

Abstract
Genome-wide association studies (GWASs) have enabled the discovery of common genetic variation contributing to normal and pathological traits and clinical drug responses, but recognizing the precise targets of these associations is now the major challenge. Here, we review recent approaches to the functional follow-up of GWAS loci, including fine mapping of GWAS signal(s), prioritization of putative functional SNPs by the integration of genetic epidemiological and bioinformatic methods, and in vitro and in vivo experimental verification of predicted molecular mechanisms for identifying the targeted genes. The majority of GWAS-identified variants fall in noncoding regions of the genome. Therefore, this review focuses on strategies for assessing likely mechanisms affected by noncoding variants; such mechanisms include transcriptional regulation, noncoding RNA function, and epigenetic regulation. These approaches have already accelerated progress from genetic studies to biological knowledge and might ultimately guide the development of prognostic, preventive, and therapeutic measures.
Main Text
Introduction
Since the advent of high-density genotyping arrays, researchers have used genome-wide associations studies (GWASs) to identify over 1,000 loci associated with a multitude of physiological traits.1 These studies exploit the nonrandom coinheritance of genetic variants (linkage disequilibrium [LD]) to simultaneously assay hundreds of thousands of markers for an association with any given trait.2 Given that the SNPs on genotyping chips are “tags” for haplotypes on which the directly functional variants reside, the next major challenge lies in moving from associated tag SNPs to finding the strongest candidate causal variants and then identifying their target gene(s). We define a causal variant as one that influences a molecular or cellular process to affect a human phenotype. Contrary to early expectations, few GWAS-identified variants are predicted to disrupt protein-coding regions, and approaches to determining their functional consequences are reviewed elsewhere.3 The vast majority of GWAS tag SNPs lie in intergenic or intronic regions (approximately 88%1) and therefore are likely to influence gene regulation (assuming that the same is true for the correlated candidate causal SNPs). There are now a limited number of studies that have pursued the function of GWAS hits, some of which have been defined by fine mapping (Table 1). However, the relevance of those studies that lack comprehensive fine mapping is questionable. Here, we review recent approaches to post-GWAS fine mapping and functional evaluation of noncoding variants. Our proposed functional pipeline for the follow-up of GWAS loci addresses the prioritization of putative functional SNPs (Figure 1). We describe the integration of genetic data and statistical analysis, computational approaches, and the use of specific assays to determine the contribution of short-listed variants to the regulation of particular genes and pathways.
Table 1.
Functional Genetic Variants and/or Target Genes Successfully Identified at GWAS Signals
| Disease or Phenotype | Locus | Functional SNPs or Regions | Target Gene(s) | Fine Mappinga | Key Methods | References |
|---|---|---|---|---|---|---|
| Low-density lipoprotein cholesterol levels (MIM 605028) | 1p13 | rs12740374 | SORT1 (MIM 602458) | − | eQTL, reporter assays, EMSAs, mouse models | Musunuru et al.4 |
| Fetal hemoglobin levels | 2p16 | rs1427407, rs7606173 | BCL11A (MIM 606557) | − | 3C, ChIP-seq, reporter assays, transgenic mouse models, allele-specific expression, TALENs | Bauer et al.5 |
| Chronic obstructive pulmonary disease (MIM 606963) | 4q31 | rs1542725 | HHIP (MIM 606178) | ++ | 3C, ChIP, reporter assays, EMSAs | Zhou et al.6 |
| Prostate cancer (MIM 176807) | 5p15 | rs12653946 | IRX4 (MIM 606199) | ++ | eQTL, reporter assays, ChIP, EMSAs | Nguyen et al.7 |
| Breast cancer (MIM 114480), ovarian cancer (MIM 167000) | 5p15 | breast and ovarian cancer risk regions | TERT (MIM 187270) | ++ | FAIREs, reporter assays, splicing assays | Bojesen et al.8 |
| Colorectal cancer (MIM 120435) | 8q23 | rs16888589 | EIF3H (MIM 603912) | ++ | 3C, reporter assays, EMSAs, allele-specific expression, in vivo transgenic assays | Pittman et al.9 |
| Colorectal cancer | 8q24 | rs6983267 | c-MYC (MIM 190080) | + | 3C, EMSAs, reporter assays, allele-specific ChIP, microarrays, transgenic mouse models. | Tuupanen et al.,10 Wright et al.11 |
| Prostate, breast, and colorectal cancer | 8q24 | prostate, breast, and colorectal cancer risk regions | c-MYC | − | 3C, reporter assays, ChIP | Ahmadiyeh et al.,12 Sotelo et al.13 |
| CAD (MIM 608320) | 9p21 | rs10811656 or rs10757278 | CDKN2A (MIM 600160), CDKN2B (MIM 600431), MTAP (MIM 156540), IFNA21 (MIM 147584) | ++ | 3C-DSL, ChIP, FISH | Harismendy et al.14 |
| CAD | 9p21 | CAD risk region | CDKN2A, CDKN2B | − | targeted mouse models, allele-specific expression | Visel et al.15 |
| Breast cancer | 10q26 | rs7895676, rs2981578 | FGFR2 (MIM 176943) | − | eQTL, EMSAs, ChIP, reporter assays | Meyer et al.16 |
| Breast cancer | 11q13 | rs554219, rs78540526, rs75915166 | CCND1 (MIM 168461) | ++ | 3C, reporter assays, EMSAs, allele-specific ChIP | French et al.17 |
| Renal cancer (MIM 144700) | 11q13 | renal cancer risk region | CCND1 | − | ChIP, FAIREs, 3C, three-dimensional FISH, allele-specific expression | Schodel et al.18 |
| Asthma (MIM 600807) and autoimmune disease (MIM 109100) | 17q12 17-q21 | rs12936231, rs80667378 | ZPBP2 (MIM 608499), GSDMB (MIM 611221), ORMDL3 (MIM 610075) | ++ | FAIREs, allele-specific ChIP, EMSAs, reporter assays | Verlaan et al.19 |
| Prostate cancer | 17q24 | rs8072254, rs1859961 | SOX9 (MIM 608160) | − | 3C, reporter assays, ChIP-seq, allele-specific ChIP | Zhang et al.20 |
Abbreviations are as follows: 3C, chromatin conformation capture; 3C-DSL, 3C with DNA selection and ligation; CAD, coronary artery disease; ChIP, chromatin immunoprecipitation; ChIP-seq, ChIP sequencing; eQTL, expression quantitative-trait loci; EMSA, electrophoretic mobility shift assay; FAIRE, formaldehyde-assisted isolation of regulatory element; FISH, fluorescence in situ hybridization; and TALEN, transcription-activator-like effector nuclease.
Symbols are as follow: −, no fine mapping; +, fine mapping using imputation from the GWAS tag SNPs; and ++, dense genotyping and imputation or resequencing of LD block.
Figure 1.
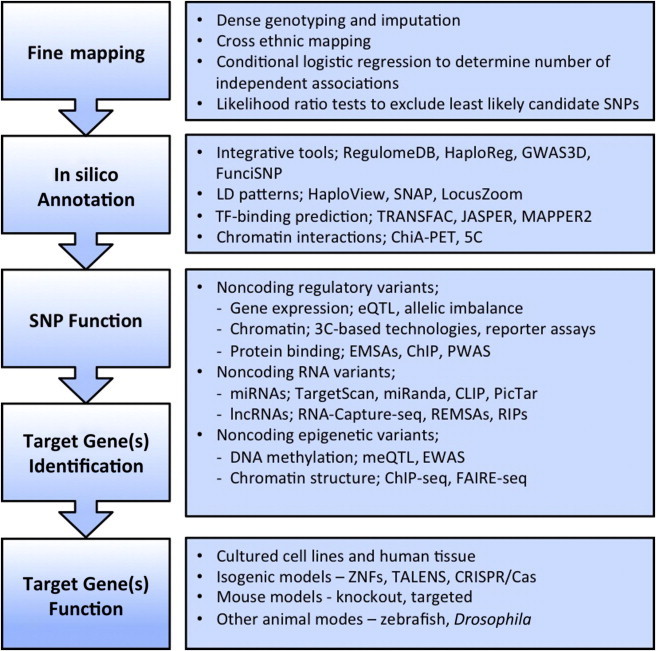
Workflow for Functionally Analyzing and Interpreting GWAS Loci
Fine Mapping of Associated Loci
GWASs reveal associations between specific genomic loci and genetic traits or diseases via a set of marker SNPs designed to tag all known common variants in the genome.21 Human history and ancestry have caused complete segments of DNA, termed haplotype blocks, to be shared within populations.22 A typical haplotype block is illustrated in Figure 2B, where segments of correlated SNPs are separated by recombination hot spots (the gaps between the gray-scale triangular matrices). Patterns of block structure differ between ethnic groups, and on average, European-ancestry populations have more highly correlated SNPs and longer haplotype blocks (Figure 2B, top panel) than do populations of African or Asian ancestry (Figure 2B, lower panels).23 It is possible to infer which type of segment a person carries by typing selected markers within each haplotype block, obviating the need to type the majority of variants in the human genome.2,23 Modern GWAS genotyping chips typically contain 300,000–5,000,000 SNPs, chosen for their correlation with, and thus ability to tag, as many other human genetic variants as possible. GWAS tag SNPs are not selected for having likely functional consequences, and if any are subsequently recognized, this is simply serendipitous. The conclusion that can be drawn at the end of a successful GWAS is that one or more genetic variants within the locus, marked by the associated tag SNP, must have biological functions that drive the observed association. In this review, we use the term “locus” to mean a haplotype block containing both the GWAS tag SNP and the directly causal variant (Figure 2).
Figure 2.
