse IgG1 Alexa Fluor 568, goat anti-rabbit Alexa Fluor 488, and goat anti-rabbit Alexa Fluor 568.
Scale bars represent 5 μm in (A)–(E).
(F) WDR34 interacts with DYNLL1 in vitro, as shown by coimunoprecipitation. HEK293T cells were transfected with plasmids encoding tGFP-tagged WDR34 (Origene) and either Flag-tagged DYNLL1 (pEF-Flag-DYNLL145) (left) or a Flag-only control plasmid (right) using Fugene-6 reagent (Promega). Cells were lysed with nondenaturing cell lysis buffer (Cell Signaling Technology). For IP lanes (top and middle), the lysate was incubated with anti-Flag M2-beads (Sigma) and beads were washed according the manufacturers’ protocol. After blotting, the membrane was blocked for 1 hr in 5% skimmed milk then incubated with anti-tGFP antibody 1:500 (Origene) (top) or anti-M2-Flag antibody (Sigma) (middle); the secondary was an HRP-coupled anti-mouse antibody (Cell Signaling Technology). The top panel shows coprecipitation of WDR34-tGFP in the DYNLL1-Flag lane but not in the Flag-only control lane. Middle panel: Both DYNLL1-Flag and Flag only were present in the IP reactions. Bottom panel: WDR34-tGFP input was similar for both the DYNLL1-Flag and the Flag-only reaction.
Because most of the individuals carrying mutations in WDR34 did not survive past infancy and no samples were available for further functional analysis, we generated stable murine chondrocyte precursor (ATDC5) Wdr34 knockdown cell lines by using lentiviral-based shRNAs. We produced four lentiviruses expressing different shRNAs directed against Wdr34, in addition to a control nonsilencing shRNA, by using the OpenBiosystems pGIPZ mouse shRNA library: Wdr34shRNA1: V2LMM_104963, pGIPZ RMM4431-98763533; Wdr34shRNA2: V2LMM_74890, pGIPZ RMM4431-98973537; Wdr34shRNA3: V3LMM_423739, pGIPZ RMM4431-101258631; Wdr34shRNA4: V3LMM_423740, pGIPZ RMM4431-101266083; control shRNA (nonsilencing), pGIPZ RHS4480. Murine chondrocyte precursor cells (ATDC5) were transduced with the lentiviral particles according to the manufacturer’s protocol at an MOI of 5 with an equivalent transduction efficiency controlled for by GFP expression from the shRNA plasmids. Stably transduced lines were generated after 3 weeks of 3 μg/ml puromycin selection. Consistent with the WDR34 staining pattern observed in other cell types, the staining in ATDC5 cells was concentrated around the basal body area. In two cell lines created with two of the four independent Wdr34 shRNAs (Wdr34shRNA1 and Wdr34shRNA4), a 70%–80% level of Wdr34 transcript knockdown was achieved, as verified by real-time PCR (Figure S6A). High-resolution immunofluorescence analysis with the WDR34 antibody also revealed an apparent reduction in staining in these two cell lines, with a specific decrease in the peribasal staining (Figures S6B–S6E).
In contrast to the staining pattern we observed for endogenous human and mouse WDR34 (Figures 3A–3C), overexpression of turboGFP (tGFP)-tagged human WDR34 in IMCD3 cells, followed by detection with an anti-tGFP antibody, resulted in a mainly axonemal and faint nuclear staining in low-expressing cells (Figure 3E), with ubiquitous staining of the entire cell seen in high-expressing cells (data not shown). In contrast to this GFP antibody, the WDR34 antibody was not able to detect axonemal GFP-WDR34 in low-level overexpressing cells but detected the overexpressed protein in the highly expressing cells (data not shown), indicating that the lack of axonemal staining observed with this antibody might reflect an antibody sensitivity issue. Alternatively, it is possible that the epitope recognized by this antibody is masked in the axonemal pool of WDR34, as has been proposed for other ciliary proteins.38 However, because axonemal proteins are dynamically organized, it is also possible that the axonemal localization of endogenous WDR34 might depend on specific stimuli or activation processes. For example, Smoothened, an integral component of the hedgehog pathway, can normally be detected in the cilium only after activation of Hh signaling with the chemical agonist SAG or Hh-conditioned medium, but overexpression leads to axonemal localization regardless of activation of the pathway.39,40 We did not detect significant amounts of overexpressed GFP-tagged WDR34 at the ciliary base or around the centrioles, potentially suggesting that the peribasal body localization might be hampered by the GFP tag.
