

Abstract
Most ancient specimens contain very low levels of endogenous DNA, precluding the shotgun sequencing of many interesting samples because of cost. Ancient DNA (aDNA) libraries often contain <1% endogenous DNA, with the majority of sequencing capacity taken up by environmental DNA. Here we present a capture-based method for enriching the endogenous component of aDNA sequencing libraries. By using biotinylated RNA baits transcribed from genomic DNA libraries, we are able to capture DNA fragments from across the human genome. We demonstrate this method on libraries created from four Iron Age and Bronze Age human teeth from Bulgaria, as well as bone samples from seven Peruvian mummies and a Bronze Age hair sample from Denmark. Prior to capture, shotgun sequencing of these libraries yielded an average of 1.2% of reads mapping to the human genome (including duplicates). After capture, this fraction increased substantially, with up to 59% of reads mapped to human and enrichment ranging from 6- to 159-fold. Furthermore, we maintained coverage of the majority of regions sequenced in the precapture library. Intersection with the 1000 Genomes Project reference panel yielded an average of 50,723 SNPs (range 3,062–147,243) for the postcapture libraries sequenced with 1 million reads, compared with 13,280 SNPs (range 217–73,266) for the precapture libraries, increasing resolution in population genetic analyses. Our whole-genome capture approach makes it less costly to sequence aDNA from specimens containing very low levels of endogenous DNA, enabling the analysis of larger numbers of samples.
Introduction
With the advent of next-generation sequencing techniques and the rapidly declining cost of sequencing, the field of hominin paleogenetics has begun to transition from focusing on PCR-amplified mitochondrial DNA and Y chromosomal markers to shotgun sequencing of the whole genome.1–8 The use of autosomal DNA is advantageous because it provides information about the genome as a whole, whereas the mitochondrial DNA (mtDNA) and Y chromosome, as nonrecombining markers, represent only a single maternal or paternal lineage. Whole-genome sequencing of single ancient genomes, including Neandertals,1 Denisovan,7,9 a Paleo-Eskimo,2 the Tyrolean Iceman,4 and an Australian Aborigine,3 have transformed our understanding of human migrations and revealed previously unknown admixture among ancient populations.
Importantly, most of these specimens were exceptional in their levels of preservation: the Neandertal and Denisovan bones, found in caves, contained ∼1%–5%1 and 70%7,9 endogenous DNA, respectively, and the Paleo-Eskimo and Aborigine genomes were obtained from hair specimens, which generally contain lower levels of contamination10 but are not available in most archaeological contexts. Indeed, sequencing libraries derived from bones and teeth from temperate environments typically contain <1% endogenous DNA,6 with the remaining ∼99% primarily consisting of DNA from environmental contaminants such as bacteria and fungi. Although some samples with 1%–2% endogenous DNA can still, with sufficient sequencing, yield enough information for population genetic analyses,5,6 the required amount of sequencing of specimens with less endogenous DNA is costly and thus untenable for many researchers. Ancient DNA (aDNA) researchers have begun to address this issue for hominin genomes by using targeted capture to enrich for only the mtDNA, selected regions of the genome, or a single chromosome.8,11–13 However, because of the highly fragmented nature of aDNA, an ideal enrichment technique would target as much of the endogenous genome as possible so as not to discard any potentially informative sequences.
In the present study, we use a method we call whole-genome in-solution capture (WISC) as an unbiased means to increase the proportion of endogenous DNA in aDNA sequencing libraries. To target as much of the remaining endogenous DNA as possible, we created human genomic DNA “bait” libraries from a modern reference individual with adapters containing T7 RNA polymerase promoters (see Material and Methods). We then performed in vitro transcription of these libraries with biotinylated UTP, producing RNA baits covering the entire human genome. Analogous to current exome capture technologies,14 these baits were hybridized to aDNA libraries in solution and pulled down with magnetic streptavidin-coated beads. The unbound, predominantly nonhuman DNA was then washed away, and the captured endogenous human DNA was eluted and amplified for sequencing. Figure 1 shows a schematic overview of the WISC process, including the creation of the RNA bait libraries. By using both baits and adaptor-blocking oligos made from RNA, we were able to remove any residual baits and blockers by RNase treatment prior to PCR amplification.
Figure 1.
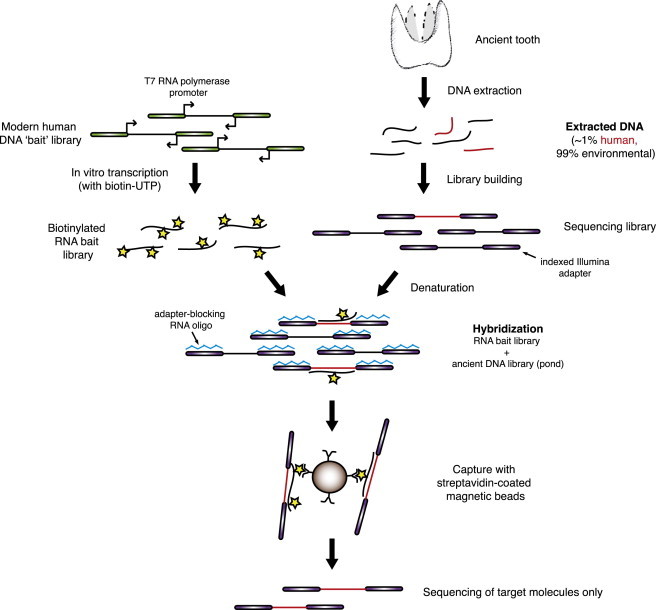
Schematic of the Whole-Genome In-Solution Capture Process
To generate the RNA “bait” library, a human genomic library is created via adapters containing T7 RNA polymerase promoters (green boxes). This library is subjected to in vitro transcription via T7 RNA polymerase and biotin-16-UTP (stars), creating a biotinylated bait library. Meanwhile, the ancient DNA library (aDNA “pond”) is prepared via standard indexed Illumina adapters (purple boxes). These aDNA libraries often contain <1% endogenous DNA, with the remainder being environmental in origin. During hybridization, the bait and pond are combined in the presence of adaptor-blocking RNA oligos (blue zigzags), which are complimentary to the indexed Illumina adapters and thus prevent nonspecific hybridization between adapters in the aDNA library. After hybridization, the biotinylated bait and bound aDNA is pulled down with streptavidin-coated magnetic beads, and any unbound DNA is washed away. Finally, the DNA is eluted and amplified for sequencing.
Material and Methods
Ancient Specimens
The four Bulgarian teeth used in this study were obtained from four different excavations.
Sample P192-1 was found at the site of a pit sanctuary near Svilengrad, Bulgaria, excavated between 2004 and 2006.15 The pits are associated with the Thracian culture and date to the Early Iron Age (800–500 BC) based on pottery found in the pits. A total of 67 ritual pits, including 16 pits containing human skeletons or parts of skeletons, were explored during the excavations. An upper wisdom tooth from an adult male was used for DNA analysis.
Sample T2G2 was found in a Thracian tumulus (burial mound) near the village of Stambolovo, Bulgaria. Two small tumuli dating to the Early Iron Age (850–700 BC) were excavated in 2008.16 A canine tooth from an inhumation burial of a child (c.12 years old) inside a dolium was used for DNA analysis.
Sample V2 was found in a flat cemetery dating to the Late Bronze Age (1500–1100 BC) near the village of Vratitsa, Bulgaria. Nine inhumation burials were excavated between 2003 and 2004.17 A molar from a juvenile male (age 16–17) was used for DNA analysis.
