

Abstract
Dravet syndrome is a severe epilepsy syndrome characterized by infantile onset of therapy-resistant, fever-sensitive seizures followed by cognitive decline. Mutations in SCN1A explain about 75% of cases with Dravet syndrome; 90% of these mutations arise de novo. We studied a cohort of nine Dravet-syndrome-affected individuals without an SCN1A mutation (these included some atypical cases with onset at up to 2 years of age) by using whole-exome sequencing in proband-parent trios. In two individuals, we identified a de novo loss-of-function mutation in CHD2 (encoding chromodomain helicase DNA binding protein 2). A third CHD2 mutation was identified in an epileptic proband of a second (stage 2) cohort. All three individuals with a CHD2 mutation had intellectual disability and fever-sensitive generalized seizures, as well as prominent myoclonic seizures starting in the second year of life or later. To explore the functional relevance of CHD2 haploinsufficiency in an in vivo model system, we knocked down chd2 in zebrafish by using targeted morpholino antisense oligomers. chd2-knockdown larvae exhibited altered locomotor activity, and the epileptic nature of this seizure-like behavior was confirmed by field-potential recordings that revealed epileptiform discharges similar to seizures in affected persons. Both altered locomotor activity and epileptiform discharges were absent in appropriate control larvae. Our study provides evidence that de novo loss-of-function mutations in CHD2 are a cause of epileptic encephalopathy with generalized seizures.
Main Text
Epileptic encephalopathies (EEs) are severe, intractable childhood epilepsies with concomitant cognitive impairment and other associated comorbidities. Although a broad range of exogenous factors can lead to EE, in a significant subset of affected individuals the etiology remains unidentified and might be endogenous. Consequently, a genetic cause is assumed.1 Among the various EEs of genetic origin, Dravet syndrome (MIM 607208) has emerged as one of the best-defined phenotypes and the one with the highest mutation detection yield. Children with Dravet syndrome are prone to repetitive and prolonged epileptic seizures in the setting of fever.2 Although these fever-induced seizures start around the age of 6 months (range = 3–16 months),3 other seizure types occur during the course of the disease, and developmental plateauing is typical in the second year of life.
Taking into account SCN1A abnormalities due to single or multiple exons deletions, de novo mutations in SCN1A (MIM 182389) are known to cause Dravet syndrome in ∼75% of affected probands.4 Despite this high probability of identifying a mutation in an individual with a typical phenotype, up to 25% of affected persons do not carry SCN1A mutations, suggesting the involvement of other genes. Mutations in PCDH19 (MIM 300460) can result in a Dravet-syndrome-like phenotype in females and explain some of the cases without an SCN1A mutation.5 For a relevant subset of probands, however, a causative mutation cannot be identified in either gene. We therefore explored the involvement of mutations in additional genes in these probands by using whole-exome sequencing in proband-parent trios. This approach provides the possibility of querying the entire exome for de novo mutations in simplex cases, which has been proven to be successful for intellectual disability, schizophrenia, autism, and some forms of epilepsy or epilepsy-related disorders.6,7
We first studied nine Dravet-syndrome-affected individuals in whom SCN1A sequence mutations or copy-number variations had been excluded. These probands were selected on the basis of broad inclusion criteria, extending the onset age of seizures to an age between 3 months and 2 years of life. Further inclusion criteria consisted of (1) the presence of both febrile and afebrile seizures; (2) multiple seizure types including tonic-clonic, hemiclonic, myoclonic, absence, and/or focal seizures; (3) pharmacoresistant epilepsy at least during childhood or frequent status epilepticus; (4) normal development prior to epilepsy onset, although some minor degree of developmental delay could be present; and (5) slowing or stagnation of development after onset of seizures and absence of epileptogenic lesions on brain MRI.
Signed informed consent was obtained from all study participants or their legal representatives. The local ethical committees of the University of Antwerp and Antwerp University Hospital, University of Kiel, and other collaborating centers approved this study. Genomic DNA was extracted from peripheral blood according to standard procedures.
We performed whole-exome sequencing on genomic DNA of the nine selected individuals with Dravet syndrome and both unaffected parents at the Wellcome Trust Sanger Institute (Hinxton, Cambridgeshire). In brief, genomic DNA (∼3 μg) was fragmented by sonication, and fragments with a length of 150–200 bp were purified. After a paired-end DNA library was prepared from the DNA fragments (with the TruSeq DNA Sample Preparation Kit from Illumina), targeted enrichment was performed with the SureSelect Human All Exon 50Mb Kit (Agilent Technologies). Captured DNA was then sequenced on a HiSeq2000 (Illumina) as paired-end 75 bp reads according to the manufacturer’s protocol. Sequencing reads passing quality filtering were aligned to the human reference genome (hg19, UCSC Genome Browser) with the Burrows-Wheeler Aligner.8 The Genome Analysis Toolkit (GATK)9 was used to recalibrate base quality scores, realign around indels, and mark duplicate reads. Independent variant calling was performed on the mapped reads with SAMtools10 mpileup, GATK UnifiedGenotyper, and Dindel.11 The GenomeComb12 program was used for annotating, comparing, and filtering the data. For de novo variant calling, the DeNovoGear13 program by Conrad and colleagues was used and double-checked by GenomeComb analysis.