The localization of WDR34 around the ciliary base, as well as around centrioles and spindle poles in dividing cells, parallels the mainly peribasal body localization of its Chlamydomonas homolog FAP133, with only small amounts of FAP133 detected within the axonemal flagella fraction.6 It is known that ciliary targeting of Chlamydomonas FAP133 depends on the IFT dynein heavy chain homolog of human DYNC2H16 and we therefore investigated whether WDR34 localization is changed in fibroblasts from two affected JATD individuals previously published7 and one (more severely affected) SRPS individual carrying biallelic DYNC2H1 mutations. Individual 001 (JATD-2 in Schmidts et al.7) carries a homozygous missense substitution in the microtubule binding stalk domain of DYNC2H1 (c.9044A>G [p.Asp3015Gly]), whereas individual 26 (JATD-3 in Schmidts et al.7) harbors two heterozygous substitutions affecting AAA domain 3 and 5 of DYNC2H1 (c.9817C>T [p.Glu3273∗] and c.7442G>A [p.Arg2481Gln]). Individual 35 affected with SRPS harbors a heterozygous nonsense substitution in DYNC2H1 (c.536G>A [p.Trp179∗]) in the N terminus, in addition to two missense changes (c.8726G>T [p.Cys291Phe] in proximity of the DHC-N1 domain and c.10343T>C [p.Cys3448Pro] affecting the 5th AAA domain). We were not able to detect any differences in WDR34 localization in any of the DYNC2H1-deficient cells compared to human control fibroblasts (Figure S7). These data do not prove that DYNC2H1 is dispensable for WDR34 targeting to the base of the cilium, because it could be that the particular DYNC2H1 mutations in these three individuals may not specifically affect interactions with WDR34, in contrast to the Chlamydomonas FAP133-null mutation, possibly reflecting their presumed hypomorphic nature. Furthermore, because we are not able to detect significant amounts of endogenous axonemal WDR34 with the commercial antibody used, it remains unclear whether axonemal localization of WDR34 might be affected in these DYNC2H1-deficient cells.
Reduced cilia number and/or length has previously been reported in ciliary chondrodysplasias such as SRPS,12,13,41 but we found that the proportion of ciliated cells and cilia length were not significantly affected in any of the Wdr34-knockdown ATDC5 lines we generated, including lines 1 and 4 that had 70%–80% reduced Wdr34 expression (Figures S8A–S8D). Staining for mitotic cells with a phosphorylated-histone H3 (PH3) marker of cell proliferation was also unchanged (Figures S8E–S8H). These results suggest that at the achieved level of knockdown, WDR34 is not sufficient to result in a ciliogenesis defect, similarly to results reporting no loss or malformation of cilia in the Ift80-deficient hypomorphic Jeune syndrome mouse model where it is considered that a low level of Ift80 expression allows ciliogenesis but does not rescue hedgehog signaling defects.16 Also, we have previously observed normal cilia number and length in fibroblasts from JATD-affected individuals with DYNC2H1 mutations but found that retrograde IFT is impaired in those cells;7 however, we did not observe any significant accumulation of IFT88 particles at the ciliary tip indicative of a retrograde IFT defect in Wdr34 knockdown ATDC5 cells in comparison to controls (Figures S8I–S8N). This suggests either that WDR34 is not essential for retrograde IFT of IFT88, or, more likely, that the knockdown levels were insufficient to cause a phenotype.