Sample K8 was found in the Yakimova Mogila Tumulus, which dates to the Iron Age (450–400 BC), near Krushare, Bulgaria. An aristocratic inhumation burial containing rich grave goods was excavated in 2008.18 A molar from one individual, probably male, was used for DNA analysis.
Other specimens are as follows.
Sample M4 is an ancient hair sample obtained from the Borum Eshøj Bronze Age burial in Denmark. The burial comprised three individuals in oak coffins, commonly referred to as “the woman,” “the young man,” and “the old man.” The M4 sample is from the latter. The site was excavated in 1871–1875 and the coffins dated to c.1350 BC.19
Samples NA39-50 were obtained from pre-Columbian Chachapoyan and Chachapoya-Inca remains dating between 1000 and 1500 AD. They were recovered from the site Laguna de los Condores in northeastern Peru.20 Bone samples were used for DNA analysis.
DNA Extraction and aDNA Library Preparation
All DNA extraction and initial library preparation steps (prior to amplification) were performed in the dedicated clean labs at the Centre for GeoGenetics in Copenhagen, Denmark, via established procedures to prevent contamination, including the use of indexed adapters and primers during library preparation.2,21,23 The lab work was conducted over an extended time period and by a number of different researchers, which is why the exact protocols vary somewhat between samples.
Bulgarian Samples
The surface of each tooth was wiped with a 10% bleach solution and then UV irradiated for 20 min. Part of the root was then excised and the inside of the tooth was drilled to produce approximately 200 mg of powder. DNA was isolated with a previously described silica-based extraction method.24 The purified DNA was subjected to end repair and dA-tailing with the Next End Prep Enzyme Mix (New England Biolabs) according to the manufacturer’s instructions. Next, ligation to Illumina PE adapters (Illumina) was performed by mixing 25 μl of the end repair/dA-tailing reaction with 1 μl of PE adapters (5 μM) and 1 μl of Quick T4 DNA Ligase (NEB). The mixture was incubated at 25°C for 10 min and then purified with a QIAGEN MinElute spin column according to the manufacturer’s instructions (QIAGEN). Finally, the libraries were amplified by PCR by mixing 5 μl of the DNA library template with 5 μl 10× PCR buffer, 2 μl MgCl2 (50 mM), 2 μl BSA (20 mg/ml), 0.4 μl dNTPs (25 mM), 1 μl each primer (10 μM, inPE + multiplex indexed23), and 0.2 μl of Platinum Taq High Fidelity Polymerase (Invitrogen/Life Technologies). The PCR conditions were as follows: 94°C/5 min; 25 cycles of 94°C/30 s, 60°C/20 s, 68°C/20 s; 72°C/7 min. The resulting libraries were purified with QiaQuick spin columns (QIAGEN) and eluted in 30 μl EB buffer.
Peruvian Bone Samples
DNA was isolated from seven bone samples via a previously described silica-based extraction method.24 DNA was further converted into indexed Illumina libraries with 20 μl of each DNA extract with the NEBNext DNA Library Prep Master Mix Set for 454 (NEB) according to the manufacturer’s instructions, except that SPRI bead purification was replaced by MinElute silica column purification (QIAGEN). Illumina multiplex blunt end adapters were used for ligation at a final concentration of 1.0 μM in a final volume of 25 μl. The Bst Polymerase fill-in reaction was inactivated after 20 min of incubation by freezing the sample. Library preparation was followed by a two-step PCR amplification. Amplification of purified libraries was done with Platinum Taq High Fidelity DNA Polymerase (Invitrogen) with a final mixture of 10× High Fidelity PCR Buffer, 50 mM magnesium sulfate, 0.2 mM dNTP, 0.5 μM Multiplexing PCR primer 1.0, 0.1 μM Multiplexing PCR primer 2.0, 0.5 μM PCR primer Index, 3% DMSO, 0.02 U/μl Platinum Taq High Fidelity Polymerase, 5 μl of template, and water to 25 μl final volume.23 Three PCR reactions were done for each library with the following PCR conditions: a 3 min activation step at 94°C, followed by 14 cycles of 30 s at 94°C, 20 s at 60°C, 20 s at 68°C, with a final extension of 7 min at 72°C. All three reactions per library were purified with QIAGEN MinElute columns and pooled into one single reaction. A second PCR was performed with the same conditions as before but with 22 cycles. One reaction per library was then performed with 10 μl from the purified pool of the three previous reactions. Libraries were run on a 2% agarose gel and gel purified with a QIAGEN gel extraction kit according to the manufacturer’s instructions.
Danish Hair Sample
DNA was extracted from 70 mg of hair with phenol-chloroform combined with MinElute columns from QIAGEN as previously described.3 While fixed on silica filters, the DNA was purified sequentially with AW1/AW2 wash buffers (QIAGEN Blood and Tissue Kit), Salton buffer (MP Biomedicals), and PE buffer, before being eluted in 60 μl EB buffer (both QIAGEN). Then, 20 μl of DNA extract was built into a blunt-end NGS library with the NEBNext DNA Sample Prep Master Mix Set 2 (E6070) and Illumina specific adapters.23 The libraries were prepared according to manufacturer’s instructions, with a few modifications outlined below. The initial nebulization step was skipped because of the fragmented nature of ancient DNA. End-repair was performed in 25 μl reactions with 20 μl of DNA extract. This was incubated for 20 min at 12°C and 15 min at 37°C and purified with PN buffer with QIAGEN MinElute spin columns and eluted in 15 μl. After end-repair, Illumina-specific adapters (prepared as in Meyer and Kircher23) were ligated to the end-repaired DNA in 25 μl reactions. The reaction was incubated for 15 min at 20°C and purified with PB buffer on QIAGEN MinElute columns before being eluted in 20 μl EB Buffer. The adaptor fill-in reaction was performed in a final volume of 25 μl and incubated for for 20 min at 37°C followed by 20 min at 80°C to inactivate the Bst enzyme. The entire DNA library (25 μl) was then amplified and indexed in a 50 μl PCR reaction, mixing with 5 μl 10× PCR buffer, 2 μl MgSO4 (50 mM), 2 μl BSA (20 mg/ml), 0.4 μl dNTPs (25 mM), 1 μl of each primer (10 μM, inPE forward primer + multiplex indexed reverse primer), and 0.2 μl Platinum Taq High Fidelity DNA Polymerase (Invitrogen). Thermocycling was carried out with 5 min at 95°C, followed by 25 cycles of 30 s at 94°C, 20 s at 60°C, and 20 s at 68°C, and a final 7 min elongation step at 68°C. The amplified library was then purified with PB buffer on QIAGEN MinElute columns, before being eluted in 30 μl EB.
Preparation of RNA Bait Libraries
Creation of Human Genomic DNA Libraries with T7 Adapters
Five micrograms of human DNA (HapMap individual NA21732, a Masai male) was sheared on a Covaris S2 instrument with the following conditions: 8 min at 10% duty cycle, intensity 5, 200 cycles/burst, frequency sweeping. The resulting fragmented DNA (∼150–200 bp average size, range 100–500) was subjected to end repair and dA-tailing by a KAPA library preparation kit (KAPA) according to the manufacturer’s protocol. Ligation was also performed with this kit, but with custom adapters. T7 adaptor oligos 1 and 2 (5′-GATCTTAAGGCTAGAGTACTAATACGACTCACTATAGGG∗T-3′ and 5′-P-CCCTATAGTGAGTCGTATTAGTACTCTAGCCTTAAGATC-3′) were annealed by mixing a 12.5 μl of each 200 μM oligo stock with 5 μl of 10× buffer 2 (NEB) and 20 μl of H2O. This mixture was heated to 95°C for 5 min, then left on the bench to cool to room temperature for approximately 1 hr.