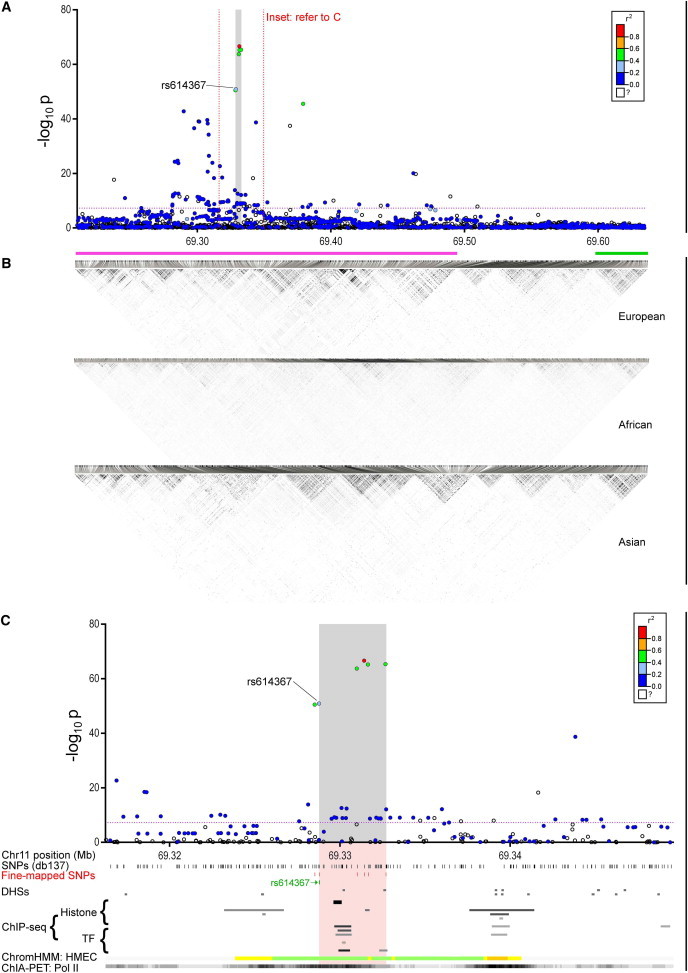
Integrated Genetic and Genomic Data at the 11q13 Breast Cancer Susceptibility Locus
(A) Manhattan plot displaying the strength of genetic association (−log10 p) versus chromosomal position (Mb). Each dot represents a genotyped or imputed SNP. Dot colors signify the degree of pairwise correlation (r2) with the top SNP, as presented in the color key. White dots depict SNPs for which r2 values are unknown. The gray shaded stripe represents the iCHAV, encompassing a physical area bound by SNPs that are statistically indistinguishable by stepwise conditional analysis and including the GWAS lead SNP rs614367. The purple dotted line represents the threshold for genome-wide significance (p = 5 × 10−8).
(B) Linkage disequilibrium plots depicting pairwise correlation between SNPs genotyped in the 1000 Genomes Project for European (CEU), African (YRI), and Asian (CHB) populations. The plots are in grayscale, for which white and black signify r2 = 0 and 1, respectively. The pink and green bars denote haplotype blocks described in the text in relation to transethnic fine mapping.
(C) Inset from panel (A). The UCSC Genome Browser was used for visualizing ENCODE data tracks, which are indicative of regulatory function. The pink stripe indicates the genomic region corresponding to the iCHAV at 11q13, and the locations of the fine-mapped SNPs are shown as red marks. Regions of open chromatin, indicative of putative regulatory signals, are detected as DNaseI hypersensitive sites (DHSs) and are marked. ChIP-seq data for histone marks associated with regulatory regions and specific TFs relevant to breast cancer are shown. The ENCODE ChromHMM track represents integrated analysis of chromatin states based upon histone ChIP-seq data from human mammary epithelial cells (HMECs). Color coding is as follows: green, weak transcription; yellow and orange, enhancer; red, promoter; blue, insulator; gray, repressed. RNA Pol II ChIA-PET data from MCF7 cells are represented as a grayscale bar; darker regions indicate more frequent interactions.
Successful fine-scale mapping, intended to identify the truly functional variants underlying observed GWAS signals, requires as a starting point a complete catalog of all variants in the associated locus. The working principle is that the functional variants being sought must be contained in the initial list. For a number of years after the publication of successful GWASs, generating such a catalog was the rate-limiting step. During this period, targeted sequencing of the locus in DNA from sufficient subjects was needed for ensuring the identification of all variants that could conceivably explain the association.24 Most common variants are found from the sequencing of relatively few DNA samples, but progressively more samples must be sequenced for identifying increasingly rarer variants. This process has been technically demanding, time consuming, and very costly; consequently, very few loci have been examined by this route. The public availability of the 1000 Genomes Project data25 has provided the necessary breakthrough to grant all researchers access to sufficiently comprehensive sequence data from which to compile their catalogs. This data set currently comprises 1,092 subjects, drawn from each major human ancestry, and has sufficient sequencing depth for all discoverable polymorphic variants (minor allele frequency > 1%) to be found.25
Once a comprehensive SNP catalog becomes available, the next requirement is to genotype these SNPs in DNA from studies in which the phenotype of interest has been accurately measured. The key to success is statistical power, most often developed by collaborating international consortia with a shared interest (e.g., Wellcome Trust Case Control Consortium,26 Genetic Investigation of Anthropometric Traits,16 and Breast Cancer Association Consortium [BCAC]27). These consortia have also generated their own genotyping chips for fine-scale-mapping loci they have discovered (e.g., Immunochip,28 Metabochip,29 and iCOGs).30 To date, the majority of GWASs and mapping studies have been performed in European-ancestry populations, which tend to have longer haplotype blocks containing many highly correlated SNPs.23 This pattern provides a GWAS advantage of requiring fewer tag SNPs for genome-wide coverage but a subsequent fine-mapping disadvantage due to associated loci containing many correlated SNPs, most of which are not directly causal. Typically, study sample sizes ranging from 10,000 to 100,000 have been required for teasing apart the small differences in risk associated with different SNPs.31 The aim is to separate variants displaying the greatest associations (the best candidate causal variants) from those with strong but significantly lesser associations so that the majority of variants in a given locus can be rationally excluded from further consideration. The ability to discriminate between gradations in degree of association is a function of study power, which in turn is affected by the magnitude of the effect of the causal SNP and the sample size.31 In addition to available sample size, other limiting factors are the cost of genotyping and difficulties in developing successful assays for every genetic variant under consideration, because no single genotyping platform can analyze all possible variant types. Imputation techniques (such as IMPUTE2), which use known correlations between SNPs (e.g., from the 1000 Genomes Project data) to estimate the probable genotype of any SNP in a subject,31 have overcome this hurdle. This approach aims to fill in all the missing genotype data (with a known degree of accuracy) in any data set and can be used when the relevant SNPs are not included on the platform or because the genotyping failed. After imputation, a complete data set containing all genotyped or imputed SNPs within a locus should be compiled for all subjects.
Loci vary enormously both in physical length and in the number of common genetic variants that they contain. It is not unusual for a locus to be >100 kb long and to contain >1,000 variants, of which hundreds might display significant associations with the phenotype of interest. Each causal variant lies within an independent set of correlated, highly trait-associated variants, which we have termed an “iCHAV.” An iCHAV resulting from an association study is broadly analogous to the “linkage peak” identified via a familial linkage study. For example, Figure 2B (green bar) illustrates a tight ∼30 kb haplotype block in all ethnicities at the right-hand side of the diagram and a less defined 0.3 Mb block comprising several smaller blocks (Figure 2B, purple bar) containing an iCHAV (Figure 2C, gray area) in Europeans. Exclusion of noncasual variants begins with the identification of the number of iCHAVs within a given locus, typically by forward conditional logistic regression analysis. Haplotype analysis can also be useful for examining the interrelationship of multiple putatively causal SNPs.32 Such analysis proved particularly revealing in the fine-scale mapping of the 11q13 breast cancer locus (Table 1).17 Indeed, fine mapping revealed several more strongly associated variants than the original GWAS tag SNP, rs614367 (Figure 2A).17,33 If a single iCHAV is identified, the next stage is straightforward: the SNP showing the strongest phenotypic association (the one with the greatest effect size and most significant association p value) is assumed to be the best candidate for causation, and relative to this one, other variants within the same iCHAV can be excluded from further consideration by means of a likelihood ratio test.31 As a rule of thumb, just as in a Bonferroni correction, it is considered safe to exclude all variants with likelihood ratios >100 times worse than the strongest SNP in the iCHAV.31 This stage removes from further consideration all SNPs simply associated with a phenotype because they are carried on the same haplotype as the strongest candidates because of shared ancestral history. Under ideal circumstances, i.e., with a sample size large enough for adequate power, it might be possible to exclude all but the most strongly associated variant. In practice, because of the high degree of shared ancestry in European populations, there can be many very highly correlated variants in any given iCHAV, leading to difficulty identifying single putative causal variants from genetic epidemiological studies alone. More often, at the end of this stage there remain 10–50 highly correlated, strong candidates, all with good likelihood ratios relative to the best hit,8,17,24,34 in one or more iCHAVs underlying the original GWAS association.
Under appropriate conditions, it can be possible to use studies from different ethnicities to further reduce the remaining number of candidates.35 The necessary conditions are that the same association must be observable in collected study samples from more than one ethnicity and that the patterns of SNP correlation must differ between the different ethnic groups. Given adequate sample sizes, it should thus be possible to exclude more candidates by using similar studies from different ethnic groups, but in practice the haplotype structure at the locus of interest is not always favorable.36
We began this type of fine-scale mapping of breast cancer loci within the BCAC with the working hypothesis that a single functional variant, in a single iCHAV, would explain each GWAS hit. However, among the nine breast cancer loci we examined in sufficient detail, and in reports from others, there is now strong evidence of the existence of multiple, independent functional variants at many GWAS-discovered loci.8,17,28,37–40 In some loci, these are carried on separate haplotype backgrounds (and separate iCHAVs), indicating that they result from separate mutational events. In other loci, multiple, apparently functional variants occur together on the same haplotype background (within a single iCHAV), generating additive effects on cancer risk.17,36 The discovery of multiple causal variants at loci originally thought to contain only one will explain more “missing heritability”41 than previously calculated from the GWAS results alone.39 Moreover, early findings indicate that independent variants within a given locus can disrupt the same target gene by differing mechanisms and are consequently associated with differing magnitudes of risk.8,17 Clearly, these data are contingent on comprehensive fine mapping for ensuring that the genuinely causal variant(s) is captured prior to functional assessment. With the list of SNPs that cannot be eliminated from consideration by statistical fine-scale mapping studies as a starting point, the next challenge is to elucidate the mechanism by which each candidate variant might influence the expression of its target gene(s). Functional evaluation of noncoding, regulatory variants requires the stepwise application of an array of computational approaches, including database searches and application of in silico tools, as well as a subsequent range of molecular experimental techniques (Figure 1).