To further investigate the cellular role of WDR34, we developed a computer-based interaction network for the protein, prompted by the findings from high-throughput affinity capture-MS that have identified WDR34 as a direct interaction partner of the cytoplasmic dynein-1 light chain DYNLL1 (also known as LC8 [MIM 601562]) in humans.42 Although only the IFT dynein-2 complex enters the ciliary axoneme in mammals, both cytoplasmic dynein-1 and IFT dynein-2 complexes are found at the centrioles. Dynein-1 directs motor-based movement of cytoplasmic cargos along microtubules, which plays an important role during mitosis; mutations in genes encoding proteins of the dynein-1 complex such as DYNC1H1 (MIM 600112) cause severe congenital neuronal phenotypes in humans associated with deficient cytoplasmic microtubular transport.43 We derived data from BioGRID v.3.2.9844 and manual input, visualizing connections with Cytoscape v.3.0.0.,42 to generate a model of the putative interactions of WDR34 with the dynein-1 and dynein-2 complexes. This shows all human dynein proteins and their neighbors (Figure S9A), as well as interactions between the human dynein proteins and WDR34 in first vertex degree with nodes and edges colored to highlight WDR34 (green), DYNLL1 (red), the other dynein proteins (orange), as well as the putative association between WDR34 and DYNC2H1 (blue) (Figure S9B). To provide further experimental evidence for an interaction between WDR34 and DYNLL1 in mammalian cells, we overexpressed both GFP-WDR34 and pEF-Flag-DYNLL145 in HEK293T cells, immunoprecipitated with a FLAG antibody and immunoblotted with either GFP or FLAG antibodies to detect WDR34 or DYNLL1, respectively. This revealed coimmunoprecipitation of DYNLL1 and WDR34 (Figure 3F), confirming that both proteins are part of the same mammalian protein complex in vitro. These results are in agreement with previous studies on Chlamydomonas flagella proteins, showing that the WDR34/FAP133 associates with DYNLL1/LC8.6
This interaction data combined with the network described above support the proposed role of WDR34 in the IFT dynein-2 complex, in addition to potentially linking WDR34 to components of the cytoplasmic dynein-1 complex, via DYNLL1. Interestingly, another WD repeat-containing Chlamydomonas flagellar protein, FAP163, the homolog of WDR60,12 is a dynein intermediate chain required for retrograde intraflagellar transport that is closely related to FAP133.26 The staining pattern we detected here for WDR34 is similar to that reported for WDR60, and it is possible that WDR34 and WDR60 belong to the same protein complex; however, accurate characterization of the spatiotemporal organization of such a complex remains to be determined. The WDR34 staining observed at the nuclear membrane in some cells (Figure 3D) potentially links WDR34 function with the cell cycle because components of the dynein-1 complex are known to play a role in nuclear envelope breakdown during mitosis.46 Although DYNLL1 is thought to be associated with both dynein-1 and dynein-2 complexes and mutations in the dynein-1 complex component TCTEX1 (MIM 601554) have been shown to result in a ciliary phenotype,47 to our knowledge no mutation has been previously identified in a gene encoding a component associated with both the dynein-1 and IFT dynein-2 motor complexes.
This evidence that WDR34 may have broader roles beyond that as a component of the IFT dynein-2 retrograde motor is consistent with previous work showing that it also acts as inhibitor of TAK1 (MIM 601426), a component of the NF-κB pathway in mammalian cells.36 TAK1 influences multiple pathways such as TGF-β, p38-MAP-kinase, and IKK, and TGF-β in turn can act as a cilia-independent hedgehog pathway activator by directly activating Gli transcription factors.48 Therefore, although it seems likely that WDR34 functions within the IFT dynein-2 and/or IFT-A complex to influence IFT-mediated hedgehog signaling, it is also conceivable that WDR34 influences signaling via TGF-β independently of its ciliary role.
In summary, here we describe mutations causing Jeune asphyxiating thoracic dystrophy in WDR34, which encodes the homolog of the FAP133 WD40-containing dynein intermediate chain associated with the retrograde intraflagellar transport motor in Chlamydomonas. In this study the mutational survey indicates that mutations in WDR34 may account for up to 10% of all JATD cases, which would make it the second most commonly mutated gene after DYNC2H1 in this disease.7,19 Our evidence based on its subcellular localization and protein interactions supports a role for WDR34 as a component of the vertebrate dynein-IFT machinery, potentially as a previously unrecognized subunit of the mammalian IFT dynein-2 complex. We also find that WDR34 might be involved in the dynein-1 complex not commonly associated with human ciliopathies. Together, these findings are important for the clinical genetics setting, in addition to implicating an additional player in the complex IFT machinery, thereby providing a better understanding of the mammalian dynein complexes in broader cilia biology and disease.