One microliter of this T7 adaptor stock was used for the ligation reaction, again according to the library preparation kit instructions (KAPA). The libraries were then size selected on a 2% agarose gel to remove unligated adapters and select for fragments ∼200–300 bp in length (inserts ∼120–220 bp). After gel extraction with a QIAquick Gel Extraction kit (QIAGEN), the libraries were PCR amplified in four separate reactions with the following components: 25 μl 2× HiFi HotStart ReadyMix (KAPA), 20 μl H2O, 5 μl PCR primer (5′-GATCTTAAGGCTAGAGTACTAATACGACTCACTATAGGG∗T-3′, same as T7 oligo 1 above, 10 μM stock), and 5 μl purified ligation mix. The cycling conditions were as follows: 98°C/1 min, 98°C/15 s; 10 cycles of 60°C/15 s, 72°C/30 s; 72°C/5 min. The reactions were pooled and purified with AMPure XP beads (Beckman Coulter), eluting in 25 μl H2O.
In Vitro Transcription of Bait Libraries
To transcribe the bait libraries into biotinylated RNA, we assembled the following in vitro transcription reaction mixture: 5 μl amplified library (∼500 ng), 15.2 μl H2O, 10 μl 5× NASBA buffer (185 mM Tris-HCl [pH 8.5], 93 mM MgCl2, 185 mM KCl, 46% DMSO), 2.5 μl 0.1 M DTT, 0.5 μl 10 mg/ml BSA, 12.5 μl 10 mM NTP mix (10 mM ATP, 10 mM CTP, 10 mM GTP, 6.5 mM UTP, 3.5 mM biotin-16-UTP), 1.5 μl T7 RNA Polymerase (20 U/μl, Roche), 0.3 μl Pyrophosphatase (0.1 U/μl, NEB), and 2.5 μl SUPERase-In RNase inhibitor (20 U/μl, Life Technologies). The reaction was incubated at 37°C overnight, treated for 15 min at 37°C with 1 μl TURBO DNase (2 U/μl, Life Technologies), and then purified with an RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions, eluting twice in the same 30 μl of H2O. A single reaction produced ∼50 μg of RNA. The size of the RNA was checked by running ∼100 ng on a 5% TBE/Urea gel and staining with ethidium bromide. For long-term storage, 1.5 μl of SUPERase-In was added, and the RNA was stored at −80°C.
Preparation of RNA Adaptor-Blocking Oligos
All of the aDNA libraries that we used for testing the enrichment protocol contained indexed multiplex adapters (see “DNA Extraction and Library Preparation” above). To block these sequences and prevent nonspecific binding during capture, we created adaptor-blocking RNA oligos, which can be produced in large amounts and are easy to remove by RNase treatment when capture is complete. The following oligonucleotides were annealed as described above: T7 universal promoter (5′-AGTACTAATACGACTCACTATAGG-3′) + either Multiplex-block-P5 (5′-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATTCCTATAGTGAGTCGTATTAGTACT-3′) or Multiplex-block-P7 (5′-AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNATCTCGTATGCCGTCTTCTGCTTGCCTATAGTGAGTCGTATTAGTACT-3′), the latter containing random nucleotides at the site of the index sequence, which allows the same adaptor-blocking oligos to be used for all libraries.
For each of these double-stranded oligonucleotide solutions, 700 ng was subjected to in vitro transcription with a T7 High-Yield RNA Synthesis kit (NEB) according to the manufacturer’s instructions. After treatment with 1 μl of TURBO DNase (37°C/15 min), the RNA was purified with an RNeasy Mini kit according to the manufacturer’s instructions, except that 675 μl of ethanol (instead of 250 μl) was added at step 2 of the protocol to ensure the retention of small RNAs. The RNA was eluted in 30 μl H2O, to which 1.5 μl of SUPERase-In was added prior to storage at −80°C.
DNA Capture
Hybridization
The in-solution capture method was adapted from a protocol for exome capture.14 For the ancient DNA “pond” (the mixture to which the RNA bait will be hybridized), 27 μl of each aDNA library (81–550 ng depending on the library) was mixed with 2.5 μl human Cot-1 DNA (1 mg/ml, Life Technologies) and 2.5 μl salmon sperm DNA (10 mg/ml, Life Technologies) in 200 μl PCR tubes. The RNA baits and adaptor-blocking oligos were mixed in a separate 1.5 ml tube as follows: for each capture, 1 μl (500 ng) biotinylated RNA bait library, 3 μl SUPERase-In, 2 μl P5 multiplex block RNA (100 μM stock, see above), and 2 μl P7 multiplex block RNA (100 μM stock, see above). The DNA pond was heated in a thermal cycler to 95°C for 5 min, followed by 65°C for 5 min. When the DNA had been at 65°C for 2.5 min, the RNA bait mix was heated to 65°C for 2.5 min in a heat block. After the pond DNA had been at 65°C for 5 min, 26 μl of prewarmed hybridization buffer (10× SSPE, 10× Denhardt’s, 10 mM EDTA, 0.2% SDS, and 0.01% Tween 20) was added, followed by 8 μl RNA bait/block mix to produce a 66 μl total reaction. The reaction was mixed by pipetting, then incubated at 65°C for ∼66 hr.
Pulldown
For each capture reaction, 50 μl of Dynabeads MyOne Streptavidin C1 beads (Life Technologies) was mixed with 200 μl bead wash buffer (1 M NaCl, 10 mM Tris-HCl [pH 7.5], 1 mM EDTA, and 0.01% Tween 20), vortexed for 30 s, then separated on a magnetic plate for 2 min before supernatant was removed. This wash step was repeated twice and after the last wash the beads were resuspended in 134 μl bead wash per sample. Next, 134 μl of bead solution was added to the 66 μl DNA/RNA hybridization mix, the solution was vortexed for 10 s, and the mix was incubated at room temperature for 30 min, vortexing occasionally. The mixture was then placed on a magnet to separate the beads and the supernatant was removed. The beads were incubated in 165 μl low-stringency buffer (1× SSC/0.1%SDS/0.01% Tween 20) for 15 min at room temperature, followed by three 10 min washes at 65°C in 165 μl prewarmed high-stringency buffer (0.1× SSC/0.1% SDS/0.01% Tween 20). Hybrid-selected DNA was eluted in 50 μl of 0.1 M NaOH for 10 min at room temperature, then neutralized by adding 50 μl 1 M Tris-HCl (pH 7.5). Finally, the DNA was concentrated with 1.8× AMPure XP beads, eluting in 30 μl H2O.
Amplification
The captured pond was PCR amplified by combining the 30 μl of captured DNA with 50 μl 2× NEB Next Master Mix, 0.5 μl each primer (200 μM stocks of primer P5, 5′-AATGATACGGCGACCACCGA-3′, and P7, 5′-CAAGCAGAAGACGGCATACGA-3′), 0.5 μl RNase A (7,000 U/ml, QIAGEN), and 18.5 μl H2O. Cycling conditions were as follows: 98°C/30 s; 15–20 cycles of 98°C/10 s, 60°C/30 s, 72°C/30 s; 72°C/2 min. The reactions were purified with 1.8× (180 μl) AMPure XP beads and eluted in 30 μl H20.
Library Pooling and Multiplex Sequencing
Captured libraries were pooled in equimolar amounts (determined by analysis on an Agilent Bioanalyzer 2100) and sequenced on either a MiSeq (postcapture Bulgarian libraries, 2 × 150 bp reads) or HiSeq (precapture Bulgarian libraries (2 × 90 bp reads) and all other libraries (2 × 101 bp reads). For the postcapture libraries, 10% PhiX (a viral genome with a balanced nucleotide representation) was spiked in to compensate for the low complexity of the libraries, which can cause problems with cross-talk matrix calculation, cluster identification, and phasing during the sequencing run.