Using Publically Available Data to Guide Functional Analyses
Many noncoding SNPs reside within regulatory sequences and influence gene expression through transcriptional, posttranscriptional, and posttranslational mechanisms. Transcription is a complex process dependent on a coordinated interplay between protein-DNA and protein-protein interactions (reviewed in Kadonaga42). RNA polymerase II (RNA Pol II) assembles at gene promoters with the basal transcription machinery, and numerous transcription factors (TFs) and accessory molecules are recruited to alter the function of RNA Pol II by associating with regulatory sequences. The accessibility of these factors is dependent on chromatin structural changes mediated by posttranslational histone modifications, such as methylation and acetylation.43 Importantly, regulatory signals can act over long genomic distances and are brought into contact with target promoters via three-dimensional DNA folding. Computational approaches to assigning potential regulatory function to noncoding SNPs have been greatly enhanced by the recent emergence of several large-scale genome-wide data sets. Data made publically available by projects such as ENCODE, Nuclear Receptor Cistrome (NRCistrome), and the National Institutes of Health (NIH) Roadmap Epigenomics Project can be routinely mined with a range of tools for the annotation of noncoding variants with a potential impact on regulatory mechanisms (Table 2). Regulatory function can be predicted on the basis of particular genomic features, such as histone modifications, open chromatin, and TF binding, which are measured with targeted biochemical assays and high-throughput sequencing technologies (Figure 2C). Mapping these data to a region of interest can then facilitate the design of additional functional analyses.
Table 2.
Computational Tools and Resources for the Analysis of GWAS Loci
| Feature | Description | Significance | Experimental Approach | Bioinformatic Tools and Online Resourcesa |
|---|---|---|---|---|
| Open chromatin | nucleosome-depleted chromatin | sequences harboring regulatory signals | DNase-seq, FAIRE sequencing | ENCODE, NIH Roadmap Epigenomics Project, RegulomeDB, HaploReg, FunciSNP |
| TF-binding prediction | short DNA consensus recognition sequence characteristic of a particular DNA-binding protein | computationally predicted TF recognition site | position weight matrices | TRANSFAC, JASPAR, MAPPER2 |
| DNA-protein interaction | short DNA sequence associated with a DNA-binding protein after precipitation with a specific antibody | physical protein-nucleic-acid binding (note: no direct evidence of activity) | ChIP-seq, DNase footprinting | ENCODE, NRCistrome, RegulomeDB, HaploReg |
| DNA methylation | methylation of cytosine residues in CpG dinucleotides | repression of gene expression | methylation array, bisulphite sequencing, MeDIP-seq, MRE-seq | ENCODE, NIH Roadmap Epigenomics Project, MethDB, EpiGraph |
| RNA expression | detection and measurement of transcribed RNA | coding RNA, noncoding RNA, alternative splicing | RNA-seq, RNA-PET, CAGE | ENCODE, Gene Expression Omnibus, Galaxy |
| Histone modifications | specific posttranslational modifications of particular histone protein residues are associated with various regulatory activities | H3K4me1: promoters and enhancers H3K4me2: promoters and enhancers H3K4me3: promoters H3K79me2: transcription transition H3K27ac: active regulatory region H3K9ac: promoters H3K9me1: active chromatin H3K9me3: repressed chromatin |
ChIP-seq | ENCODE, NIH Roadmap Epigenomics Project, NRCistrome, RegulomeDB, HaploReg, ChromHMM, GWAS3D, Segway, ChroMoS |
| Chromatin interactions | long-range physical interactions between distal genomic regions | contact between regulatory motifs, such as tissue-specific enhancers and promoters | 3C, 4C, 5C, 6C, Hi-C, ChIA-PET | GWAS3D, Hi-C Project, ChIA-PET Browser |
Abbreviations are as follows: 3C, chromosome conformation capture; 4C, circular 3C; 5C, carbon-copy 3C; 6C, combined 3C-ChIP-cloning; CAGE, cap analysis gene expression; DNase-seq, DNaseI hypersensitive site sequencing; MeDIP-seq, methylated DNA immunoprecipitation sequencing; MRE-seq, methylation-sensitive restriction enzyme sequencing; and RNA-PET, RNA paired-end-tag sequencing.
Nonexhaustive list of examples.
A range of computational tools, including RegulomeDB, HaploReg, and FunciSNP, are available for specifically querying these data sets (Table 2). With these, the generation of hypotheses to test candidate-SNP effects can be prioritized for experimental assessment. These programs test the potential impact of sequence variation on several genomic features, including gene and isoform annotations, expression quantitative-trait loci (eQTL), chromatin immunoprecipitation sequencing (ChIP-seq), DNaseI hypersensitive site (DHS) sequencing, chromatin interactions, evolutionary sequence conservation, and TF-binding motifs. It is important to incorporate fine-mapping genetic data (i.e., the best candidate SNPs) into these analyses, and this can be easily achieved with GWAS3D, which is capable of analyzing an entire GWAS output. Because these tools are freely available online, the combined outputs should be routinely used for triaging variants for further characterization (Table 2). It should be noted that many tools rely on ENCODE data, which are not exhaustive (i.e., limited TFs and cell types have been assayed), so there is also a high probability of false negatives, for example, where missing data might lead to the absence of valid results. Conversely, these programs do not take into account tissue specificity, which could lead to false positives, such as where SNPs might influence signals in irrelevant cell types. Overall, with the consideration of the specific biological question, computational approaches currently represent a useful starting point to guide the design of functional assays.
Potentially interesting results from such tools can be further interrogated via manual investigation of specific loci in the UCSC Genome Browser, which is able to display customizable ENCODE and NIH Roadmap Epigenomics Project data. Experimental matrices that display all available data are a starting point for loading various “tracks” into the browser. Users are also able to upload data, including SNP positions of interest or experimental data retrieved from repositories such as the NCBI Gene Expression Omnibus and NRCistrome. Other notable features include “sessions,” or configurations of track sets that can be shared between users, and the “table browser” feature, which provides a means by which the database can be queried for automated analyses.
Analysis of LD patterns is facilitated through the use of several key resources, including the HapMap and 1000 Genomes projects. Downloaded genotype data covering specific loci can be visualized and manipulated with HaploView44 for investigating haplotype structure and frequency (Figure 2B). LD patterns can be compared between ethnicities, enabling the determination of variant segregation in different populations. Several online resources, including the SNP Annotation and Proxy Search tools at the Broad Institute and LocusZoom, enable plotting of association data with LD information. Figure 2A demonstrates how pairwise correlation r2 coefficients can be related to a lead SNP via color coding.
Several elegant examples utilizing these computational approaches have recently been published. Maurano et al. showed that GWAS signals were overrepresented in regulatory regions by assessing the frequency of variant occurrence in DHSs generated by the ENCODE and NIH Roadmap Epigenomics consortia.45 Furthermore, assessment of putative functional variants at the 17q24.3 prostate cancer risk locus with the use of ENCODE and NIH Roadmap Epigenomics Project data facilitated the identification of a prostate-specific enhancer.20 We used mammary-cell-specific ChIP-seq signals from ENCODE and NRCistrome to assess potential regulatory functions at the 11q13 breast cancer risk locus, where we ultimately showed that CCND1 is a target of that association (Figure 2C).17 These resources are extremely useful provided that the cell and tissue types most relevant to the disease state under study have been assayed. For example, limited data are currently publically available for ovarian tissues, precluding the approaches outlined above in preliminary investigations for the correlation of ovarian cancer risk alleles in regulatory signals. Furthermore, there are limits to the availability of tissues at each developmental stage or under exposures to different environmental conditions.
Pathway-based analyses are another strategy being used for prioritizing genes from GWAS-identified regions (reviewed in Wang et al.46). These approaches typically examine whether a group of genes in the same biological pathway are jointly associated with a GWAS trait. The advantage of using these approaches is that important biological pathways underlying the GWAS trait can be uncovered. However, caution should be taken because these types of analyses are based on the often incorrect assumption that SNPs can be assigned to genes on the basis of proximity or the LD block in which they reside. In addition, prior biological knowledge about the genes and pathways is also required, thus undermining the agnostic nature of the GWAS approach.
eQTL
Levels of gene expression are highly heritable,47,48 and specific genomic regions containing variants that influence gene expression are known as eQTL. Multiple studies have provided strong evidence that GWAS signals are enriched with eQTL in a tissue-specific manner,49,50 highlighting their utility in understanding the mechanisms underlying GWAS hits. Many resources, including online databases such as GeneVar, are now available for eQTL analyses. Importantly, eQTL annotation is carried out in an unbiased fashion; hence, associations between alleles and target genes require no prior knowledge of functional mechanisms. Although GWAS variants can be associated with expression (ideally in a relevant tissue type), additional functional assays are required for confirming the mechanistic relevance to the disease or trait. A recent approach interrogated cancer-derived samples by using RNA sequencing (RNA-seq) and correlated allelic imbalance (AI) to breast cancer risk genotypes.51 Importantly, the analysis adjusted for copy number and methylation, significant confounders when gene expression is examined in tumor material.
Given that somatic alterations present in tumor cells can greatly affect expression, subtle genotype-associated influences can be undetectable.52 It is therefore ideal to measure eQTL effects in normal cells, representative of the cell of origin for the disease under study. This is particularly important because it is estimated that 50%–90% of eQTL are tissue dependent,50,53 and trait-associated variants tend to exert more tissue-specific effects.54,55 Not surprisingly, Fu et al. also showed that SNPs that fall in regulatory regions of the genome are also more likely to confer tissue specificity.54 To date, most eQTL data sets are derived from only a limited number of source cell types, including monocytes,56 lymphoblastoid cells,53 and brain cells.57 In an effort to address this issue, a large NIH-funded project known as Genotype Tissue Expression has been initiated with the aim of characterizing eQTL in more than 60 different normal tissues from 900 individuals.