Acknowledgments
We are very grateful to the families for their participation in this study and to all participating physicians. We thank Sharon Song and Lisa Anderson for technical support (exome capture, sequencing) and Ideke J.C. Lamers (cell biology). We thank ESDN, Andrea Superti-Furga, and Sheila Unger (University of Lausanne) in supporting diagnoses of individual SKDP-78.3. We thank Lotte Bang Pedersen (University of Copenhagen) and Dominic Norris (MRC Harwell, UK) for very helpful discussions and Hamsa Puthalakath (La Trobe University, Australia) for the kind gift of pEF-Flag-DYNLL1. This study makes use of data generated by the UK10K Consortium who we thank, in particular Matthew Hurles and participants of the UK10K RARE group. Funding for UK10K was provided by the Wellcome Trust under award WT091310. A full list of investigators who contributed to the generation of the data is available from http://www.uk10k.org/publications.html. This study was supported by Dutch Kidney Foundation grants CP11.18 (M.S., P.L.B., H.H.A.) and IP11.58 (H.H.A.), the European Community’s Seventh Framework Programme FP7/2009; 241955, SYSCILIA (P.L.B.), the Netherlands Organization for Scientific Research NWO Veni-91613008 (H.H.A.), and NIHR (Moorfields Eye Hospital BRC) (A.T.M.). Confocal microscopy at UQ was carried out at the Institute for Molecular Bioscience Dynamic Imaging Facility for Cancer Biology, developed with the generous support of the Australian Cancer Research Foundation. P.J.S. is supported by the British Heart Foundation, P.L.B. is a Wellcome Trust Senior Fellow, C.W. is a University of Queensland (UQ) Vice-Chancellor’s Senior Research Fellow, C.R.C. is supported by a UQ International PhD Scholarship, A.M.M.-L. is supported by a University of Queensland postgraduate scholarship, M.A.B. is supported by an NHMRC senior fellowship, and M.S. is supported by an Action Medical Research UK Clinical Training Fellowship (RTF-1411). H.M.M. is supported by Action Medical Research UK and the Newlife Foundation for Disabled Children.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Miriam Schmidts, Email: miriam.schmidts.10@ucl.ac.uk.
Hannah M. Mitchison, Email: h.mitchison@ucl.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
FEVA (Family-Based Exome Variants Analysis), http://www.exome.info
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UK10K Consortium, http://www.uk10k.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Goetz S.C., Anderson K.V. The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pedersen L.B., Veland I.R., Schrøder J.M., Christensen S.T. Assembly of primary cilia. Dev. Dyn. 2008;237:1993–2006. doi: 10.1002/dvdy.21521. [DOI] [PubMed] [Google Scholar]
- 3.Liem K.F., Jr., Ashe A., He M., Satir P., Moran J., Beier D., Wicking C., Anderson K.V. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol. 2012;197:789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ocbina P.J., Eggenschwiler J.T., Moskowitz I., Anderson K.V. Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mikami A., Tynan S.H., Hama T., Luby-Phelps K., Saito T., Crandall J.E., Besharse J.C., Vallee R.B. Molecular structure of cytoplasmic dynein 2 and its distribution in neuronal and ciliated cells. J. Cell Sci. 2002;115:4801–4808. doi: 10.1242/jcs.00168. [DOI] [PubMed] [Google Scholar]
- 6.Rompolas P., Pedersen L.B., Patel-King R.S., King S.M. Chlamydomonas FAP133 is a dynein intermediate chain associated with the retrograde intraflagellar transport motor. J. Cell Sci. 2007;120:3653–3665. doi: 10.1242/jcs.012773. [DOI] [PubMed] [Google Scholar]
- 7.Schmidts M., Arts H.H., Bongers E.M., Yap Z., Oud M.M., Antony D., Duijkers L., Emes R.D., Stalker J., Yntema J.B., UK10K Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 2013;50:309–323. doi: 10.1136/jmedgenet-2012-101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taschner M., Bhogaraju S., Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83:S12–S22. doi: 10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber C., Cormier-Daire V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:165–174. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 10.Ho N.C., Francomano C.A., van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am. J. Med. Genet. 2000;90:310–314. doi: 10.1002/(sici)1096-8628(20000214)90:4<310::aid-ajmg9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 11.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McInerney-Leo A.M., Schmidts M., Cortés C.R., Leo P.J., Gener B., Courtney A.D., Gardiner B., Harris J.A., Lu Y., Marshall M., UK10K Consortium Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am. J. Hum. Genet. 2013;93:515–523. doi: 10.1016/j.ajhg.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidts M., Frank V., Eisenberger T., Al Turki S., Bizet A.A., Antony D., Rix S., Decker C., Bachmann N., Bald M. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum. Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rix S., Calmont A., Scambler P.J., Beales P.L. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum. Mol. Genet. 2011;20:1306–1314. doi: 10.1093/hmg/ddr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haycraft C.J., Zhang Q., Song B., Jackson W.S., Detloff P.J., Serra R., Yoder B.K. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 18.Tüysüz B., Bariş S., Aksoy F., Madazli R., Ungür S., Sever L. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. Am. J. Med. Genet. A. 2009;149A:1727–1733. doi: 10.1002/ajmg.a.32962. [DOI] [PubMed] [Google Scholar]