Mapping and Data Analysis
Prior to mapping, paired-end reads were merged and adapters were trimmed with the program SeqPrep with default settings, including a length cutoff of 30 nt. The merged reads and trimmed unmerged reads were mapped separately to the human reference genome (UCSC Genome Browser hg19) with BWA v.0.5.9,25 with seeding disabled (-l 1000). Duplicates were then removed from the combined bam file with samtools26 (v.0.1.18) and reads were filtered for mapping qualities ≥30.
For the postcapture libraries, we noted that there were a small number of fragments with the exact same lengths and mapping coordinates (primarily mapping to the mtDNA) in multiple libraries. Because we performed the captures and amplifications separately for each library prior to sequencing, the most parsimonious explanation for this observation is that the high clonality of the libraries led to mixed clusters on the sequencer and some misassignments of index sequences, despite the spike-in of PhiX described above. This phenomenon has been previously reported for multiplexed libraries and is probably exacerbated by high levels of clonality.27 To correct for this issue, any potentially cross-contaminating fragments (defined as those with the same lengths and mapping coordinates in more than one library) were removed bioinformatically with an in-house bash script and BEDTools.28
For downsampling experiments, the initial fastq file was reduced to the desired number of reads and then the reads were mapped as described above. Overlap between the pre- and postcapture libraries was assessed with BEDTools. Coverage plots were created with Integrative Genomics Viewer.29 DNA damage tables were generated with mapDamage 2.0.30 Overlap with repetitive regions of the genome was determined by intersecting with the RepeatMasker table for hg19 (UCSC Genome Browser) via BEDTools. For mtDNA haplogroup assignments, all trimmed and merged reads were separately aligned to the revised Cambridge reference sequence (rCRS)31 with the same pipeline described above for the full genome. Mutations were identified with MitoBamAnnotator32 and haplogroups were assigned with mthap v.0.19a based on PhyloTree Build 15.33 Sex identification was performed with a previously published karyotyping tool for shotgun sequencing data.34
Variant Calling and Principal Component Analysis
For variant calling, sites were overlapped with SNPs from the 1000 Genomes Project Phase 1 data set (v.3), filtering for base qualities ≥30 in the ancient samples and removing related individuals from 1000 Genomes. For PCAs with Native Americans, low-coverage sequenced genomes from ten additional individuals (Mayan individuals HGDP00854, HGDP00855, HGDP00856, HGDP00857, HGDP00860, HGDP00868, HGDP00877; Karitiana individuals HGDP00998 and BI16; and Aymara individual TA6) were also included in the intersection (M. Raghavan, M. DeGiorgio, O.E. Cornejo, S. Rasmussen, S. Shringarpure, A. Eriksson, A. Albrechtsen, I. Moltke, K. Harris, D. Meltzer, M. Metspalu, M. Karmin, K. Tambets, M.W. Sayres, A.M.-E., K.S., H. Rangel-Villalobos, D.P., D.L., P. Norman, P. Parham, M.R., T.S. Korneliussen, P. Skoglund, T.V.O. Hansen, F.C. Nielsen, T.L. Pierre, M. Crawford, T. Kivisild, R. Malhi, R. Villems, M. Jakobsson, F. Balloux, A. Manica, C.D.B., R. Nielsen, E.W., unpublished results). Because of low coverage in the ancient samples, most positions were covered by 0 or 1 read; for positions covered by more than one read, a random read was sampled and the site was made homozygous. For PCA analysis, SNPs were filtered for minor allele frequencies ≥5% and PCAs were constructed with smartpca.35 Principal components were computed with only the modern samples, and the ancient individual was then projected onto the PCA. PCA plots were created with R v.2.14.2.
Results
We tested WISC on 12 human aDNA libraries derived from non-frozen-preserved specimens: four Iron and Bronze Age human teeth from Bulgaria, seven pre-Columbian human mummies from Peru, and one Bronze Age human hair sample from Denmark. The DNA was extracted and the libraries built in a dedicated clean room (see Material and Methods). Shotgun sequencing prior to capture indicated that all libraries contained low levels of endogenous DNA (average 1.2%, range 0.04%–6.2%; see Table 1). To allow for direct comparison, the numbers of reads in the pre- and postcapture libraries were adjusted to be equal prior to mapping by taking the first n reads from the respective raw fastq files (Table 1). In the case of the hair and bone libraries, the results for 1 million reads are shown for ease of comparison with the tooth libraries. Prior to mapping, the paired-end reads were merged where possible, any remaining adaptor sequence was trimmed from the merged and unmerged reads, and reads containing only adaptor sequence (i.e., adaptor dimers) were discarded. As shown in Table 1, whole-genome capture decreased the number of reads discarded at this step, reducing the sequencing capacity taken up by these uninformative sequences, which are common contaminants in aDNA sequencing libraries.
Table 1.
Results of Sequencing 12 Ancient Samples Before and After WISC
| ID | Pre- or Postcapture | Read Pairs (#) | Read Pairs Discarded (Contain Adaptor) (#) | Individual Reads after Merging and Trimming (#) | Mapped Human Reads (%) | Fold Enrichment in # Mappeda | Unique Reads (%) | Fold Enrichment in # Uniquesb | Duplicate Reads (of Mapped) (%) | Precapture Reads Present in Postcapture (%) | Positions Covered (#) | Reads in Repeats (%) | Fold mtDNA Coverage | SNPs Overlapping with 1000G (#) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bulgaria 1500–500 BC Tooth | ||||||||||||||
| V2 | pre | 1,390,960 | 98,697 | 1,331,130 | 0.3% | 0.3% | 9% | 38,908 | 34% | 0.01 | 5,281 | |||
| post | 1,390,960 | 30,681 | 1,446,302 | 20.2% | 70 | 2.7% | 10 | 87% | 46% | 4,077,324 | 45% | 0.4 | 40,583 | |
| P192-1 | pre | 819,844 | 118,493 | 705,234 | 4.3% | 3.9% | 9% | 2,248,978 | 35% | 0.3 | 30,081 | |||
| post | 819,844 | 14,993 | 829,256 | 23.2% | 6 | 7.8% | 2 | 66% | 52% | 5,000,399 | 45% | 2 | 67,221 | |