Our analysis revealed a heterozygous de novo mutation in CHD2 (MIM 302119; RefSeq accession number NM_001271.3), encoding chromodomain helicase DNA binding protein 2 (CHD2; RefSeq NP_001262.3), in two out of nine probands. One individual carried a nonsense mutation, c.4971G>A (p.Trp1657∗), in exon 38; the second person carried a splice-site mutation, c.1810−2A>C, affecting the splice acceptor site of exon 16. Presence or absence of the mutations was confirmed on genomic DNA of the probands or parents, respectively, by bidirectional Sanger sequencing using the ABI BigDye Terminator v.3.1 cycle sequencing kit on an ABI 3730xl automated DNA Analyzer (Applied Biosystems).
To obtain further genetic evidence of pathogenicity, we performed a mutation analysis of all 39 coding exons and intron-exon boundaries of CHD2 by using bidirectional sequencing (primer sequences are available upon request) in a cohort of 150 EE probands similar to the two individuals carrying a CHD2 mutation: all selected probands had infantile- or childhood-onset epilepsy with subsequent developmental delay. All individuals had normal brain MRI and at least one of the following seizure types: tonic-clonic seizures, myoclonic seizures, (atypical) absence seizures, or atonic seizures. In this cohort, we identified a third person carrying a de novo mutation in CHD2 (c.1396C>T [p.Arg466∗]) in exon 13.
The two identified premature stop codons are predicted to result in degradation of the mutant transcript by means of nonsense-mediated mRNA decay (NMD). The effect of the splice-site mutation on transcript level is unclear, given that both exon skipping and (partial) intron retention are possible. Most likely, the splice acceptor mutation results in skipping of exon 16 and a subsequent out-of-frame deletion of exon 16, leading to a premature stop codon in exon 17 (p.Thr604Valfs2∗). Therefore, NMD could also be active in this case. To test the NMD hypothesis, we extracted RNA from fresh blood (QIAGEN RNeasy Micro Kit) of the three probands and a parent as a control individual, removed contaminated DNA by Ambion DNA-free DNase treatment (Life Technologies), and synthesized cDNA by Superscript III reverse transcriptase (Life Technologies). For the two nonsense mutations, we performed bidirectional sequencing on cDNA with primers flanking the mutation. With these experiments, we were able to detect the mutation with a 50/50 ratio, proving that the aberrant allele was not degraded by NMD. These results were confirmed by Sybr-Green-based quantitative PCR (qPCR) experiments (Sigma-Aldrich) with six primer pairs complementary to CHD2 cDNA (data not shown). For the splice-site mutation, we performed qPCR and developed sequencing primers flanking exon 16 on cDNA. Also here, qPCR showed the presence of both alleles. Additionally, the sequencing experiment showed an abundance of alternative splicing events in the probands compared to the control individual, but the consequence of the alternative splicing remained unknown. Although skipping of exon 16 appeared to be a logical consequence, additional investigations revealed more complex alternative splicing events. Whereas definite identification of all alternatively spliced transcripts was not possible, we were able to rule out NMD. We assumed that either the aberrant proteins were degraded by the proteasome or the shorter proteins were not fully functional; both of these cases would result in loss of function. In support of the pathogenicity of these mutations, none of the identified mutations have been observed in the 1000 Genomes Project, National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server (EVS), or dbSNP (build 137). Furthermore, splice-site, frameshift, or nonsense variations are absent in the EVS and 1000 Genomes Project cohorts.