Several factors must be considered when eQTL data are used. The majority of identified eQTL are cis-acting, arbitrarily defined as regulation of genes within 1 Mb, given that their effect sizes are usually relatively large and can be detected with smaller sample sizes.58 However, genetic variants can also affect the expression of genes that reside further away or are on different chromosomes (trans-eQTL).59 Notably, Fehrmann et al. identified independent trans-associated SNPs affecting similar genes, suggesting that independent GWAS associations might influence similar biological pathways.60 Haplotype might also influence eQTL effects:61 because LD patterns are population specific, association between variants that tag a haplotype could lead to the ambiguous identification of the true casual SNP. Furthermore, the target genes of eQTL associations could be coding or noncoding RNAs,62 although small RNA expression is not measurable by certain platforms. Recent RNA-seq experiments have revealed that genotype can also influence alternative isoform production,63 and variation in mRNA stability is also known to be under the control of correlated alleles.64 As with GWASs for any complex trait, the analysis of eQTL across diverse populations will enable more accurate mapping of regulatory variants.48,53,65 Variation in levels of specific protein isoforms have also recently been shown to be heritable.66 It should be noted that identifying an eQTL provides only indirect evidence of a link between genotype and gene transcription. Elucidating the involved mechanisms will then rely on a range of molecular approaches, which we describe below.
Regulatory Variation at GWAS Loci
The recent fine mapping and functional characterization of GWAS variants have indicated that cis-regulation is a common mechanism underlying these associations.4,8,17 The most frequent elements affected are transcriptional enhancers and silencers. These elements are typically located more than 1 kb from their target genes and regulate transcription through long-range interactions, mediated by the formation of chromatin loops.35 The ability to identify the target gene(s) of cis-regulatory variants is key to understanding the mechanism by which GWAS variants act. The identification of eQTL can be used for predicting the target genes; however, this strategy usually only provides indirect evidence of an association, and experimental approaches are necessary for confirming its mechanistic relevance.
A more direct approach is to use chromatin conformation capture (3C), a technique that converts chromatin interactions into specific ligation products, which are then quantified individually by real-time PCR.67 3C has already been used for successfully identifying the target gene of several regulatory variants identified through GWASs. For example, we fine mapped the 11q13 breast cancer risk locus and showed that the strongest signal mapped to a transcriptional enhancer that distally regulates the CCND1 promoter, located 125 kb away (Figure 2).17 SNPs associated with chronic obstructive pulmonary disease at 4q31 also lie within a transcriptional enhancer and physically interact with the HHIP promoter, located 85 kb away.6 In both of these cases, the target promoter of the regulatory elements is the closest gene. However, recent analysis by the ENCODE Consortium shows that only 27% of the distal regulatory elements have an interaction with the nearest promoter,68 suggesting that the nearest gene is often not the target of a given GWAS association. For example, the likely causal variant for a GWAS association at 1p13 with low-density lipoprotein cholesterol is rs12740374, which lies in a transcriptional enhancer and alters the activity by creating a C/EBP TF-binding site.4 On the basis of the strong correlation with expression in the liver, the likely target gene of this cis-regulatory variant is the fourth-closest gene, SORT1. ENCODE data also suggest that the average number of local target genes of a distal regulatory element is 2.5, indicating that genetic variants located in cis-regulatory elements might influence transactivation of multiple promoters and therefore directly affect the expression of more than one gene.68 Indeed, our unpublished data indicate that the variants responsible for the association at the 11q13 breast cancer risk region affect at least one more gene in addition to CCND1.
3C has two principle limitations. First, it is not possible to distinguish relevant nearby chromatin interactions (within ∼20 kb) above background interactions caused by random collisions. Second, 3C can only detect specific interactions between prespecified regions because it relies on PCR primers designed across interacting regions. To overcome these limitations, researchers have developed several variations of the 3C method so as to provide unbiased approaches to identifying the target gene of regulatory elements. Circular 3C (also called 4C) allows the entire genome to be screened for sequences that contact a specific DNA or “bait” region by means of inverse PCR with bait primers from a circular intermediate of 3C. Limitations of 4C include (1) the inability to identify interactions around the “bait” region, (2) the lack of resolution (∼100 kb to 1 Mb), (3) that the choice of enzymes used might preclude the identification of some interactions, and (4) that trans-interactions (interchromosomal) and distal cis-interactions (>500 kb from the bait) need to be further validated by independent methods such as fluorescence in situ hybridization. Despite these limitations, using the 4C method, Patel et al. showed that aberrant TAL1 (MIM 187040) expression in human T cell acute lymphoblastic leukemia (T-ALL [MIM 613065]) is mediated by a T-ALL-specific interchromosomal interaction between the TAL1 promoter on chromosome 1 and a regulatory element called TIL16 (TAL-1 interacting locus on chromosome 16) on chromosome 16.69 These observations imply that the target gene(s) of any given GWAS hit could also be located on a different chromosome. To date, however, 4C approaches have not been applied to GWAS loci, and until such time, it is difficult to assess the contribution of interchromosomal interactions to common disease.
Other genome-wide variants of the 3C method provide unbiased methods for identifying long-range chromatin interactions. Carbon-copy 3C (also known as 5C) detects all chromatin interactions across large genomic regions by using multiplex PCR in combination with high-throughput sequencing or microarrays.70 Using a similar technique, coined 3C with DNA selection and ligation, Harisemendy et al. showed that cis-regulatory variants associated with coronary artery disease (CAD) interact with four genes, including IFNA21, located more than 900 kb away. The CAD risk alleles disrupted a binding site for STAT1, a well-known effector of interferon signaling, and treatment of cells with interferon-γ increased the frequency of the interaction between the enhancers and IFNA21.14
Chromatin interaction analysis by paired-end-tag sequencing (ChIA-PET), another variation of 3C, detects chromatin interactions bound by a specific protein.71 Fullwood et al. originally developed ChIA-PET to map chromatin interactions bound by estrogen receptor (ER) from breast cancer cells treated with estrogen. By mining these data, we identified chromatin interactions between the CCND1 promoter and a transcriptional enhancer harboring genetic variants associated with ER-positive breast cancer (Figure 2C).17 Additional ChIA-PET data sets for CTCF (CCCTC-binding factor), RNA Pol II, and H3K4Me2 (a chromatin modification associated with enhancers) are now available for several different cell lines.72–74 An obvious limitation of ChIA-PET is the fact that any given TF is only expected to be involved in a subset of chromatin interactions, so ChIA-PET data sets will thus not include all promoter-enhancer interactions. However, it might be possible to identify the majority of promoter-enhancer interactions by using antibodies against the general TFs (such as RNA Pol II) or chromatin modifications (such as H3K4Me1 and H3K4Me2) together with deeper sequence read depth.72,73 Given that cis-regulatory elements are highly tissue specific, future chromatin-interaction profiles generated in multiple cell lines will be an invaluable resource for post-GWAS studies.
Once the target gene(s) of a regulatory element has been established, the impact of SNP(s) on the transactivation of a specific promoter can be tested via standard reporter assays. In these assays, regulatory elements are cloned into a promoter-driven reporter construct and transiently transfected into relevant cell lines. The effect of individual SNPs, or preferably the risk haplotype, can then be compared to the common allele or haplotype constructs. Importantly, the effect of the SNP(s) might vary depending on the promoter used to drive reporter expression. Viral promoters are commonly used for assessing enhancer activity; however, it should be noted that promoter-specific effects can be overlooked. The choice of cell type is also important because cis-regulatory elements are highly tissue- and cell-type specific. For example, a recent study revealed very different activities of eleven enhancers across four mammary epithelial cell lines, emphasizing the importance of conducting these assays in various cellular contexts.75 Reporter assays can also be used for mapping DNA regions harboring regulatory activity. This is particularly useful when limited information regarding the regulatory potential is available. For example, functional enhancer mapping of a 40 kb region at the 8q21 ovarian cancer risk locus identified two regulatory regions that harbor risk-associated SNPs.76
DNA variants can also affect chromatin looping, a mechanism distinct from transactivation.77 Allele-specific chromatin interactions can be detected by direct sequencing of 3C products.11,17 However, the SNP needs to be relatively close to a 3C restriction site for this to be feasible. The assessment of allele-specific protein binding is also important given that the majority of regulatory functions (such as chromatin looping and transactivation) are mediated through TFs and other proteins. Computational prediction of TF binding is the most widely used method for identifying candidate TFs. These predictions are based on models called position weight matrices, which quantitatively score the likelihood of observing a particular nucleotide at a specific position of the known or candidate TF-binding site. Many web-based programs such as MAPPER2 provide easily searchable platforms that combine motifs documented in databases such as JASPAR and TRANSFAC (Table 2). Recent mapping of histone modifications by means of ChIP-chip (ChIP followed by microarray hybridization) or ChIP-seq technologies provides a complementary approach to predicting TF-binding sites.78 However, ChIP assays are limited in that each experiment profiles just one TF, and it is difficult to determine the precise binding site for a factor because of the low resolution of the assay. Protein binding can be assessed with in vitro assays such as electrophoretic mobility shift assays (EMSAs), for which knowledge of the bound proteins is not required. Antibodies against TFs of interest are then used in supershift EMSA experiments for testing which proteins mediate allele-specific binding. High-throughput TF-binding methods, such as proteome-wide analysis of SNPs (PWAS), that utilize quantitative mass spectroscopy can also be used for screening SNPs for differential TF binding.79 An advantage of this technique is that multiple SNPs can be assayed and the TF can be identified in one experiment. By applying PWAS to twelve fine-mapped SNPs associated with type 1 diabetes (MIM 222100), Butter et al. identified at the IL2RA (MIM 147730) locus four SNPs that displayed preferential binding of common TFs.79 Allele-specific protein binding should also be verified by ChIP experiments because the in vitro nature of EMSAs and PWAS can generate false-positive results. The effect of these proteins on transactivation in the presence and absence of a SNP can then be tested by cotransfection in reporter assays.