- 19.Baujat G., Huber C., El Hokayem J., Caumes R., Do Ngoc Thanh C., David A., Delezoide A.L., Dieux-Coeslier A., Estournet B., Francannet C. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J. Med. Genet. 2013;50:91–98. doi: 10.1136/jmedgenet-2012-101282. [DOI] [PubMed] [Google Scholar]
- 20.de Vries J., Yntema J.L., van Die C.E., Crama N., Cornelissen E.A., Hamel B.C. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. Eur. J. Pediatr. 2010;169:77–88. doi: 10.1007/s00431-009-0991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yerian L.M., Brady L., Hart J. Hepatic manifestations of Jeune syndrome (asphyxiating thoracic dystrophy) Semin. Liver Dis. 2003;23:195–200. doi: 10.1055/s-2003-39950. [DOI] [PubMed] [Google Scholar]
- 22.Insinna C., Besharse J.C. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev. Dyn. 2008;237:1982–1992. doi: 10.1002/dvdy.21554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jonassen J.A., SanAgustin J., Baker S.P., Pazour G.J. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 2012;23:641–651. doi: 10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 25.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel-King R.S., Gilberti R.M., Hom E.F., King S.M. WD60/FAP163 is a dynein intermediate chain required for retrograde intraflagellar transport in cilia. Mol. Biol. Cell. 2013;24:2668–2677. doi: 10.1091/mbc.E13-05-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Hokayem J., Huber C., Couvé A., Aziza J., Baujat G., Bouvier R., Cavalcanti D.P., Collins F.A., Cordier M.P., Delezoide A.L. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J. Med. Genet. 2012;49:227–233. doi: 10.1136/jmedgenet-2011-100717. [DOI] [PubMed] [Google Scholar]
- 35.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao D., Wang R., Li B., Yang Y., Zhai Z., Chen D.Y. WDR34 is a novel TAK1-associated suppressor of the IL-1R/TLR3/TLR4-induced NF-kappaB activation pathway. Cell. Mol. Life Sci. 2009;66:2573–2584. doi: 10.1007/s00018-009-0059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krock B.L., Mills-Henry I., Perkins B.D. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest. Ophthalmol. Vis. Sci. 2009;50:5463–5471. doi: 10.1167/iovs.09-3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Follit J.A., Tuft R.A., Fogarty K.E., Pazour G.J. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu V.M., Chen S.C., Arkin M.R., Reiter J.F. Small molecule inhibitors of Smoothened ciliary localization and ciliogenesis. Proc. Natl. Acad. Sci. USA. 2012;109:13644–13649. doi: 10.1073/pnas.1207170109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohatgi R., Milenkovic L., Corcoran R.B., Scott M.P. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA. 2009;106:3196–3201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S.A., von der Haar S., Zenker M., Zahnleiter D., Stöss H., Beinder E. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutchins J.R., Toyoda Y., Hegemann B., Poser I., Hériché J.K., Sykora M.M., Augsburg M., Hudecz O., Buschhorn B.A., Bulkescher J. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harms M.B., Ori-McKenney K.M., Scoto M., Tuck E.P., Bell S., Ma D., Masi S., Allred P., Al-Lozi M., Reilly M.M. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78:1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark C., Breitkreutz B.J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34(Database issue):D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puthalakath H., Huang D.C., O’Reilly L.A., King S.M., Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- 46.Salina D., Bodoor K., Eckley D.M., Schroer T.A., Rattner J.B., Burke B. Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell. 2002;108:97–107. doi: 10.1016/s0092-8674(01)00628-6. [DOI] [PubMed] [Google Scholar]
- 47.Li A., Saito M., Chuang J.Z., Tseng Y.Y., Dedesma C., Tomizawa K., Kaitsuka T., Sung C.H. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat. Cell Biol. 2011;13:402–411. doi: 10.1038/ncb2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hui C.C., Angers S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.