Between the age of 14 months and 3.5 years, the three persons carrying a de novo CHD2 mutation presented with febrile seizures followed by therapy-resistant generalized seizures (Table 1). Frequent myoclonic seizures, generalized tonic-clonic seizures (GTCSs), and absences were seen in all three individuals. Proband 1 developed normally during the first year of life. The first febrile seizure occurred when he was 14 months old and was soon followed by afebrile head drops occurring several times a day. When he was 2 years old, therapy-resistant myoclonic seizures, atypical absences, and GTCSs rarely associated with fever developed. On one occasion, he had a status epilepticus. Electroencephalography (EEG) showed generalized polyspike wave discharges and, later, focal epileptic discharges. He has been treated with valproic acid, levetiracetam, phenytoin, topiramate, bromide, phenobarbital, ethosuximide, topiramate, vitamine B6, and prednisolon. He now has moderate intellectual disability (ID), dysarthria, and ataxia. Proband 2 had a normal early development. At the age of 2 years, she had a cluster of febrile seizures. At the age of 2.5 years, she developed therapy-resistant absence seizures accompanied by eyelid myoclonias, myoclonic seizures, and febrile and afebrile GTCSs. EEG showed frequent generalized spike-wave complexes and polyspikes. She has been treated with vigabatrin, valproate, bromide, ethosuximide, lamotrigine, and levetiracetam and is currently taking a combination of topiramate and valproic acid. She still has afebrile GTCSs and moderate ID. Proband 3 had slightly delayed early motor and speech development, given that he walked at the age of 1.5 years and produced his first words at the age of 2 years. At the age of 3.5 years, he had two GTCSs during an episode of high fever. During the following 2 years, he had pharmacoresistant febrile and afebrile GTCSs, hemiclonic, atonic, and myoclonic seizures, and atypical absences. He was treated with valproic acid, topiramate, and levetiracetam. After the start of clobazam at the age of 5.5 years, he became seizure free. EEG showed generalized (poly)spike-wave complexes. After seizure onset, a cognitive decline was seen but partially improved when seizures were controlled. He now has mild ID, autism spectrum disorder (ASD), attention deficit hyperactivity disorder, and mild ataxia. Brain MRI was normal in probands 1 and 2 and showed atrophic changes in proband 3.
Table 1.
Clinical and Genetic Characteristics of Persons with a CHD2 Mutation
| Proband | Sex | Age at Inclusion | Mutation | Development Prior to Epilepsy | Age of Seizure Onset | First Seizure Type | Further Seizure Types | Fever Sensitivity | EEG | Imaging | Clinical Exam | Cognitive Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | male | 6 years | c.1810−2A>C (p.?) | normal | 14 months | simple FSs | head drops, GTCSs, myoclonic seizures, atypical absences, status epilepticus | + | frequent generalized (poly)SWs | normal | ataxia, dysarthria | mild ID |
| 2 | female | 24 years | c.4971G>A (p.Trp1657∗) | normal | 2 years | cluster of FSs | myoclonic absences, myoclonic seizures, GTCSs | + | frequent generalized SWs and polyspikes | normal | normal | mild ID |
| 3 | male | 6 years | c.1396C>T (p.Arg466∗) | subtle delay in motor and speech development | 3 years, 6 months | two FSs during single fever episode | GTCSs and hemiclonic, atonic, myoclonic, atypical absences | + | generalized (poly)SWs | nonspecific atrophy | mild ataxia | mild ID, ASD, ADHD |
Abbreviations are as follows: ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; FS, febrile seizure; GTCS, generalized tonic-clonic seizure; ID, intellectual disability; and SW, spike-wave complex.
In addition to our three individuals with a CHD2 mutation, one simplex case with ID and absence epilepsy and one simplex ASD case both carrying a mutation in CHD2 have been reported in the literature.14,15 Recently, two other research groups have also shown the involvement of CHD2 mutations in EEs.16,17 Furthermore, several individuals affected by ID and generalized epilepsy and carrying a multigenic chromosomal deletion of 15q26.2, including CHD2, have been described (Table 2).18–22 The presumed haploinsufficiency of CHD2 in these probands prompted us to screen all individuals without a previous detected mutation from both studied cohorts for copy-number variants in CHD2 with the multiplex amplicon quantification (MAQ) technique, an in-house-developed technique based on a semiquantified multiplex PCR. The multiplex PCR reaction consisted of six target amplicons located in the genomic region of CHD2 and four reference amplicons randomly located on different chromosomes. The MAQ analysis was performed as described previously.23 A partial or full deletion or duplication of the gene was not identified in any of the remaining studied persons (7 of the initial cohort and 149 of the second-stage cohort).
Table 2.