| T2G2 | pre | 1,596,526 | 20,644 | 1,633,734 | 0.05% | 0.05% | 14% | 45,111 | 33% | 0.3 | 597 | |||
| post | 1,596,526 | 16,168 | 1,870,076 | 7.4% | 159 | 0.3% | 8 | 96% | 15% | 303,848 | 30% | 16.1 | 4,068 | |
| K8 | pre | 1,817,223 | 76,872 | 1,980,966 | 1.0% | 0.8% | 14% | 1,506,968 | 35% | 0.06 | 19,960 | |||
| post | 1,817,223 | 15,322 | 2,537,422 | 36.0% | 48 | 3.4% | 5 | 90% | 90% | 7,093,382 | 37% | 0.3 | 94,394 | |
| Denmark ∼1350 BC Hair | ||||||||||||||
| M4 | pre | 1,000,000 | 210,491 | 828,494 | 0.5% | 0.5% | 7% | 364,855 | 35% | 0.06 | 5,115 | |||
| post | 1,000,000 | 26,695 | 1,269,181 | 36.6% | 114 | 2.2% | 8 | 94% | 70% | 3,152,432 | 37% | 0.6 | 40,340 | |
| Peru ∼900–1500 AD Bone | ||||||||||||||
| NA39 | pre | 1,000,000 | 50,534 | 1,192,685 | 1.1% | 1.0% | 14% | 1,066,246 | 34% | 5.3 | 14,751 | |||
| post | 1,000,000 | 6,472 | 1,419,774 | 59.5% | 62 | 2.1% | 3 | 96% | 56% | 4,301,252 | 37% | 19.9 | 40,048 | |
| NA40 | pre | 1,000,000 | 89,763 | 1,010,267 | 0.72% | 0.7% | 2% | 642,917 | 36% | 0.05 | 9,119 | |||
| post | 1,000,000 | 24,214 | 1,191,241 | 26.5% | 44 | 7.7% | 13 | 70% | 42% | 17,253,987 | 38% | 2.7 | 129,872 | |
| NA41 | pre | 1,000,000 | 76,485 | 1,358,860 | 0.30% | 0.3% | 10% | 334,441 | 33% | 0.01 | 4,621 | |||
| post | 1,000,000 | 12,319 | 1,628,753 | 23.2% | 92 | 1.3% | 6 | 94% | 75% | 1,966,403 | 36% | 0.6 | 26,118 | |
| NA42 | pre | 1,000,000 | 74,460 | 1,117,389 | 6.2% | 4.9% | 20% | 5,197,492 | 36% | 3.5 | 73,266 | |||
| post | 1,000,000 | 14,847 | 1,341,546 | 41.0% | 8 | 7.9% | 2 | 80% | 57% | 16,609,757 | 37% | 10.9 | 147,243 | |
| NA43 | pre | 1,000,000 | 116,780 | 966,013 | 0.18% | 0.2% | 11% | 113,616 | 38% | 0.01 | 1,553 | |||
| post | 1,000,000 | 81,544 | 1,036,263 | 7.4% | 45 | 0.6% | 4 | 91% | 68% | 579,192 | 40% | 0.4 | 6,337 | |
| NA47 | pre | 1,000,000 | 92,800 | 973,662 | 0.13% | 0.1% | 4% | 93,784 | 38% | 0.01 | 1,279 | |||
| post | 1,000,000 | 32,741 | 1,107,880 | 9.1% | 77 | 0.8% | 7 | 90% | 58% | 833,067 | 42% | 0.5 | 9,393 | |
| NA50 | pre | 1,000,000 | 126,605 | 1,001,135 | 0.035% | 0.03% | 3% | 15,135 | 40% | 0 | 217 | |||
| post | 1,000,000 | 37,653 | 1,292,570 | 1.7% | 61 | 0.3% | 10 | 78% | 24% | 377,875 | 43% | 0.5 | 3,062 | |
The first four samples were adjusted to have identical numbers of pre- and postcapture reads, based on the number of reads obtained from MiSeq sequencing of the postcapture libraries. The last eight samples were adjusted to 1 million reads each for ease of comparison with the first four samples. Prior to mapping, overlapping paired-end reads were computationally merged, and adapters were trimmed from both merged and unmerged reads (note that the number of reads listed after merging and trimming includes both forward and reverse reads for pairs that were not merged). Mapped reads were filtered for mapping qualities ≥30. Overlap with repeats was determined by intersection with the RepeatMasker annotation of human genome repeats. 1000G: 1000 Genomes reference panel.
Does not vary with amount of sequencing
Varies with library complexity and amount of sequencing
After capture, we observed enrichments ranging from 6-fold to 159-fold for number of reads mapping to the human genome at MAPQ ≥ 30, resulting in 1.6%–59.2% of reads mapping after capture. For unique fragments, we observed enrichments of 2-fold to 13-fold (Table 1); however, the fraction of unique reads changes with different amounts of sequencing and also is sensitive to the level of complexity of the original library (Figures 2A and 2B and Figure S1 available online). The level of enrichment was negatively correlated with the amount of endogenous DNA present in the precapture library—the higher the amount prior to capture, in general, the lower the degree of enrichment (e.g., samples P192-1 and NA42; see Table 1). This phenomenon has previously been observed for the enrichment of pathogen DNA in clinical samples.36 The number of unique reads increased in all cases; however, even after sequencing of 1 million reads, most of the unique molecules in the postcapture libraries had already been observed, as evidenced by the high levels of clonality (66%–96%) in these libraries. We generally captured a large proportion (15%–90%) of the endogenous fragments observed in the precapture libraries (Table 1). This number also increased with additional sequencing (see Figure 2C and discussion below). We observed only a slight increase in the percent of fragments falling within known repetitive regions of the genome (Table 1), with the average increasing from 36% precapture to 39% postcapture. There was no obvious correlation with the amount of starting DNA in the sample. Thus, at least for libraries containing very low levels of endogenous DNA, biased enrichment of repetitive sequences does not appear to be a problem. In the postcapture libraries, the unmapped fraction had a similar composition of environmental (primarily bacterial) sequences to the precapture library (data not shown).
Figure 2.
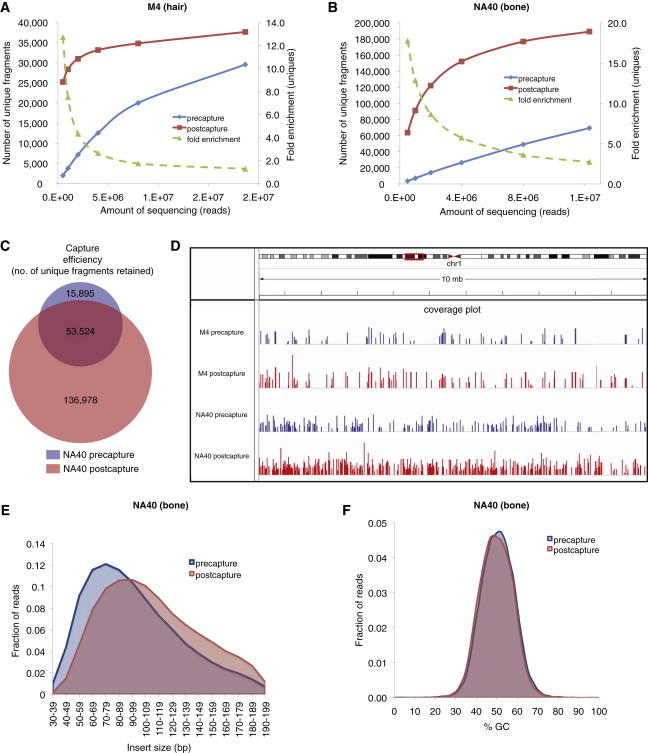
Results of Increased Sequencing of Samples M4 and NA40
(A) Yield of unique fragments for M4 (Bronze Age hair) precapture (blue) and postcapture (red) libraries with increasing amounts of sequencing. The fold enrichment in number of unique reads with increasing amounts of sequencing is plotted in green, with values on the secondary y axis.
(B) Yield of unique fragments for NA40 (Peruvian bone) precapture (blue) and postcapture (red) libraries with increasing amounts of sequencing. The fold enrichment in number of unique reads with increasing amounts of sequencing is plotted in green, with values on the secondary y axis.
(C) Venn diagram showing the overlap between the NA40 pre- and postcapture libraries based on sequencing of 12.3 million reads.
(D) Coverage plot of the M4 and NA40 libraries based on sequencing of 18.6 million and 12.3 million reads, respectively. Shown is a random 10-megabase segment of chromosome 1. Coverage was calculated in 1 kb windows across the region.
(E) Insert size distribution for NA40 pre- and postcapture libraries.
(F) Percent GC content of reads for NA40 pre- and postcapture libraries.
