

Abstract
Many ion channel genes have been associated with human genetic pain disorders. Here we report two large Chinese families with autosomal-dominant episodic pain. We performed a genome-wide linkage scan with microsatellite markers after excluding mutations in three known genes (SCN9A, SCN10A, and TRPA1) that cause similar pain syndrome to our findings, and we mapped the genetic locus to a 7.81 Mb region on chromosome 3p22.3–p21.32. By using whole-exome sequencing followed by conventional Sanger sequencing, we identified two missense mutations in the gene encoding voltage-gated sodium channel Nav1.9 (SCN11A): c.673C>T (p.Arg225Cys) and c.2423C>G (p.Ala808Gly) (one in each family). Each mutation showed a perfect cosegregation with the pain phenotype in the corresponding family, and neither of them was detected in 1,021 normal individuals. Both missense mutations were predicted to change a highly conserved amino acid residue of the human Nav1.9 channel. We expressed the two SCN11A mutants in mouse dorsal root ganglion (DRG) neurons and showed that both mutations enhanced the channel’s electrical activities and induced hyperexcitablity of DRG neurons. Taken together, our results suggest that gain-of-function mutations in SCN11A can be causative of an autosomal-dominant episodic pain disorder.
Main Text
Pain serves as a defense system to protect the body against further injury and promotes healing of damaged tissues,1 but chronic pain often represents a severe and debilitating condition.2 Up to 10% of the worldwide population is affected with pain, and chronic pain significantly diminishes the quality of life in many affected individuals.1–4 Nociceptors detect noxious stimuli and produce the sensation of pain, and such a pain-modulating signal is conveyed to the central nervous system (CNS) by means of action potentials.2,4 Monogenic heritable pain disorders have provided the molecular insights into human pain and suggested new analgesic drug targets.1,3
The voltage-sensitive sodium channels are essential for the generation of action potentials in excitable cells.5 Sodium channels in mammals consist of an α subunit that encodes the core protein of the channel and auxiliary β subunits that modify the channel function.5,6 Thus far, ten different genes (SCN1A [MIM 182389], SCN2A [MIM 182390], SCN3A [MIM 182391], SCN4A [MIM 603967], SCN5A [MIM 600163], SCN8A [MIM 600702], SCN9A [MIM 603415], SCN10A [MIM 604427], SCN11A [MIM 604385], and SCN7A [MIM 182392]) of α subunits (Nav1.1–1.9 and Nax) have been identified in mammals.4,5,7 Each α subunit of sodium channels consists of four domains (DI–DIV) and each domain is composed of six transmembrane segments (S1–S6). S1–S4 are believed to form the voltage-sensing part of the channel, and S5–S6 and a membrane re-entering p-loop form the channel pore.5
Gain-of-function mutations in SCN9A have been reported in different painful disorders including primary erythermalgia (PE [MIM 1330220]),8,9 paroxysmal extreme pain disorder (PEPD [MIM 167400]),10,11 and small fiber neuropathy (SFN [MIM 133020]).4,12 Loss-of-function mutations in the same gene have been identified in congenital insensitivity to pain (CIP [MIM 243000]) and anosmia.13,14 Gain-of-function mutation in the transient receptor potential subfamily A member 1 gene (TRPA1 [MIM 604775]) leads to familial episodic pain syndrome.15 Recently, gain-of-function mutations in SCN10A have been associated with painful neuropathy.12
The SCN11A is preferentially expressed in nociceptive neurons of dorsal root ganglia (DRG) and trigeminal ganglia.16 Similar to Nav1.8 and Nav1.5, Nav1.9 is also a tetrodotoxin-resistant (TTX-r) channel and is activated by relatively negative membrane potentials.17 Nav1.9 knockout mice exhibited analgesic phenotype,18,19 further investigation in which showed that inflammatory mediators such as formalin, carrageenan, CFA and prostanoids,18 prostaglandin E2, bradykinin, interleukin-1β, and ATP19 diminished the pain hypersensitivity. Nav1.9 is therefore a major effector of peripheral inflammatory pain hypersensitivity. Up till now, it has been unclear whether mutations in SCN11A are associated with human genetic pain disorders.