Clinical Phenotype of Probands Reported in Literature with CHD2 Deletions or Mutations
|
Reference |
||||||||
|---|---|---|---|---|---|---|---|---|
| Capelli et al.18 | Dhamija et al.19 | Veredice et al.20 | Li et al.21 | Lund et al.22 | Rauch et al.14 | Carvill et al.16 | Allen et al.17 | |
| Number of individuals | 1 | 1 | 1 | 1 | 1 | 1 | 6 | 1 |
| Genetic findings | de novo 0.5 Mb deletion including CHD2 and RGMA | de novo 0.9 Mb deletion including CHD2 and three other genes | de novo 5 Mb deletion including CHD2 and 55 other genes | 3.3 Mb deletion including CHD2 and 17 other genes (no segregation analysis) | 2 Mb deletion including CHD2 and seven other genes. Also carried five additional deletions and duplications, including a total of 100 genes (paternal DNA not available) | de novo frameshift mutation c.1809 del (p.Thr604Leufs∗19) | four de novo frameshift and two de novo missense alterations: p.Glu1412Glyfs∗64, p.Arg121∗, p.Gly491Valfs∗13, p.Arg1644Lysfs∗22, p.Trp548Arg, p.Leu823Pro | de novo splice mutation c.1502+1G>A |
| Age at seizure onset | 2 years | 3.5 years | 6 months | not specified | 4 years | 5 years | 1–3 years | 6 months |
| Seizure type at onset | not specified | CPSs | febrile generalized clonic SE | two episodes of FSs | atypical ASs, MSs | ASs | atypical ASs, AtSs, MSs, GTCSs, FSs, FDSs | unknown |
| Further seizure types | not specified | therapy-resistant ASs with eyelid flutter, TSs, MSs, GTCSs | therapy-resistant massive MSs with head drop, eyelid MSs, prolonged hemiclonic FSs | none | TSs, MSs, atypical ASs, nonconvulsive SE | not specified | FSs, AtSs, MSs, GTCSs, NCS, SE, TSs, HSs, FDSs, MAs, atypical ASs | MSs, FDSs, GTCSs, atypical ASs, AtSs |
| Fever sensitivity | not specified | no | yes | yes | no | not specified | one patient | no |
| EEG | generalized spike waves and focal discharges | generalized spike waves, PPR | irregular generalized spike waves, PPR | not specified | generalized slow spike waves and runs of fast spikes | not specified | generalized (poly)spike waves, slow spike waves, multifocal discharges, generalized paroxysmal fast activity, diffuse slowing | slow background, generalized spike waves |
| MRI | normal | normal | vermis hypoplasia, cisterna magna | normal | partial agenesis of vermis | unknown | unknown | normal |
| Development prior to epilepsy | not specified | delayed | delayed | delayed | delayed | delayed | normal or delayed | normal |
| Developmental outcome | globally delayed, severe speech impairment | mild ID | mild ID | mild to moderate ID, speech impairment | severe ID | mild ID | moderate to severe ID | unspecified delay |
| Other clinical findings | ataxia, relative microcephaly, mild facial dysmorphisms | microcephaly, short stature, mild facial dysmorphisms | microcephaly, congenital hypothyroidism, bicuspid aortic valve, hypotonia | microcephaly, short stature, mild facial dysmorphisms | short stature, hypertelorism, epicantal fold, micropenis, single palmar creases | Duane anomaly | ||
Abbreviations are as follows: AS, absence seizure; AtS, atonic seizure; CPS, complex partial seizure; FDS, focal dyscognitive seizure; FS, febrile seizure; GTCS, generalized tonic-clonic seizure; HS, hemiclonic seizure; ID, intellectual disability; MA, myoclonic absence; MS, myoclonic seizure; NCS, nonconvulsive status epilepticus; PPR, photo paroxysmal response; SE, status epilepticus; and TS, tonic seizure.
To establish additional evidence of the implication of CHD2 in the development of epilepsy, we examined the functional consequence of CHD2 haploinsufficiency by knocking down chd2 in zebrafish by using targeted morpholino (MO) antisense oligomers.24 All zebrafish experiments carried out were approved by the ethics committee of the University of Leuven (Ethische Commissie van de KU Leuven, approval number P05090) and by the Belgian Federal Public Service of Health, Food Chain Safety, and Environment (Federale Overheidsdienst Volksgezondheid, Veiligheid van de Voedselketen en Leefmileu, approval number LA1210199). In order to mimic loss-of-function mutations, we designed a MO (E2I2 MO, 5′-GATCAGACTGGCCTTTTTGTGTACC-3′) to target the splice donor site of exon 2 and interfere with normal pre-mRNA splicing of zebrafish chd2 (ENSDART00000127730). Targeting of the exon 2-intron 2 boundary should result in abnormal exon 2 splicing, leading to its complete or partial deletion together with its flanking introns. This should result in an mRNA shorter than the wild-type transcript (Figure 1). A control MO (randomized 25 N oligomer) was used as a negative control (ctrl MO). All MOs were designed and synthesized by GeneTools. Gene knockdowns were achieved through microinjection of MOs into 1- to 2-cell-stage embryos from the AB (wild-type) strain according to the method previously described.25 In order to mimic haploinsufficiency, we titrated the amount of E2I2 MO to 9 ng per injection so as to reduce correctly spliced chd2 mRNA levels by approximately 50%. The same amount of ctrl MO was injected into sibling control embryos. To evaluate the level of knockdown in zebrafish embryos and larvae, we performed qPCR on splice-blocked pre-mRNA. Total mRNA was purified from each of 10 ctrl-MO- and E2I2-MO-injected embryos and larvae between 1 and 5 days postfertilization (dpf) with TRIzol reagent (Life Technologies) according to the recommended protocol. Reverse transcription of 2 μg total RNA to single-stranded cDNA was performed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The PCR reaction was performed with Phusion Polymerase (ThermoScientific) and chd2-specific primers (5′-AAGACGAAGGCTCGACTCAA-3′ and 5′-TGCAGGCTCTTGTCTACTGC-3′) for detecting the amplicon with or without the partially deleted exon 2. PCR products were visualized with standard agarose gel electrophoresis. As expected, the ctrl MO did not have any effect on splicing and resulted in the predicted wild-type mRNA corresponding to a 384 bp PCR product (Figure 1). chd2 knockdown using the E2I2 MO resulted in two products: (1) a 384 bp band corresponding to the wild-type mRNA and (2) a shorter product (∼300 bp) probably corresponding to an abnormally spliced mRNA, suggesting a partial knockdown. The discrepancy between the length of predicted (149 bp) versus observed (∼300 bp) abnormally spliced mRNA was most likely a result of the activation of a cryptic splice site in exon 2 instead of splicing to the 5′ end of the upstream splice site (this could cause a partial deletion of exon 2). This abnormal product appeared after 1 dpf and was sustained until 5 dpf. Adult zebrafish (Danio rerio) of the AB strain (Zebrafish International Resource Center, Eugene) were maintained at 28.5°C on a 14/10 hr light/dark cycle under standard aquaculture conditions, and fertilized eggs were collected via natural spawning. Embryos were raised in embryo medium consisting of 1.5 mM HEPES, pH 7.6, 17.4 mM NaCl, 0.21 mM KCl, 0.12 mM MgSO4, and 0.18 mM Ca(NO3)2) in an incubator on a 14/10 hr light/dark cycle at 28.5°C. For all experiments described, larvae of 1–5 dpf were used.