Importantly for aDNA studies, which have historically relied on identifying mtDNA haplogroups from ancient samples, >1× coverage of the mtDNA was achieved with 1 million reads for 5 of the 12 postcapture libraries (Table 1). For these five samples, we were able to tentatively call mtDNA haplogroups (Table S1). Intersection with the 1000 Genomes Project reference panel37 demonstrated that capture increased the number of unique SNPs between 2- and 14-fold (Table 1), increasing the resolution of principal component analysis plots involving these individuals (see Discussion below). We did not observe any bias in X chromosome capture resulting from the use of a male Masai individual (NA21732) for the capture probes: the proportion of reads mapped to the X chromosome remained approximately the same before and after capture (Table S2). Furthermore, for the 17 total SNPs that changed alleles between the eight pre- and postcapture libraries sequenced to higher levels (0–6 SNPs per sample), only ten SNPs changed from not matching to matching NA21732 after capture (Table S3). Thus, at least for modern humans, divergence between the probe and target on the population level does not appear to produce significant allelic bias in the postcapture library. However, it is possible that more noticeable effects could be seen for indels or copy number variants if high enough coverage were obtained.
To determine how many new unique fragments are discovered with increasing amounts of sequencing, we sequenced the hair and bone libraries to higher coverage (∼8–18 million reads via multiplexed Illumina HiSeq sequencing). Figures 2A and 2B show the results of increasing levels of sequencing of libraries NA40 (Peruvian bone) and M4 (Danish hair), which are generally representative of the patterns we saw for the remaining six libraries (see Figure S1). For NA40, although the yield of unique fragments from the precapture library increased in a linear manner, the yield from the postcapture library increased rapidly with initial sequencing and began to plateau after approximately four million reads (Figure 2A). Similarly, there was a rapid initial increase in unique fragments up to approximately five million reads sequenced for both the pre- and postcapture M4 libraries; this increase then slowed with sequencing up to 18.7 million reads (Figure 2B). The results from the remaining six libraries are shown in Figure S1. These plots also demonstrate that the fold enrichment in unique reads decreases with increasing amounts of sequencing (Figures 2A, 2B, and S1), as the precapture library begins to be sampled more exhaustively. Thus, WISC allowed us to access the majority of unique reads present in the postcapture library with even low levels of sequencing, such as those obtainable with a single run on an Illumina MiSeq.
We next examined how efficiently we were able to capture endogenous molecules present in the precapture library with higher levels of sequencing. As shown in Figure 2C, for library NA40, 77% (53,524) of unique fragments in the precapture library were also sequenced in the postcapture library with 12,285,216 reads sequenced; note that this fraction was 42% for 1 million reads sequenced (Table 1). Furthermore, an additional 136,978 unique fragments were sequenced after capture with the same amount of sequencing (Figure 2C). These fragments were generally evenly distributed across the genome; Figure 2D shows a coverage plot for libraries M4 and NA40 at a random 10 Mb region of chromosome 1. The size of the fragments in the postcapture libraries tended to be slightly larger (Figure 2E), probably because of the stringency of the hybridization and wash steps—which could be decreased but would, we predict, result in lower levels of enrichment—and some loss during purifications, resulting in the preferential retention of longer fragments. Because aDNA is highly fragmented compared to modern contaminants, we tested whether the overall DNA damage patterns (an increase in C-to-T and G-to-A transitions at the ends of fragments, diagnostic of ancient DNA38) also changed with the change in fragment size after capture. We observed that the overall DNA damage patterns remained similar in the pre- and postcapture libraries (Table S4), both for the libraries as a whole and when they were partitioned by size (<70 bp and >70 bp). The patterns for libraries V2, K8, and M4 are not typical of ancient DNA, possibly because of favorable preservation conditions, sample contamination prior to capture, or both (Table S4). Finally, the GC content of reads in the postcapture library was slightly decreased (Figure 2F), as previously observed for in-solution exome capture.14
The ultimate goal of sequencing DNA from ancient samples is usually to identify informative variation for population genetics analyses. We used the SNPs identified by intersections with the 1000 Genomes reference panel (see Table 1 and discussion above) to perform principal component analysis (PCA). Only SNPs with a minor allele frequency ≥5% were used for this analysis. Figure 3 shows the pre- and postcapture PCAs for samples V2 (Bulgarian), M4 (Danish hair), and NA40 (Peruvian mummy); the PCAs for the remaining samples are shown in Figure S2. As expected, the two European samples fell into the European clusters on the PCA both before capture (Figures 3A and 3C) and after capture (Figures 3B and 3D). However, the increased number of SNPs after capture allows for improved resolution of the subcontinental affiliation of each ancient sample (Figures 3B and 3D). PCAs with only the European populations in 1000 Genomes further resolve the placement of some of these samples after capture (Figure S3). For the Peruvian mummies, we also included 10 Native American individuals from Central and South America in the PCA (Figures 3E and 3F). Interestingly, all of the mummies fell between the Native American populations (KAR, MAY, AYM) and East Asian populations (JPT, CHS, CHB), as would be expected for a nonadmixed Native American individual (Figures 3E, 3F, and S2). These mummies belonged to the pre-Columbian Chachapoya culture, who, by some accounts, were unusually fair-skinned,39 suggesting a potential for pre-Columbian European admixture. However, based on our preliminary results, these individuals appear to have been ancestrally Native American.
Figure 3.
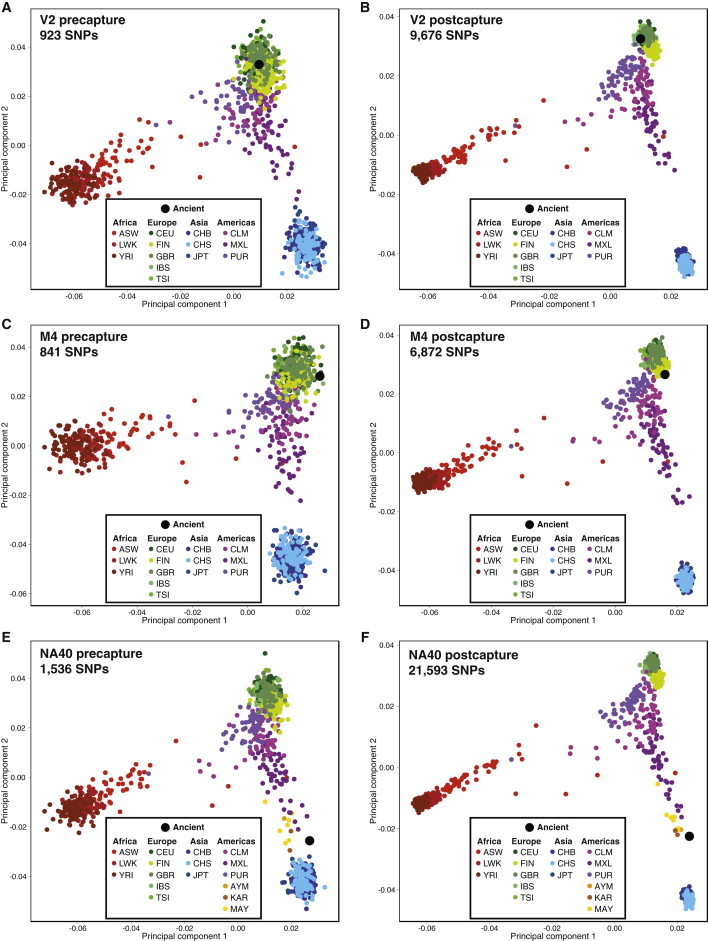
Principal Component Analysis of Pre- and Postcapture Samples Based on Sequencing One Million Reads Each
Principal component analysis of SNPs overlapping between the 1000 Genomes reference panel and each ancient individual, with Native American individuals also included in (E) and (F). The principal components were calculated with the modern individuals only, and the ancient individual was then projected onto the plot. Shown are (A) V2 (Bulgarian tooth) precapture and (B) postcapture; (C) M4 (Bronze Age hair) precapture and (D) postcapture; and (E) NA40 (Peruvian bone) precapture and (F) postcapture. Population key: ASW, Americans of African ancestry in SW USA; AYM, Aymara from the Peruvian Andes; CEU, Utah residents (CEPH) with Northern and Western European ancestry; CHB, Han Chinese in Beijing, China; CHS, Southern Han Chinese; CLM, Colombians from Medellin, Columbia; FIN, Finnish in Finland; GBR, British in England and Scotland; IBS, Iberian population in Spain; JPT, Japanese in Tokyo, Japan; KAR, Karitiana from the Brazilian Amazon; LWK, Luhya in Webuye, Kenya; MAY, Mayan from Mexico; MXL, Mexican ancestry from Los Angeles, USA; PUR, Puerto Ricans from Puerto Rico; TSI, Toscani in Italy; YRI, Yoruba in Ibadan, Nigeria.