Noncoding RNAs at GWAS Loci
Many post-GWAS studies have focused on cis-regulatory variation to explain disease associations. However, it is becoming increasingly clear that risk variants can also affect the expression or function of noncoding RNAs (ncRNAs). These regulatory ncRNAs can be divided into two broad categories according to size: (1) small RNAs (<200 bp), such as microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), and piwi-interacting RNAs (piRNAs); and (2) long noncoding RNAs (lncRNAs; >200 bp), which are often spliced transcripts transcribed from both DNA strands.
miRNAs typically regulate gene expression through binding to 3′ UTRs of target mRNAs to direct their posttranscriptional repression.80 Recent studies have indicated that sequence variation in target genes can modulate miRNA activity. For example, Kulkarni et al. demonstrated that variation within the 3′ UTR of HLA-C (MIM 142840) affects binding by miR-148 and thus results in differential cell-surface expression levels of HLA-C allotypes.81 Their findings provided the mechanism for a linked HIV-control-associated SNP located 35 kb upstream of HLA-C.82 Predicting potential target genes is the major challenge in exploring miRNA function, given that a single miRNA can potentially regulate hundreds of different genes. A variety of computational tools, including TargetScan and PicTar, are available for miRNA target prediction, and several databases have also been developed for linking polymorphisms in predicted miRNA-binding sites with complex traits. It should be noted that most of these programs rely on target-prediction algorithms that only assess the possibility of interaction, and so all predictions need to be experimentally validated. With the increasing availability of data from deep-sequencing and crosslinking-immunoprecipitation projects,83 a few groups have now tried to characterize the relationship among genetic variation, miRNA, and gene expression on a more genome-wide scale.84,85 Collectively, the results indicate that genetic variants within the 3′ UTRs of susceptibility genes at miR-binding sites are associated with disease risk and should be routinely considered in post-GWAS functional studies.
Although it is clear that lncRNAs are important for regulating gene expression, their role in influencing disease susceptibility is only now being realized. Several studies have shown that lncRNAs are transcribed from genomic regions associated with disease risk. ANRIL (MIM 613149), an antisense lncRNA expressed from the CDKN2B-CDKN2A locus, spans 126 kb and appears to be a hot spot of GWAS hits.86 SNPs within this locus are associated with multiple genetic diseases, including diabetes, coronary heart disease, glaucoma (MIM 231300), and multiple cancers. Although it cannot be ruled out that these SNPs affect other genes at this locus, the disease-associated variants are more strongly correlated with ANRIL expression in peripheral blood than with that of two nearby genes, CDKN2A and CDKN2B.86 Fine mapping of this region and functional studies in the relevant cell types will be required for confirming that ANRIL is the main target of these genetic associations. Another example is the thyroid-specific lncRNA PTCSC3 (MIM 614821), which is transcribed from a locus associated with papillary thyroid carcinoma (PTC [MIM 188550]). SNP rs944289, which is significantly associated with PTC, lies in the promoter of PTCSC3 and is associated with increased expression in normal thyroid tissue.87 Conversely, rs944289 is associated with decreased PTCSC3 expression in PTC, suggesting that the effects of this SNP differ in normal and malignant cells. Lastly, genetic variants within a gene desert at 8q24 are associated with multiple cancers, including breast, prostate, and colorectal cancer.37 The most likely target gene at this locus is c-MYC (although it has been difficult to demonstrate this link), but it is worth noting that several lncRNAs, as well as a miRNA cluster (miR-1204-1208), are produced from this locus. These ncRNAs could either work alone or in concert with c-MYC to explain the association.
As more RNA sequencing data in relevant tissues become available, it is likely that other lncRNAs will be identified at risk loci identified by GWASs. In addition, new technologies for identifying rare transcripts, a common feature of lncRNAs, will be important given that it is now clear that exhaustive sequencing of the human transcriptome has not been achieved with standard RNA-seq. For example, Mercer et al. developed a technique termed RNA-CaptureSeq, which achieves deeper sequencing of transcripts derived from targeted regions.88 As in targeted genomic sequencing, cDNAs are hybridized onto tiling arrays prior to sequencing, thus enriching for a specific fraction of the transcriptome and allowing deeper sequencing with the same amount of sequence reads. With this technology, targeted capture of ∼50 human genes identified 200 new protein isoforms and 163 lncRNAs at or near protein-coding genes, providing evidence that the human transcriptome is more complex than previously thought. Application of this technology in relevant tissues could uncover entirely new mechanisms for the genetic associations identified by GWASs.
A battery of techniques will then be required for elucidating how individual SNPs affect lncRNA function. Similar to protein-coding genes, some SNPs are likely to fall in regulatory elements of lncRNAs, just as rs944289 falls within the promoter of the PTCSC3 lncRNA.87 These SNPs could be analyzed with methods similar to those described above. Other SNPs could fall within the lncRNA transcript itself and therefore influence lncRNA function by affecting secondary structure and/or protein binding. Allele-specific protein binding can be tested by RNA mobility shift assays, in vitro assays that do not require prior knowledge of the bound proteins. Alternatively, if the RNA-binding protein is known or predicted, RNA immunoprecipitation (RIP) using an antibody against the RNA-binding protein could be performed and allele-specific binding could be assessed. Interestingly, using RIP, Olshavsky et al. showed that allele-specific binding of the RNA-binding protein SRSF1 (also known as ASF/SF2) affects the splicing of CCND1;89 such a mechanism could also apply to lncRNAs. It is also possible that genetic variants within the transcript could affect the stability of the lncRNA.90
Epigenetic Contribution to Complex Traits
Epigenetic modifications represent an additional layer of complexity to understanding gene regulation and might explain some of the failure to identify target genes of complex diseases. Epigenetic changes such as DNA methylation and histone modifications are required for normal gene regulation and are therefore obvious candidate targets for GWAS associations.91 DNA methylation is perhaps the most extensively studied epigenetic modification. Aberrant promoter methylation of tumor-suppressor genes is frequently observed in cancer and firmly established as an important mechanism for gene inactivation.92 Although not well understood, gene-body methylation can suppress transcriptional noise and might also be involved in splicing regulation.93 Evaluating the link among genetic variants, DNA methylation, and disease predisposition is currently a very active area in research. There are several examples of cis-acting genetic alterations giving rise to epimutations. In hereditary nonpolyposis colorectal cancer, MLH1 (MIM 120436) hypermethylation and transcriptional silencing have been linked to a SNP within its 5′ UTR.94 Similarly, heritable DAPK1 (MIM 600831) methylation in individuals with familial chronic lymphocytic leukemia is associated with a point mutation upstream of its promoter.95 Moreover, there is also evidence that altered DNA methylation of distal transcriptional enhancers might also play a role in cancer predisposition.96 Further insight into this potentially causative relationship has been made possible with the development of high-throughput DNA methylation arrays.97 Similar to eQTL, methylation quantitative-trait loci (metQTL) can be used for associating SNPs with CpG methylation in any tissue or cell type of interest. Most studies to date have found that correlations with cis-genotypes are more frequent, but there is also some evidence of trans-regulation cross multiple tissues.98
Risk-associated SNPs can also disrupt the structural organization of chromatin in the nucleus. Chromatin-state signatures correlate with tissue-specific gene expression; therefore, allelic differences in chromatin could potentially change expression patterns. Consistent with this, changes in allele-specific binding of CTCF (an important chromatin organizer) can affect the expression of several genes in the ORMDL3 locus and contribute to the risk of asthma and autoimmune disease.19 Furthermore, common variants at TCF7L2 (MIM 602228) confer risk of type 2 diabetes (MIM 125852) by altering cis-regulation and local chromatin structure in islet cells.99 In addition, Cowper-Sal-lari et al. recently used a systematic approach to explore the effect of genetic variation on allele-specific TF binding and chromatin structure. They showed that the majority of SNPs associated with breast cancer risk are enriched with FOXA1- and ER-binding sites, as well as H3K4me1 histone modification.100 FOXA1 is a pioneer factor central for opening compacted chromatin, nucleosome repositioning, and ER function. These studies further emphasize the complexity of regulatory variation.
The clear relationship between genetic and epigenetic variation for complex diseases has provided a strong argument for integrating GWASs with epigenome-wide association studies (EWASs). As the name suggests, EWASs utilize large-scale epidemiologic studies and high-throughput arrays as an unbiased strategy to systematically study epigenetic control.101 Currently, DNA methylation is the most suitable mark for EWASs and has been used, with some success, in the study of both nonmalignant diseases and epigenetic perturbations in cancer. It should be noted, though, that like GWASs, EWASs have a number of limitations and considerations in both their design and their interpretation. Some of the main challenges include the choice of tissue to be sampled and the matter of tissue heterogeneity, obtaining adequate sample size for achieving the necessary statistical power, and replication in independent study populations. Environmental factors are another variable because they can also directly or indirectly induce epigenetic changes.102 Despite these issues, Bell et al. successfully combined GWAS and EWAS data to identify variant-CpG-restricted haplotype-specific methylation within the FTO (MIM 610966) obesity susceptibility locus.103 Their study highlights the potential for integrating genetic and epigenomic approaches as a post-GWAS strategy for dissecting the functional consequences of GWAS loci.
In Vitro and In Vivo Models for Assessing SNP Function
The approaches described above only identify candidate target genes. The next step in the functional analysis pipeline is to validate each gene and to explore the fundamental mechanisms of disease (Figure 1). Human disease models can be based on either in vitro studies in cultured cell lines and/or primary tissues or in vivo models of disease development. Established human cancer cell lines have been extensively used for testing the function of candidate genes at risk loci. The primary advantages of these models are their ease of manipulation, homogeneity, and extended replicative capacity. Furthermore, hundreds of cell lines representing the major forms of cancer are commercially available, and many of them have been genetically characterized.104 However, some important limitations must be recognized, especially when these models are used for studying genetic associations. The most relevant limitation is that any functional effects could be obscured or falsely elevated by cellular genomic instability. In addition, karyotypic abnormalities and variability due to extensive passaging and different culture conditions must also be considered. Better models for evaluating GWAS-identified genetic variations are normal, nonaberrant tissues and cell lines. The main challenges impeding the establishment of these in vitro models are limited access to clinical samples and difficulties in culturing and manipulating primary cells. However, several reasonable models of normal tissue, including MCF10As for breast, PNT2 for prostate, EndoC-βH1 for pancreatic beta cells, and lymphoblastoid cell lines for blood traits, are already available.