Figure 1.
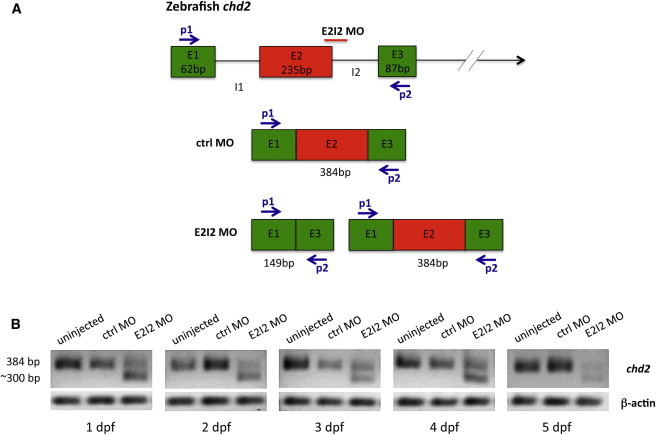
chd2 Knockdown: E2I2 MO Causes Missplicing of Zebrafish chd2
(A) Schematic cartoon showing predicted splicing events in the pre-mRNA of chd2 E2I2-MO- and ctrl-MO-injected fish. The exons are represented by colored boxes (E1, E2, and E3), and the introns are represented by solid black lines (I1 and I2). The splice MO binding site (E2I2 MO) and the primer binding sites (P1 and P2) used for qPCR are indicated. According to the predictions, PCR products of 384 and 149 bp were expected for ctrl-MO- and E2I2-MO-injected larvae, respectively. In the case of a partial knockdown, both products should be present.
(B) qPCR analysis of MO-knockdown larvae. Normal splicing in ctrl-MO-injected larvae resulted in the predicted 384 bp product, indicating the presence of wild-type mRNA. Abnormal splicing in E2I2-MO-injected larvae resulted in two products: 384 bp (corresponding to the wild-type mRNA) and ∼300 bp (an abnormal mRNA), indicating partial knockdown. The larger-than-expected size of the PCR product could have been a result of the activation of a cryptic splice site. The aberrant mRNA was present from 1–5 dpf and was accompanied by the wild-type 384 bp fragment.
E2I2-MO-injected larvae exhibited prominent morphological and behavioral alterations. The E2I2-MO-injected larvae showed from 2 dpf onward multiple developmental abnormalities: pericardial edema, microcephaly, body curvature, absent swim bladder, and stunted growth (Figure 2). The ctrl-MO-injected larvae were morphologically indistinguishable from the uninjected ones. Visual observation of chd2 knockdown larvae by means of high-magnification stereomicroscopy revealed that all 4 dpf larvae displayed abnormal movement patterns with frequent whirlpool-like events. Occasionally, we also noticed pectoral-fin and jaw twitching and whole-body trembling. To confirm whether this behavior could have been a result of seizure activity, we performed field-potential recordings on larval tecta as described previously.26 Already from 4 dpf, E2I2-MO-injected larvae displayed epileptiform discharges (Figure 3). Discharges consisted of multiple upward spikes of amplitudes several times larger than those of ctrl-MO-injected or uninjected larvae and with occasional ictal-like patterns. This spiking pattern resembled preictal discharges observed in immature hippocampi of a mouse model of temporal lobe epilepsy.27 These experimental findings suggest that loss of CHD2 results in an epilepsy phenotype.
Figure 2.