Discussion
We have developed a whole-genome in-solution capture method, WISC, that can be used to highly enrich the endogenous contents of aDNA sequencing libraries, thus reducing the amount of sequencing required to sample the majority of unique fragments in the library.
Previous methods for targeted enrichment of aDNA libraries have focused only on a subset of the genome (e.g., the mitochondrial genome, a single chromosome, or a subset of SNPs).8,11–13 Although these methods have generated useful information while reducing sequencing costs, they all involve discarding a large proportion of potentially informative sequences, often from samples that already contain a reduced representation of the genome.
Excluding initial library costs (which are the same for all methods) and sequencing, the cost to perform WISC is approximately $50/sample, primarily because of the cost of the streptavidin-coated beads used for capture. In contrast, in-solution exome capture via a commercial kit is approximately $1,000/sample, and we calculate the previously reported chromosome 21 capture method8 to have an initial cost of approximately $5,000 (to purchase the nine one-million-feature DNA arrays used to generate the RNA probes), plus a cost of ∼$50/sample for the actual capture experiments. Finally, if one desired to array-synthesize probes tiled across the entire genome—i.e., a similar approach to the chromosome 21 capture but for the whole genome—we calculate that it would cost ∼$300,000–$400,000 to purchase the necessary arrays. All of these methods would reduce sequencing costs to a large extent compared to sequencing the precapture library, but, as noted above, several do so at the cost of discarding potentially informative sequences.
With regard to the data generated, the most similar method to WISC for aDNA capture is chromosome 21 capture.8 That method was performed on libraries from a single specimen from the Tianyuan Cave in China that contained 0.01%–0.03% endogenous DNA. Prior to collapsing duplicates, the chromosome 21-capture libraries contained 46.8% endogenous DNA (∼4.4 million out of ∼9.4 million reads ≥35 bp; the five libraries were sequenced on an entire lane of Illumina GAIIx, but the exact number of reads generated is not stated).8 WISC-enriched libraries contained 1.6%–59.2% endogenous DNA after capture, although it should be noted that most of our libraries started with higher levels of endogenous DNA than did the Tianyuan libraries. After the removal of duplicate reads, the Tianyuan libraries had 8.4% uniques (789,925), whereas the WISC libraries contained 0.3%–7.9% unique reads. It is difficult to directly compare these numbers because the underlying complexities of the libraries differ; however, at least with regard to the total yield of target DNA, these two methods appear to perform similarly. Future studies directly comparing these methods will be required to determine which one retrieves the highest number of informative variants with the least amount of sequencing.
Our test libraries, like many aDNA libraries created from similar specimens,5,6 did not contain sufficient endogenous DNA to cover the entire genome, making it impossible to call genotypes for these samples; indeed, >99.9% of sites were covered by 0 or 1 read. Identifying SNPs from these samples is further complicated by the presence of DNA damage, specifically C-to-T and G-to-A transitions.38 Thus, in order to more confidently identify SNPs, we intersected our data set with a list of known SNPs from the 1000 Genomes reference panel. The likelihood that a damaged SNP will be found at the exact same position and with a matching allele as a SNP from the reference set is quite low, and thus we were able to leverage the identified SNPs to perform informative population genetics analyses without filtering out large subsets of the data (Figures 3, S2, and S3). A similar approach was taken by two previous studies.5,6 It should be noted that a reference panel, preferably with full genome sequence data (although this is not essential), is required for this type of analysis of poorly preserved specimens with low levels of genome coverage. However, because WISC reduces the required amount of sequencing required per library, multiple individuals from the same population can be analyzed, a key consideration for studies focusing on the spatial and temporal distribution of ancient populations.
As shown in Table 1, we also obtained >1× coverage of the mtDNA for five of the libraries. This number is lower than the typical enrichment achieved when targeting the mtDNA alone via capture,11 but this is not surprising given that a wider range of sequences is being targeted. A similar phenomenon was observed in the capture of nuclear and organellar DNA from ancient maize.40 We were able to tentatively call mtDNA haplogroups for these samples (Table S1). The two Bulgarian Iron Age individuals (P192-1 and T2G5) fell into haplogroups U3b and HV(16311), respectively. Haplogroup U3 is especially common in the countries surrounding the Black Sea, including Bulgaria, and in the Near East, and HV is also found at low frequencies in Europe and peaks in the Near East.41 The three Peruvian mummies fell into haplogroups B2, M (an ancestor of D), and D1, all derived from founder Native American lineages and previously observed in both pre-Columbian and modern populations from Peru.42
In our experiments, capture yield was limited by the degree of complexity of the starting libraries and could potentially be increased by improved aDNA extraction and library preparation methods.9,43,44 A recently published novel method for single-stranded aDNA library preparation has enabled researchers to obtain high-coverage ancient genomes from ancient hominins9,44 by retaining many small, damaged DNA fragments that would have been lost in conventional library preparation methods. Although this method is a breakthrough for the field of aDNA, it does not necessarily decrease the cost of sequencing samples with low endogenous DNA contents, because the single-stranded library still contains high levels of contaminating DNA. We predict that the combination of this method and WISC may substantially increase the complexity and endogenous DNA contents of aDNA libraries. However, it will probably be necessary to reduce the stringency of the WISC hybridization conditions in order to retain more of these smaller fragments during capture.
Finally, because it is not necessary to design an array for our method (i.e., a sequenced genome is not required), WISC could also be used to capture DNA from specimens of extinct species by creating baits from the genome of an extant relative. The effect of sequence divergence between species on capture efficiency remains to be determined, but chimpanzee-targeted probes have successfully been used to capture human and gorilla sequences.45 In addition, WISC has applications in other contexts, such as the enrichment of DNA in forensic, metagenomic, and museum specimens.
Acknowledgments
The authors would like to thank members of the C.D.B. lab, especially P. Underhill and S. Shringarpure, for helpful discussion, and M.C. Yee and A. Adams for assistance with experiments. Support for this work was provided by National Institutes of Health grants HG005715 and HG003220 and an NRSA Postdoctoral Fellowship (NHGRI) to M.L.C. The sample M4 was obtained and DNA extracted as part of “The Rise” project funded by the European Research Council under the European Union’s Seventh Framework programme (FP/2007-2013)/ERC Grant Agreement n. 269442 - THE RISE. Portions of this manuscript are subject to one or more patents pending. C.D.B. consults for Personalis, Inc., Ancestry.com, Invitae (formerly Locus Development), and the 23andMe.com project “Roots into the Future.” None of these entities played any role in the design of the research or interpretation of the results presented here.