Ideally, we need to create panels of genetically matched “normal cells” to provide isogenic systems to study disease mechanisms. Significant progress toward reaching this goal has been made with the development of programmable nucleases, such as zinc finger nucleases (ZFNs) and transcription-activator-like effector nucleases (TALENS), which are promising new tools that enable targeted genome modifications.105,106 In a genome, these enzymes induce site-specific double-strand breaks (DSBs), which cells then respond to with various repair mechanisms. Many types of genomic alterations, including point mutations, can be introduced with ZFNs or TALENs, thus potentially enabling studies to determine the functional significance of individual sequence variants. There are several recent examples of interrogation of putative causal genes in appropriate human cell types. For example, Soldner et al. used engineered ZFNs to generate isogenic pluripotent stem cells to study risk variants for Parkinson disease (MIM 168600).107 The lines differed by two single point mutations in α-synuclein (MIM 163890) but maintained a pluripotent state, the ability to form teratomas, and the ability to differentiate into dopaminergic neurons in vitro. Another technical challenge for post-GWAS research is the difficulty of studying locus heterogeneity, the joint effect of multiple independent variants across different loci. This limitation might soon be overcome by means of an alternative genome editing system called CRISPR/Cas (Clustered Regularly Interspaced Short Palindromic Repeats),108 which relies on the RNA-guided DNA endonuclease, Cas9, inducing targeted DSBs into genomic DNA. The method also allows for the simultaneous introduction of multiple guide RNAs, resulting in multiplex genome editing in mammalian cells,109 providing proof of principle of the potential of this system for GWAS functional studies.
Currently, one of the biggest challenges in post-GWAS validation is being able to accurately evaluate the effects of SNPs and their associated genes in an intact organism. The mouse is usually the mammalian model of choice because of its high degree of genome similarity, numerous techniques for genetic manipulation, and capacity to mimic human multifactorial disease phenotypes.110 Mouse models have mainly been used for studying the function of the most compelling or nearest candidate gene to the GWAS SNP for a range of complex diseases. For example, GWAS-identified SNPs in the vicinity of NR5A2 (MIM 604453) are associated with pancreatic cancer (MIM 260350) risk.111 Two recent papers used loss-of-function mouse models to show that NR5A2 is a key regulator of pancreatic acinar cell plasticity and that loss of NR5A2 cooperates with KRAS in preneoplastic transformation of the pancreas.112,113 However, a caveat for many of these studies is the lack of evidence linking the functional SNPs to expression of candidate target genes. This issue has been partly addressed for the 8q24 susceptibility locus. Several studies have indicated that the 8q24 cancer risk SNP rs6983267 resides in an enhancer element (called MYC-335) that can control expression of the oncogene c-MYC.10,114 Sur et al. subsequently generated Myc-335-knockout mice and showed a modest decrease in c-MYC in the colon, but more interestingly, these mice were resistant to intestinal tumorigenesis when challenged with the APCmin (MIM 611731) mutation.115 Although these results clearly show the importance of the MYC-335 region in intestinal tumorigenesis, the relevance of c-MYC itself remains uncertain, and they also highlight the need for unbiased approaches to identifying target gene(s).
As discussed earlier, because most disease-associated SNPs confer only modest effects on risk, the relevance of a complete versus heterozygous knockout must always be considered. TALEN-, ZFN-, and CRISPR/Cas-based methods are the most promising approaches to addressing heterozygous allele combinations. Indeed, these technologies have already successfully generated monoallelic and biallelic modifications in mice.116 Currently, only one group has used ZFNs to study GWAS loci;99 however, the speed with which ZNF-, TALEN-, and CRISPR/Cas-engineered mice can be created will greatly accelerate GWAS-driven functional research. It should be noted that there are some important “mouse traps” that should be considered when mouse models are used for delineating human genotype-phenotype relationships. These include species-species differences in gene function, poor evolutionary conservation in noncoding regions, changes in cellular microenvironments and immunity, the genetic background of the mice, and the presence of specific microbiota.110 Future mouse models will also most likely require a more thorough mapping of the genetic variants already present and the introduction of multiple genetic variations to the model so that the human genetic landscape can be more faithfully recapitulated.
Zebrafish and Drosophila are other increasingly popular model organisms for studying the functions of normal and disease-associated alleles. Both models have a number of advantages for post-GWAS analysis over their rodent counterparts; these include the ease of genetic manipulation, the ability to rapidly produce large numbers of organisms of a specific genotype, and the capacity to study tissue-specific gene expression in live animals. Again, most studies to date have used these organisms to study the function of the nearest candidate gene(s) to the sentinel SNP without first establishing that the gene is the target of that SNP.117 For zebrafish, transient disruption of normal gene function can be achieved by microinjection of mRNA, DNA, or morpholinos into early embryos.35 Although not as straightforward, regulatory elements can also be assessed by the generation of transgenic zebrafish by means of reporter constructs.118 In Drosophila, transgenic RNAi is the most common method used for downregulating orthologs of candidate genes.119 A disadvantage of using nonmammalian animals to model human disease is the need for functionally similar orthologs. A recent study attempted to address this limitation by performing post-GWAS functional assays in both zebrafish and Drosophila.120 Of the 49 candidate genes, results were available for the orthologs of 12 genes in zebrafish and 25 genes in Drosophila, and orthologs from six genes were available in both species.120 Clearly, not all downstream mechanistic studies will be feasible in these organisms, but for many of the simpler in vitro strategies we have described, they could help prioritize hypotheses for testing in more complex organisms.
Conclusions
GWASs have robustly identified thousands of disease- and trait-associated genetic variants. However, significant obstacles have hampered our ability to pinpoint casual variants, identify genes affected by causal variants, and disentangle the mechanism by which genotype influences phenotype. The critical first step is to undertake mapping at increased marker density with imputation to capture all nongenotyped alleles, which is vital to clarifying the most likely causal candidates prior to assessment of whether they contribute to a molecular mechanism. Even then, it should be noted that variants are unlikely to act alone, and the importance of combinatorial effects should be considered. In addition, consistency in the direction of the effect detected in genetic analyses (e.g., increased or decreased risk) and the probable mechanism (e.g., upregulation or downregulation of transcription) is ultimately required for adequately explaining the molecular association, although unknown and surprising gene functions should always be anticipated.
The emergence of several large-scale genomic data sets generated by projects such as ENCODE have revolutionized our ability to query and annotate putative cis-regulatory variants and form readily testable hypotheses. Expansion of these resources and establishing well-powered eQTL databases derived from relevant tissues and cells will enable studies to pinpoint likely causal variants at any locus. We emphasize that functional studies must then be undertaken in an unbiased manner so that undermining the agnostic approach of GWASs can be avoided. Most reports have simply implicated the nearest gene to a GWAS hit without any evidence that it is the true target of the functional variant. This is particularly relevant for regulatory variants that could be considerably distant from target gene(s). New technologies, including 3C-based techniques and the use of TALENS and ZFNs for generating isogenic cell lines, will clearly be important for identifying and validating the gene(s) directly affected by the risk-associated variants. Modeling the effect of validated variants in laboratory animals might then provide avenues for studying genetic disease.
This review provides a functional pipeline for the identification of candidate causal variants at GWAS loci. It is important to note, however, that it is difficult to unequivocally prove that a SNP is the direct cause of any given association. As opposed to linking rare mutations to disease, proving that common variants exert deleterious effects is problematic. Although experimental evidence might strongly support a plausible mechanism for an association, it is unlikely that definitive proof of causality, equivalent to “Koch’s postulates for genes,” will be achieved.121 Furthermore, it is becoming clear that causal variants are not always single SNPs acting alone and that combinations of variants are often required in order for effects to be explained. SNPs could also act in unanticipated cell types or could be involved in as yet undefined mechanisms.
GWASs have also received criticism for their lack of clinical translation because most effect sizes have been deemed too small to be meaningful. However, individual small effect sizes (<1.5) represent the reality of common genetic variation and do not necessarily preclude clinical utility. For example, the extremely successful cholesterol-lowering statin drugs target HMGCR, a GWAS hit for circulating lipid levels.122 This discrepancy occurs because a drug’s efficacy bears little relation to the degree of genetic variation in its target gene. Unraveling the complex mechanisms underlying GWAS associations will ultimately identify important biological pathways that could present suitable targets for drug development or repositioning of known therapeutics.123 Steps toward filling this knowledge gap, as described in this review, will bring us closer to elucidating the genetic basis of complex disease and offer opportunities for personalized medicine.
Acknowledgments
We would like to thank Georgia Chenevix-Trench, Dylan Glubb, and Karen McCue for critical reading of the manuscript. This work was supported by Australian National Health and Medical Research Council grants 1021731 (to S.L.E. and J.D.F.) and 1012023 (to J.B.). S.L.E. and J.D.F. were supported by fellowships from the National Breast Cancer Foundation Australia. A.M.D. was funded by Cancer Research UK (C8197/A10865) and the Joseph Mitchell trust. Fine-scale mapping in her laboratory was funded by Cancer Research UK grants (C1287/A10118) and (C8197/A10123) and European Commission Seventh Framework Programme Health-F2-2009-223175 “COGS” WP4 “Genomic Locus Mapping.” The funders had no role in study design, decision to publish, or preparation of the manuscript.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
The Cancer Genome Atlas, http://cancergenome.nih.gov/
ChIA-PET Browser, http://cms1.gis.a-star.edu.sg/index.php
ChromHMM, http://compbio.mit.edu/ChromHMM/
Collaborative Oncological Gene-environment Study, http://www.nature.com/icogs/
ENCODE, http://www.genome.gov/10005107
EpiGRAPH, http://epigraph.mpi-inf.mpg.de/WebGRAPH/
FunciSNP, http://bioconductor.org/packages/2.12/bioc/html/FunciSNP.html
Galaxy, https://usegalaxy.org/
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
GeneVar, http://www.sanger.ac.uk/resources/software/genevar/
GTEx (Genotype-Tissue Expression) eQTL Browser, http://www.ncbi.nlm.nih.gov/gtex
GWAS3D, http://jjwanglab.org/gwas3d/
IMPUTE2, http://mathgen.stats.ox.ac.uk/impute/impute_v2.html
JASPAR, http://jaspar.binf.ku.dk/
LocusZoom, http://csg.sph.umich.edu/locuszoom/
MAPPER2, http://genome.ufl.edu/mapperdb
MethDB, http://www.methdb.de
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
NHGRI GWAS Catalog, http://www.genome.gov/GWAstudies
Nuclear Receptor Cistrome, http://www.cistrome.org/Cistrome/Cistrome_Project.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PicTar, http://pictar.mdc-berlin.de/
RegulomeDB, http://www.regulomedb.org
NIH Roadmap Epigenomics Project, http://www.roadmapepigenomics.org/
TargetScan 6.2, http://www.targetscan.org/
TRANSFAC, http://www.biobase-international.com/gene-regulation
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Hindorff, L.A., MacArthur, J., Morales, J., Junkins, H.A., Hall, P.N., Klemm, A.K., and Manolio, T.A. A Catalog of Published Genome-Wide Association Studies. http://www.genome.gov/gwastudies.