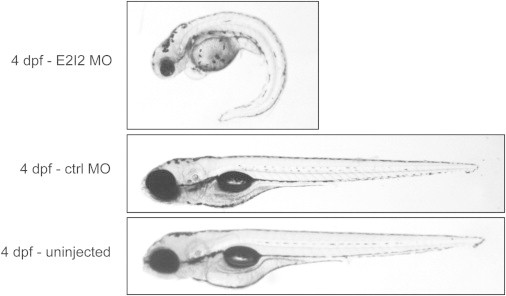
Representative Pictures of 4-Day-Old chd2 Knockdown and Control Larvae
chd2 E2I2-MO-injected larvae displayed pericardial edema, microcephaly, body curvature, absent swim bladder, and stunted growth (A). Ctrl-MO-injected (B) and uninjected (C) larvae developed normally. The experiment was performed at least three times with 100 embryos injected per condition.
Figure 3.
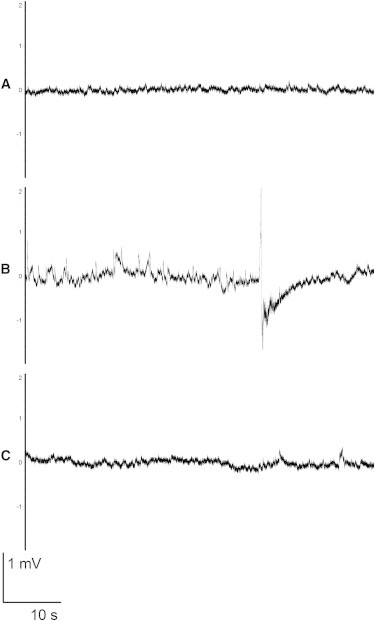
Electrographic Activity of 4-Day-Old chd2 Knockdown and Control Larvae
(A) Uninjected larva.
(B) chd2 E2I2-MO-injected larva displaying ictal-like discharges.
(C) Ctrl-MO-injected larva.
One-minute fragments are shown. The y axis shows a 4 mV span. Ictal-like events shown in (B) (more than 3 s in duration) were observed in five out of seven fish larvae in the chd2 E2I2-MO-injected group.26 Seven, six, and five fish were analyzed for the chd2 E2I2 MO, ctrl MO, and uninjected larvae, respectively.
CHD2 encodes CHD2, a protein that does not have an established link to neuronal function or hyperexcitability. CHD proteins are assumed to modify gene transcription by affecting chromatin structure through helicase function. A Chd2-deficient mouse model has been reported to show spinal abnormalities, renal dysfunction, growth retardation, and susceptibility to tumors, but not epileptic seizures.28,29 None of the features described in the mouse model were seen in the probands with de novo CHD2 mutations, suggesting that in contrast to the zebrafish model presented here, the existing mouse model insufficiently replicates the human phenotype. On the other hand, in addition to showing electrographic seizures observed in zebrafish chd2 knockdowns, these larvae displayed edemas, body curvature, and stunted growth that could be considered equivalent to the cardiovascular and renal defects, lord kyphosis, and postnatal stunted-growth phenotypes described in Chd2 mouse mutants.28,30,31 Thus, the zebrafish phenocopies aspects of both the human and the mouse spectra. Why no seizure phenotype has been reported for Chd2 mutant mice remains to be determined. Perhaps these mice have to be monitored with EEG for seizure detection and/or challenged before (presumably) spontaneous seizure behavior can be detected. Additionally, mutations in other genes encoding members of the CHD family have been identified, such as CHD7 mutations in persons with CHARGE syndrome32 and de novo mutations in CHD3, CHD7, and CHD8 in individuals with ASD.33,34 Although the exact role of CHD2 in neuronal hyperexcitability remains to be determined, our findings suggest that helicase dysfunction might be a mechanism involved in epileptogenesis and neurodevelopment.
In summary, we identified CHD2 nonsense mutations as the underlying genetic defect for a fever-sensitive myoclonic EE. In our study, individuals carrying a CHD2 mutation displayed a spectrum of fever-sensitive generalized seizures similar to those in Dravet syndrome, but seizure onset was clearly later, and unlike in Dravet syndrome, developmental delay could be seen prior to epilepsy onset. Future studies will reveal whether CHD2 mutations also account for a subset of individuals with more typical Dravet syndrome. We showed that a MO-based knockdown of chd2 in zebrafish resulted in clinical and electrographic seizures paralleling the human phenotype. Our study further suggests that helicase dysfunction in humans might specifically result in neuronal hyperexcitability in the absence of syndromic or dysmorphic features.