Contributor Information
William J. Greenleaf, Email: wjg@stanford.edu.
Carlos D. Bustamante, Email: cdbustam@stanford.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Phase 1 data set, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521
SeqPrep, https://github.com/jstjohn/SeqPrep
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.-Y. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rasmussen M., Li Y., Lindgreen S., Pedersen J.S., Albrechtsen A., Moltke I., Metspalu M., Metspalu E., Kivisild T., Gupta R. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasmussen M., Guo X., Wang Y., Lohmueller K.E., Rasmussen S., Albrechtsen A., Skotte L., Lindgreen S., Metspalu M., Jombart T. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller A., Graefen A., Ball M., Matzas M., Boisguerin V., Maixner F., Leidinger P., Backes C., Khairat R., Forster M. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun. 2012;3:698. doi: 10.1038/ncomms1701. [DOI] [PubMed] [Google Scholar]
- 5.Sánchez-Quinto F., Schroeder H., Ramirez O., Avila-Arcos M.C., Pybus M., Olalde I., Velazquez A.M., Marcos M.E., Encinas J.M., Bertranpetit J. Genomic affinities of two 7,000-year-old Iberian hunter-gatherers. Curr. Biol. 2012;22:1494–1499. doi: 10.1016/j.cub.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Skoglund P., Malmström H., Raghavan M., Storå J., Hall P., Willerslev E., Gilbert M.T., Götherström A., Jakobsson M. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 7.Reich D., Green R.E., Kircher M., Krause J., Patterson N., Durand E.Y., Viola B., Briggs A.W., Stenzel U., Johnson P.L. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Q., Meyer M., Gao X., Stenzel U., Burbano H.A., Kelso J., Pääbo S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl. Acad. Sci. USA. 2013;110:2223–2227. doi: 10.1073/pnas.1221359110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer M., Kircher M., Gansauge M.T., Li H., Racimo F., Mallick S., Schraiber J.G., Jay F., Prüfer K., de Filippo C. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert M.T., Tomsho L.P., Rendulic S., Packard M., Drautz D.I., Sher A., Tikhonov A., Dalén L., Kuznetsova T., Kosintsev P. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science. 2007;317:1927–1930. doi: 10.1126/science.1146971. [DOI] [PubMed] [Google Scholar]
- 11.Maricic T., Whitten M., Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burbano H.A., Hodges E., Green R.E., Briggs A.W., Krause J., Meyer M., Good J.M., Maricic T., Johnson P.L., Xuan Z. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briggs A.W., Good J.M., Green R.E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajkovic D., Kucan Z. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 14.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nekhrizov, G., and Tzvetkova, J. (2012). Ritual Pit Complexes in Iron Age Thrace: The Case Study of Svilengrad. In Anatolian Iron Ages 7 The Proceedings of the Seventh Anatolian Iron Ages Colloquium Held at Edirne, 19-24 April 2010. pp. 177–209.
- 16.Nekhrizov, G. (2009). Nekropol ot rannata zhelyazna epoha pri s. Stambolovo, Haskovsko. Arheologicheski otkritiya i razkopki prez 2008 g Sofia, pp. 266–271.
- 17.Leshtakov, K., Hristova, R., and Mihailov, Y. (2010). Nekropol ot kasnata bronzova epoha pri s. Vratitsa, obshtina Kameno. Yugoiztochna Bulgaria prez II - I hilyadoletie pr Hr Varna, pp. 22–37.
- 18.Dimitrova D. Tumuli Graves – Status Symbol of the Dead in the Bronze and Iron Ages in Europe. Archaeopress; Oxford: 2012. 5th-4th c. BC Thracian Orphic Tumular Burials in Sliven Region (Southeastern Bulgaria) pp. 77–84. [Google Scholar]
- 19.Mounds with preserved oak coffins dendrochronologically investigated. Acta Archaeologica. 2006;77:190–233. [Google Scholar]
- 20.Guillén S. Momies Chachapoyas du Pérou ancien. In: Schlanger N., Taylor A.-C., editors. La préhistoire des autres, Perspectives archeologiques et anthropologiques. Inrap; Paris: 2012. pp. 321–336. [Google Scholar]
- 21.Willerslev E., Cooper A. Ancient DNA. Proc. Biol. Sci. 2005;272:3–16. doi: 10.1098/rspb.2004.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer, M., and Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 2010, pdb prot5448. [DOI] [PubMed]
- 24.Yang D.Y., Eng B., Waye J.S., Dudar J.C., Saunders S.R. Technical note: improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 1998;105:539–543. doi: 10.1002/(SICI)1096-8644(199804)105:4<539::AID-AJPA10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M., Sawyer S., Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jónsson H., Ginolhac A., Schubert M., Johnson P.L., Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682–1684. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 32.Zhidkov I., Nagar T., Mishmar D., Rubin E. MitoBamAnnotator: A web-based tool for detecting and annotating heteroplasmy in human mitochondrial DNA sequences. Mitochondrion. 2011;11:924–928. doi: 10.1016/j.mito.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 33.van Oven M., Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 34.Skoglund P., Stora J., Gotherstrom A., Jakobsson M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 2013;40:4477–4482. [Google Scholar]
- 35.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melnikov A., Galinsky K., Rogov P., Fennell T., Van Tyne D., Russ C., Daniels R., Barnes K.G., Bochicchio J., Ndiaye D. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011;12:R73. doi: 10.1186/gb-2011-12-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briggs A.W., Stenzel U., Johnson P.L.F., Green R.E., Kelso J., Prüfer K., Meyer M., Krause J., Ronan M.T., Lachmann M., Pääbo S. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Church W. Chachapoya Indians. In: Birx H.J., editor. Encyclopedia of Anthropology. Sage Publications, Inc.; Thousand Oaks: 2006. pp. 469–477. [Google Scholar]
- 40.Avila-Arcos M.C., Cappellini E., Romero-Navarro J.A., Wales N., Moreno-Mayar J.V., Rasmussen M., Fordyce S.L., Montiel R., Vielle-Calzada J.P., Willerslev E., Gilbert M.T. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Sci. Rep. 2011;1:74. doi: 10.1038/srep00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karachanak S., Carossa V., Nesheva D., Olivieri A., Pala M., Hooshiar Kashani B., Grugni V., Battaglia V., Achilli A., Yordanov Y. Bulgarians vs the other European populations: a mitochondrial DNA perspective. Int. J. Legal Med. 2012;126:497–503. doi: 10.1007/s00414-011-0589-y. [DOI] [PubMed] [Google Scholar]
- 42.Fehren-Schmitz L., Warnberg O., Reindel M., Seidenberg V., Tomasto-Cagigao E., Isla-Cuadrado J., Hummel S., Herrmann B. Diachronic investigations of mitochondrial and Y-chromosomal genetic markers in pre-Columbian Andean highlanders from South Peru. Ann. Hum. Genet. 2011;75:266–283. doi: 10.1111/j.1469-1809.2010.00620.x. [DOI] [PubMed] [Google Scholar]
- 43.Dabney J., Knapp M., Glocke I., Gansauge M.T., Weihmann A., Nickel B., Valdiosera C., García N., Pääbo S., Arsuaga J.L., Meyer M. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA. 2013;110:15758–15763. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gansauge M.T., Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013;8:737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]
- 45.Good J.M., Wiebe V., Albert F.W., Burbano H.A., Kircher M., Green R.E., Halbwax M., André C., Atencia R., Fischer A., Pääbo S. Comparative population genomics of the ejaculate in humans and the great apes. Mol. Biol. Evol. 2013;30:964–976. doi: 10.1093/molbev/mst005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.