- 2.Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- 3.Cooper G.M., Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 4.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X., Baron R.M., Hardin M., Cho M.H., Zielinski J., Hawrylkiewicz I., Sliwinski P., Hersh C.P., Mancini J.D., Lu K. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum. Mol. Genet. 2012;21:1325–1335. doi: 10.1093/hmg/ddr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen H.H., Takata R., Akamatsu S., Shigemizu D., Tsunoda T., Furihata M., Takahashi A., Kubo M., Kamatani N., Ogawa O. IRX4 at 5p15 suppresses prostate cancer growth through the interaction with vitamin D receptor, conferring prostate cancer susceptibility. Hum. Mol. Genet. 2012;21:2076–2085. doi: 10.1093/hmg/dds025. [DOI] [PubMed] [Google Scholar]
- 8.Bojesen S.E., Pooley K.A., Johnatty S.E., Beesley J., Michailidou K., Tyrer J.P., Edwards S.L., Pickett H.A., Shen H.C., Smart C.E., Australian Cancer Study. Australian Ovarian Cancer Study. Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Gene Environment Interaction and Breast Cancer (GENICA) Swedish Breast Cancer Study (SWE-BRCA) Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) Epidemiological study of BRCA1 & BRCA2 Mutation Carriers (EMBRACE) Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO) Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 2013;45:371–384. doi: 10.1038/ng.2566. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pittman A.M., Naranjo S., Jalava S.E., Twiss P., Ma Y., Olver B., Lloyd A., Vijayakrishnan J., Qureshi M., Broderick P. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS Genet. 2010;6:e1001126. doi: 10.1371/journal.pgen.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuupanen S., Turunen M., Lehtonen R., Hallikas O., Vanharanta S., Kivioja T., Björklund M., Wei G., Yan J., Niittymäki I. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009;41:885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- 11.Wright J.B., Brown S.J., Cole M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol. 2010;30:1411–1420. doi: 10.1128/MCB.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadiyeh N., Pomerantz M.M., Grisanzio C., Herman P., Jia L., Almendro V., He H.H., Brown M., Liu X.S., Davis M. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sotelo J., Esposito D., Duhagon M.A., Banfield K., Mehalko J., Liao H., Stephens R.M., Harris T.J., Munroe D.J., Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. USA. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harismendy O., Notani D., Song X., Rahim N.G., Tanasa B., Heintzman N., Ren B., Fu X.D., Topol E.J., Rosenfeld M.G., Frazer K.A. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visel A., Zhu Y., May D., Afzal V., Gong E., Attanasio C., Blow M.J., Cohen J.C., Rubin E.M., Pennacchio L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer K.B., Maia A.T., O’Reilly M., Teschendorff A.E., Chin S.F., Caldas C., Ponder B.A. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.French J.D., Ghoussaini M., Edwards S.L., Meyer K.B., Michailidou K., Ahmed S., Khan S., Maranian M.J., O’Reilly M., Hillman K.M., GENICA Network. kConFab Investigators Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schödel J., Bardella C., Sciesielski L.K., Brown J.M., Pugh C.W., Buckle V., Tomlinson I.P., Ratcliffe P.J., Mole D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet. 2012;44:420–425. doi: 10.1038/ng.2204. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verlaan D.J., Berlivet S., Hunninghake G.M., Madore A.M., Larivière M., Moussette S., Grundberg E., Kwan T., Ouimet M., Ge B. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Cowper-Sal lari R., Bailey S.D., Moore J.H., Lupien M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res. 2012;22:1437–1446. doi: 10.1101/gr.135665.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschhorn J.N., Daly M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 22.Reich D.E., Cargill M., Bolk S., Ireland J., Sabeti P.C., Richter D.J., Lavery T., Kouyoumjian R., Farhadian S.F., Ward R., Lander E.S. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 23.Dunning A.M., Durocher F., Healey C.S., Teare M.D., McBride S.E., Carlomagno F., Xu C.F., Dawson E., Rhodes S., Ueda S. The extent of linkage disequilibrium in four populations with distinct demographic histories. Am. J. Hum. Genet. 2000;67:1544–1554. doi: 10.1086/316906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Udler M.S., Ahmed S., Healey C.S., Meyer K., Struewing J., Maranian M., Kwon E.M., Zhang J., Tyrer J., Karlins E. Fine scale mapping of the breast cancer 16q12 locus. Hum. Mol. Genet. 2010;19:2507–2515. doi: 10.1093/hmg/ddq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galvin B.D., Hart K.C., Meyer A.N., Webster M.K., Donoghue D.J. Constitutive receptor activation by Crouzon syndrome mutations in fibroblast growth factor receptor (FGFR)2 and FGFR2/Neu chimeras. Proc. Natl. Acad. Sci. USA. 1996;93:7894–7899. doi: 10.1073/pnas.93.15.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutt A., Salvesen H.B., Chen T.H., Ramos A.H., Onofrio R.C., Hatton C., Nicoletti R., Winckler W., Grewal R., Hanna M. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trynka G., Hunt K.A., Bockett N.A., Romanos J., Mistry V., Szperl A., Bakker S.F., Bardella M.T., Bhaw-Rosun L., Castillejo G., Spanish Consortium on the Genetics of Coeliac Disease (CEGEC) PreventCD Study Group. Wellcome Trust Case Control Consortium (WTCCC) Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voight B.F., Kang H.M., Ding J., Palmer C.D., Sidore C., Chines P.S., Burtt N.P., Fuchsberger C., Li Y., Erdmann J. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K., Breast and Ovarian Cancer Susceptibility Collaboration. Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) kConFab Investigators. Australian Ovarian Cancer Study Group. GENICA (Gene Environment Interaction and Breast Cancer in Germany) Network Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45:353–361. doi: 10.1038/ng.2563. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Udler M.S., Tyrer J., Easton D.F. Evaluating the power to discriminate between highly correlated SNPs in genetic association studies. Genet. Epidemiol. 2010;34:463–468. doi: 10.1002/gepi.20504. [DOI] [PubMed] [Google Scholar]
- 32.Chatterjee N., Chen Y.H., Luo S., Carroll R.J. Analysis of Case-Control Association Studies: SNPs, Imputation and Haplotypes. Stat. Sci. 2009;24:489–502. doi: 10.1214/09-sts297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turnbull C., Ahmed S., Morrison J., Pernet D., Renwick A., Maranian M., Seal S., Ghoussaini M., Hines S., Healey C.S., Breast Cancer Susceptibility Collaboration (UK) Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowther-Swanepoel D., Broderick P., Ma Y., Robertson L., Pittman A.M., Price A., Twiss P., Vijayakrishnan J., Qureshi M., Dyer M.J. Fine-scale mapping of the 6p25.3 chronic lymphocytic leukaemia susceptibility locus. Hum. Mol. Genet. 2010;19:1840–1845. doi: 10.1093/hmg/ddq044. [DOI] [PubMed] [Google Scholar]
- 35.Sexton T., Bantignies F., Cavalli G. Genomic interactions: chromatin loops and gene meeting points in transcriptional regulation. Semin. Cell Dev. Biol. 2009;20:849–855. doi: 10.1016/j.semcdb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Udler M.S., Meyer K.B., Pooley K.A., Karlins E., Struewing J.P., Zhang J., Doody D.R., MacArthur S., Tyrer J., Pharoah P.D., SEARCH Collaborators FGFR2 variants and breast cancer risk: fine-scale mapping using African American studies and analysis of chromatin conformation. Hum. Mol. Genet. 2009;18:1692–1703. doi: 10.1093/hmg/ddp078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghoussaini M., Song H., Koessler T., Al Olama A.A., Kote-Jarai Z., Driver K.E., Pooley K.A., Ramus S.J., Kjaer S.K., Hogdall E., UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons’ Section of Oncology. UK ProtecT Study Collaborators Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008;100:962–966. doi: 10.1093/jnci/djn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berndt S.I., Sampson J., Yeager M., Jacobs K.B., Wang Z., Hutchinson A., Chung C., Orr N., Wacholder S., Chatterjee N. Large-scale fine mapping of the HNF1B locus and prostate cancer risk. Hum. Mol. Genet. 2011;20:3322–3329. doi: 10.1093/hmg/ddr213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanna S., Li B., Mulas A., Sidore C., Kang H.M., Jackson A.U., Piras M.G., Usala G., Maninchedda G., Sassu A. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hein R., Maranian M., Hopper J.L., Kapuscinski M.K., Southey M.C., Park D.J., Schmidt M.K., Broeks A., Hogervorst F.B., Bueno-de-Mesquita H.B., GENICA Network. Kconfab Investigators. AOCS Group Comparison of 6q25 breast cancer hits from Asian and European Genome Wide Association Studies in the Breast Cancer Association Consortium (BCAC) PLoS ONE. 2012;7:e42380. doi: 10.1371/journal.pone.0042380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadonaga J.T. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell. 2004;116:247–257. doi: 10.1016/s0092-8674(03)01078-x. [DOI] [PubMed] [Google Scholar]
- 43.Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 44.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 45.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K., Li M., Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 47.Morley M., Molony C.M., Weber T.M., Devlin J.L., Ewens K.G., Spielman R.S., Cheung V.G. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q., Seo J.H., Stranger B., McKenna A., Pe’er I., Laframboise T., Brown M., Tyekucheva S., Freedman M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nica A.C., Parts L., Glass D., Nisbet J., Barrett A., Sekowska M., Travers M., Potter S., Grundberg E., Small K., MuTHER Consortium The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu J., Wolfs M.G., Deelen P., Westra H.J., Fehrmann R.S., Te Meerman G.J., Buurman W.A., Rensen S.S., Groen H.J., Weersma R.K. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 2012;8:e1002431. doi: 10.1371/journal.pgen.1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown C.D., Mangravite L.M., Engelhardt B.E. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 2013;9:e1003649. doi: 10.1371/journal.pgen.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeller T., Wild P., Szymczak S., Rotival M., Schillert A., Castagne R., Maouche S., Germain M., Lackner K., Rossmann H. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS ONE. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myers A.J., Gibbs J.R., Webster J.A., Rohrer K., Zhao A., Marlowe L., Kaleem M., Leung D., Bryden L., Nath P. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 58.Cheung V.G., Spielman R.S. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat. Rev. Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fehrmann R.S., Jansen R.C., Veldink J.H., Westra H.J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J., Smolonska A. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garnier S., Truong V., Brocheton J., Zeller T., Rovital M., Wild P.S., Ziegler A., Munzel T., Tiret L., Blankenberg S., Cardiogenics Consortium Genome-wide haplotype analysis of cis expression quantitative trait loci in monocytes. PLoS Genet. 2013;9:e1003240. doi: 10.1371/journal.pgen.1003240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar V., Westra H.J., Karjalainen J., Zhernakova D.V., Esko T., Hrdlickova B., Almeida R., Zhernakova A., Reinmaa E., Võsa U. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9:e1003201. doi: 10.1371/journal.pgen.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lalonde E., Ha K.C., Wang Z., Bemmo A., Kleinman C.L., Kwan T., Pastinen T., Majewski J. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 2011;21:545–554. doi: 10.1101/gr.111211.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pai A.A., Cain C.E., Mizrahi-Man O., De Leon S., Lewellen N., Veyrieras J.B., Degner J.F., Gaffney D.J., Pickrell J.K., Stephens M. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 2012;8:e1003000. doi: 10.1371/journal.pgen.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stranger B.E., Montgomery S.B., Dimas A.S., Parts L., Stegle O., Ingle C.E., Sekowska M., Smith G.D., Evans D., Gutierrez-Arcelus M. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu L., Candille S.I., Choi Y., Xie D., Jiang L., Li-Pook-Than J., Tang H., Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 68.Sanyal A., Lajoie B.R., Jain G., Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patel B., Kang Y., Cui K., Litt M., Riberio M.S., Deng C., Salz T., Casada S., Fu X., Qiu Y. Aberrant TAL1 activation is mediated by an interchromosomal interaction in human T-cell acute lymphoblastic leukemia. Leukemia. 2013 doi: 10.1038/leu.2013.158. Published online May 23, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dostie J., Richmond T.A., Arnaout R.A., Selzer R.R., Lee W.L., Honan T.A., Rubio E.D., Krumm A., Lamb J., Nusbaum C. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chepelev I., Wei G., Wangsa D., Tang Q., Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012;22:490–503. doi: 10.1038/cr.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rhie S.K., Coetzee S.G., Noushmehr H., Yan C., Kim J.M., Haiman C.A., Coetzee G.A. Comprehensive functional annotation of seventy-one breast cancer risk Loci. PLoS ONE. 2013;8:e63925. doi: 10.1371/journal.pone.0063925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pharoah P.D., Tsai Y.Y., Ramus S.J., Phelan C.M., Goode E.L., Lawrenson K., Buckley M., Fridley B.L., Tyrer J.P., Shen H., Australian Cancer Study. Australian Ovarian Cancer Study Group GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat. Genet. 2013;45:362–370. doi: 10.1038/ng.2564. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qiu X., Vu T.H., Lu Q., Ling J.Q., Li T., Hou A., Wang S.K., Chen H.L., Hu J.F., Hoffman A.R. A complex deoxyribonucleic acid looping configuration associated with the silencing of the maternal Igf2 allele. Mol. Endocrinol. 2008;22:1476–1488. doi: 10.1210/me.2007-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 79.Butter F., Davison L., Viturawong T., Scheibe M., Vermeulen M., Todd J.A., Mann M. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 2012;8:e1002982. doi: 10.1371/journal.pgen.1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kulkarni S., Savan R., Qi Y., Gao X., Yuki Y., Bass S.E., Martin M.P., Hunt P., Deeks S.G., Telenti A. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thomas R., Apps R., Qi Y., Gao X., Male V., O’hUigin C., O’Connor G., Ge D., Fellay J., Martin J.N. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu C., Mallick B., Long D., Rennie W.A., Wolenc A., Carmack C.S., Ding Y. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res. 2013;41:e138. doi: 10.1093/nar/gkt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gamazon E.R., Ziliak D., Im H.K., LaCroix B., Park D.S., Cox N.J., Huang R.S. Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am. J. Hum. Genet. 2012;90:1046–1063. doi: 10.1016/j.ajhg.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Richardson K., Lai C.Q., Parnell L.D., Lee Y.C., Ordovas J.M. A genome-wide survey for SNPs altering microRNA seed sites identifies functional candidates in GWAS. BMC Genomics. 2011;12:504. doi: 10.1186/1471-2164-12-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cunnington M.S., Santibanez Koref M., Mayosi B.M., Burn J., Keavney B. Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010;6:e1000899. doi: 10.1371/journal.pgen.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jendrzejewski J., He H., Radomska H.S., Li W., Tomsic J., Liyanarachchi S., Davuluri R.V., Nagy R., de la Chapelle A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA. 2012;109:8646–8651. doi: 10.1073/pnas.1205654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mercer T.R., Gerhardt D.J., Dinger M.E., Crawford J., Trapnell C., Jeddeloh J.A., Mattick J.S., Rinn J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2012;30:99–104. doi: 10.1038/nbt.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olshavsky N.A., Comstock C.E., Schiewer M.J., Augello M.A., Hyslop T., Sette C., Zhang J., Parysek L.M., Knudsen K.E. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010;70:3975–3984. doi: 10.1158/0008-5472.CAN-09-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Dijk M., Thulluru H.K., Mulders J., Michel O.J., Poutsma A., Windhorst S., Kleiverda G., Sie D., Lachmeijer A.M., Oudejans C.B. HELLP babies link a novel lincRNA to the trophoblast cell cycle. J. Clin. Invest. 2012;122:4003–4011. doi: 10.1172/JCI65171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 92.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 93.Lorincz M.C., Dickerson D.R., Schmitt M., Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 94.Hitchins M.P., Rapkins R.W., Kwok C.T., Srivastava S., Wong J.J., Khachigian L.M., Polly P., Goldblatt J., Ward R.L. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell. 2011;20:200–213. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 95.Raval A., Tanner S.M., Byrd J.C., Angerman E.B., Perko J.D., Chen S.S., Hackanson B., Grever M.R., Lucas D.M., Matkovic J.J. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129:879–890. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aran D., Sabato S., Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. doi: 10.1186/gb-2013-14-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kerkel K., Spadola A., Yuan E., Kosek J., Jiang L., Hod E., Li K., Murty V.V., Schupf N., Vilain E. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 98.Gibbs J.R., van der Brug M.P., Hernandez D.G., Traynor B.J., Nalls M.A., Lai S.L., Arepalli S., Dillman A., Rafferty I.P., Troncoso J. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Savic D., Ye H., Aneas I., Park S.Y., Bell G.I., Nobrega M.A. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. 2011;21:1417–1425. doi: 10.1101/gr.123745.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H., Lupien M. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012;44:1191–1198. doi: 10.1038/ng.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rakyan V.K., Down T.A., Balding D.J., Beck S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cortessis V.K., Thomas D.C., Levine A.J., Breton C.V., Mack T.M., Siegmund K.D., Haile R.W., Laird P.W. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Hum. Genet. 2012;131:1565–1589. doi: 10.1007/s00439-012-1189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bell C.G., Finer S., Lindgren C.M., Wilson G.A., Rakyan V.K., Teschendorff A.E., Akan P., Stupka E., Down T.A., Prokopenko I., International Type 2 Diabetes 1q Consortium Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS ONE. 2010;5:e14040. doi: 10.1371/journal.pone.0014040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Urnov F.D., Rebar E.J., Holmes M.C., Zhang H.S., Gregory P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 106.Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 107.Soldner F., Laganière J., Cheng A.W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L.I., Myers R.H., Lindquist S. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 110.Ermann J., Glimcher L.H. After GWAS: mice to the rescue? Curr. Opin. Immunol. 2012;24:564–570. doi: 10.1016/j.coi.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Petersen G.M., Amundadottir L., Fuchs C.S., Kraft P., Stolzenberg-Solomon R.Z., Jacobs K.B., Arslan A.A., Bueno-de-Mesquita H.B., Gallinger S., Gross M. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat. Genet. 2010;42:224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Flandez M., Cendrowski J., Cañamero M., Salas A., Del Pozo N., Schoonjans K., Real F.X. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRasG12V-driven pancreatic tumourigenesis. Gut. 2013 doi: 10.1136/gutjnl-2012-304381. Published online April 18, 2013. [DOI] [PubMed] [Google Scholar]
- 113.von Figura G., Morris J.P., 4th, Wright C.V., Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2013 doi: 10.1136/gutjnl-2012-304287. Published online May 3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pomerantz M.M., Ahmadiyeh N., Jia L., Herman P., Verzi M.P., Doddapaneni H., Beckwith C.A., Chan J.A., Hills A., Davis M. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sur I.K., Hallikas O., Vähärautio A., Yan J., Turunen M., Enge M., Taipale M., Karhu A., Aaltonen L.A., Taipale J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 2012;338:1360–1363. doi: 10.1126/science.1228606. [DOI] [PubMed] [Google Scholar]
- 116.Sung Y.H., Baek I.J., Kim D.H., Jeon J., Lee J., Lee K., Jeong D., Kim J.S., Lee H.W. Knockout mice created by TALEN-mediated gene targeting. Nat. Biotechnol. 2013;31:23–24. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- 117.Gieger C., Radhakrishnan A., Cvejic A., Tang W., Porcu E., Pistis G., Serbanovic-Canic J., Elling U., Goodall A.H., Labrune Y. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ragvin A., Moro E., Fredman D., Navratilova P., Drivenes O., Engström P.G., Alonso M.E., de la Calle Mustienes E., Gómez Skarmeta J.L., Tavares M.J. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl. Acad. Sci. USA. 2010;107:775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 120.den Hoed M., Eijgelsheim M., Esko T., Brundel B.J., Peal D.S., Evans D.M., Nolte I.M., Segrè A.V., Holm H., Handsaker R.E., Global BPgen Consortium. CARDIoGRAM Consortium. PR GWAS Consortium. QRS GWAS Consortium. QT-IGC Consortium. CHARGE-AF Consortium Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat. Genet. 2013;45:621–631. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chakravarti A., Clark A.G., Mootha V.K. Distilling pathophysiology from complex disease genetics. Cell. 2013;155:21–26. doi: 10.1016/j.cell.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sanseau P., Agarwal P., Barnes M.R., Pastinen T., Richards J.B., Cardon L.R., Mooser V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012;30:317–320. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]