Consortia
Additional members of the EuroEPINOMICS RES Consortium are Rik Hendrickx, Philip Holmgren, Ulrich Stephani, Hiltrud Muhle, Manuela Pendiziwiat, Silke Appenzeller, Kaja Selmer, Eva Brilstra, Bobby Koeleman, Felix Rosenow, Eric Leguern, Katalin Sterbova, Budisteanu Magdalena, Gherghiceanu Rodica, Oana Tarta Arsene, Barca Diana, Rosa Guerrero-Lopez, Laura Ortega, Albena P. Todorova, Andrey V. Kirov, Angela Robbiano, Mutluay Arslan, Uluç Yiş, and Vanja Ivanović
Acknowledgments
We thank the probands and their families for their cooperation. The research was supported by the EUROCORES program EuroEPINOMICS of the European Science Foundation; funds from the state budget of Romania, managed by the Executive Agency for Higher Education, Research, Development, and Innovation Funding for Project 6-EUROC; the Scientific and Technological Research Council of Turkey; National Science Centre Poland funding for project 800/N-ESF-EuroEPINOMICS/10/2011/0; MH CZ-DRO, University Hospital Motol, Prague (00064203); “Investissements d’avenir” ANR-10-IAIHU-06; the Academy of Finland (grant 141549) and Folkhälsan Research Foundation; Wellcome Trust grants WT089062 and 098051; Academy of Finland grant 251704; Academy of Finland Center of Excellence in Complex Disease Genetics grants 213506 and 129680; the European Community’s Seventh Framework Programme (FP7/2007-2013) ENGAGE Consortium (grant HEALTH-F4-2007- 201413); Synaptic Systems grant 242167; the Sigrid Juselius Foundation; European Commission FP7 project 261123 (gEUVADIS); Federal Ministry of Education and Research NGFNplus/EMINet 01GS08123; IonNeurONet 01GM1105A; German Research Foundation (DFG) Le1030/11-1; the German Society for Epileptology; the Fund for Scientific Research Flanders (FWO); the Flemish government Methusalem excellence grant; and the University of Antwerp. We thank the VIB Genetic Service Facility and the institute of Clinical Molecular Biology in Kiel for providing Sanger sequencing, supported in part by DFG Cluster of Excellence “Inflammation at Interfaces” and “Future Ocean.” We thank technicians S. Greve, S. Arndt, and T. Henke for technical support. G.K. is a member of the DFG-funded Cluster of Excellence “Inflammation at Interfaces.” A.S. is a FWO postdoctoral fellow. T.D. and A.K. are Institute of Science and Technology PhD fellows.
Contributor Information
Peter De Jonghe, Email: peter.dejonghe@molgen.vib-ua.be.
the EuroEPINOMICS RES Consortium:
Rik Hendrickx, Philip Holmgren, Ulrich Stephani, Hiltrud Muhle, Manuela Pendiziwiat, Silke Appenzeller, Kaja Selmer, Eva Brilstra, Bobby Koeleman, Felix Rosenow, Eric Leguern, Katalin Sterbova, Budisteanu Magdalena, Gherghiceanu Rodica, Oana Tarta Arsene, Barca Diana, Rosa Guerrero-Lopez, Laura Ortega, Albena P. Todorova, Andrey V. Kirov, Angela Robbiano, Mutluay Arslan, Uluç Yiş, and Vanja Ivanović
Web Resources
The URLs for the data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
Beyond the Ion Channel (EuroEPINOMICS blog), http://channelopathist.net/
Burrows-Wheeler Aligner (BWA), http://bio-bwa.sourceforge.net/
Decipher, http://decipher.sanger.ac.uk/
DeNovoGear, http://sourceforge.net/projects/denovogear/
EuroEPINOMICS Consortium, http://www.euroepinomics.org/
Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/
GenomeComb, http://genomecomb.sourceforge.net
Multiplex Amplicon Quantification (MAQ), http://www.multiplicom.com/multiplex-amplicon-quantification-maq
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SAMtools, http://samtools.sourceforge.net
References
- 1.Cross J.H., Guerrini R. The epileptic encephalopathies. Handb. Clin. Neurol. 2013;111:619–626. doi: 10.1016/B978-0-444-52891-9.00064-6. [DOI] [PubMed] [Google Scholar]
- 2.Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3–9. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 3.Kearney J.A., Wiste A.K., Stephani U., Trudeau M.M., Siegel A., RamachandranNair R., Elterman R.D., Muhle H., Reinsdorf J., Shields W.D. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr. Neurol. 2006;34:116–120. doi: 10.1016/j.pediatrneurol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Depienne C., Trouillard O., Saint-Martin C., Gourfinkel-An I., Bouteiller D., Carpentier W., Keren B., Abert B., Gautier A., Baulac S. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 2009;46:183–191. doi: 10.1136/jmg.2008.062323. [DOI] [PubMed] [Google Scholar]
- 5.Depienne C., Trouillard O., Bouteiller D., Gourfinkel-An I., Poirier K., Rivier F., Berquin P., Nabbout R., Chaigne D., Steschenko D. Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Hum. Mutat. 2011;32:E1959–E1975. doi: 10.1002/humu.21373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 7.de Ligt J., Veltman J.A., Vissers L.E. Point mutations as a source of de novo genetic disease. Curr. Opin. Genet. Dev. 2013;23:257–263. doi: 10.1016/j.gde.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albers C.A., Lunter G., MacArthur D.G., McVean G., Ouwehand W.H., Durbin R. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reumers J., De Rijk P., Zhao H., Liekens A., Smeets D., Cleary J., Van Loo P., Van Den Bossche M., Catthoor K., Sabbe B. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012;30:61–68. doi: 10.1038/nbt.2053. [DOI] [PubMed] [Google Scholar]
- 13.Conrad D.F., Keebler J.E.M., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 15.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.-F., Stevens C., Wang L.-S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carvill G.L., Heavin S.B., Yendle S.C., McMahon J.M., O’Roak B.J., Cook J., Khan A., Dorschner M.O., Weaver M., Calvert S. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen A.S., Berkovic S.F., Cossette P., Delanty N., Dlugos D., Eichler E.E., Epstein M.P., Glauser T., Goldstein D.B., Han Y., Epi4K Consortium. Epilepsy Phenome/Genome Project De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capelli L.P., Krepischi A.C., Gurgel-Giannetti J., Mendes M.F., Rodrigues T., Varela M.C., Koiffmann C.P., Rosenberg C. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur. J. Med. Genet. 2012;55:132–134. doi: 10.1016/j.ejmg.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Dhamija R., Patterson M.C., Wirrell E.C. Epilepsy in children—when should we think neurometabolic disease? J. Child Neurol. 2012;27:663–671. doi: 10.1177/0883073811435829. [DOI] [PubMed] [Google Scholar]
- 20.Veredice C., Bianco F., Contaldo I., Orteschi D., Stefanini M.C., Battaglia D., Lettori D., Guzzetta F., Zollino M. Early onset myoclonic epilepsy and 15q26 microdeletion: observation of the first case. Epilepsia. 2009;50:1810–1815. doi: 10.1111/j.1528-1167.2009.02078.x. [DOI] [PubMed] [Google Scholar]
- 21.Li M.M., Nimmakayalu M.A., Mercer D., Andersson H.C., Emanuel B.S. Characterization of a cryptic 3.3 Mb deletion in a patient with a “balanced t(15;22) translocation” using high density oligo array CGH and gene expression arrays. Am. J. Med. Genet. A. 2008;146:368–375. doi: 10.1002/ajmg.a.32116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lund C., Brodtkorb E., Røsby O., Rødningen O.K., Selmer K.K. Copy number variants in adult patients with Lennox-Gastaut syndrome features. Epilepsy Res. 2013;105:110–117. doi: 10.1016/j.eplepsyres.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Suls A., Claeys K.G., Goossens D., Harding B., Van Luijk R., Scheers S., Deprez L., Audenaert D., Van Dyck T., Beeckmans S. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum. Mutat. 2006;27:914–920. doi: 10.1002/humu.20350. [DOI] [PubMed] [Google Scholar]
- 24.Nasevicius A., Ekker S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 25.Summerton J., Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 26.Afrikanova T., Serruys A.-S.K., Buenafe O.E.M., Clinckers R., Smolders I., de Witte P.A.M., Crawford A.D., Esguerra C.V. Validation of the zebrafish pentylenetetrazol seizure model: locomotor versus electrographic responses to antiepileptic drugs. PLoS ONE. 2013;8:e54166. doi: 10.1371/journal.pone.0054166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derchansky M., Jahromi S.S., Mamani M., Shin D.S., Sik A., Carlen P.L. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J. Physiol. 2008;586:477–494. doi: 10.1113/jphysiol.2007.143065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulkarni S., Nagarajan P., Wall J., Donovan D.J., Donell R.L., Ligon A.H., Venkatachalam S., Quade B.J. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am. J. Med. Genet. A. 2008;146A:1117–1127. doi: 10.1002/ajmg.a.32178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marfella C.G., Henninger N., LeBlanc S.E., Krishnan N., Garlick D.S., Holzman L.B., Imbalzano A.N. A mutation in the mouse Chd2 chromatin remodeling enzyme results in a complex renal phenotype. Kidney Blood Press. Res. 2008;31:421–432. doi: 10.1159/000190788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marfella C.G., Ohkawa Y., Coles A.H., Garlick D.S., Jones S.N., Imbalzano A.N. Mutation of the SNF2 family member Chd2 affects mouse development and survival. J. Cell. Physiol. 2006;209:162–171. doi: 10.1002/jcp.20718. [DOI] [PubMed] [Google Scholar]
- 31.Nagarajan P., Onami T.M., Rajagopalan S., Kania S., Donnell R., Venkatachalam S. Role of chromodomain helicase DNA-binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene. 2009;28:1053–1062. doi: 10.1038/onc.2008.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vissers L.E.L.M., van Ravenswaaij C.M.A., Admiraal R., Hurst J.A., de Vries B.B.A., Janssen I.M., van der Vliet W.A., Huys E.H.L.P.G., de Jong P.J., Hamel B.C.J. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 33.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]