In this study, two Chinese families from Jilin-Tonghua (JLTH) and Hebei-Beijing (HBBJ) with familial episodic chronic pain were identified and characterized by the Affiliated Union Hospital of Huazhong University of Science and Technology. The pain phenotype in both families was transmitted in an autosomal-dominant pattern. The JLTH and HBBJ families each span five generations and include 43 and 26 members, respectively (Figures 1A and 1C). Nine affected individuals and seven normal members from the JLTH family and five affected individuals and eight healthy members from the HBBJ family participated in this study. Affected individuals from both Chinese families shared most common clinical features according to their descriptions. First, the intense pain was localized principally to the distal lower extremities and occasionally in the upper body, especially in the joints of fingers and arms. Second, episodic pain appeared late in the day and relapsed once every 2–5 days for a total of 9–11 recurrences (JLTH family) or 17–19 recurrences (HBBJ family) for one cycle. Third, pain was exacerbated with fatigue, such as catching a cold or performing hard exercise. The pain was relieved by oral administration of anti-inflammatory analgesic medicines. Fourth, the period of intense pain usually was accompanied by sweating. The feeling of the pain region was extremely cold and the pain could be assuaged by a hot compress. Finally, severe pain in all affected individuals shared the property of diminishing with age. The clinical symptoms of the affected individuals in both families are detailed in the Supplemental Data and Figure S1 available online. Neurological examinations showed that two probands retained intact sensitivities to joint position, light touch, pin prick, etc.
Figure 1.
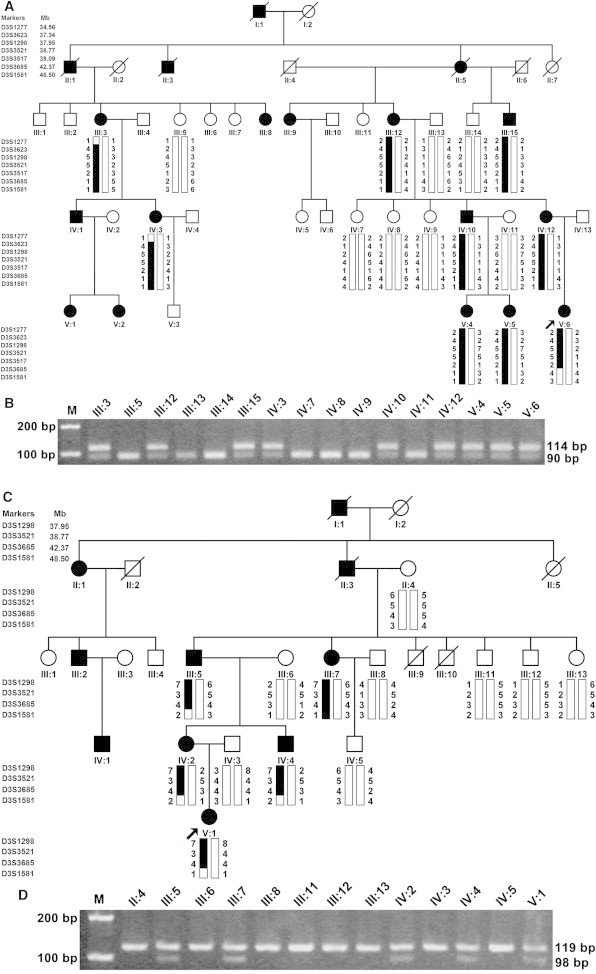
Pedigree Structures, Genotypic, and Restriction Analysis in Two Chinese Families with Episodic Pain
Individuals with pain disease are indicated by solid squares (males) or solid circles (females). Unaffected individuals are indicated by open symbols. Deceased individuals are indicated by slashes (/). The proband is indicated by an arrow.
(A) Microsatellite haplotypes spanning the linkage region on chromosome 3p22.3–p21.32 identified in the whole-genome scan from the JLTH family. Genotypes for markers D3S1277, D3S3623, D3S1298, D3S3521, D3S3517, D3S3685, and D3S1581 are shown below each symbol. The black vertical bar (including markers D3S3623, D3S1298, D3S3521, and D3S3517) indicates the haplotype cosegregating with the disease.
(B) The NciI restriction analysis showing full segregation of the c.673C>T mutation with the disease phenotype in the JLTH family.
(C) Linkage analysis from the HBBJ family showed the markers D3S1298, D3S3521, and D3S3685 cosegregating with the disease, but D3S1581 did not.
(D) The HindIII restriction analysis showing full segregation of the c.2423C>G mutation with the disease phenotype in the HBBJ family.
After the informed consent was obtained from the participants, peripheral blood samples were obtained from 14 affected and 15 unaffected family members and used for isolation of genomic DNA (Figure 1). This study was approved by the ethics committee of Huazhong University of Science and Technology (Wuhan, China) and complied with the Declaration of Helsinki. TRPA1, SCN9A, and SCN10A were first sequenced in the probands of the two families, but no pathogenic mutations were found. We then performed a genome-wide linkage scan with 382 microsatellite markers in the JLTH family, as described previously,20 as well as follow-up fine mapping. LOD scores for all genotyped markers were plotted against the physical position of each marker in Figure 1A. Positive linkage was identified only with markers (including D3S3623, D3S1298, D3S3521, and D3S3517) on chromosome 3p22.3–p21.32. The LOD scores for the chromosome 3 markers are shown in Table S1. Two markers, D3S1298 and D3S3517, generated LOD scores greater than 3, the cut-off LOD score for significant linkage, thereby suggesting the presence of a genetic locus on chromosome 3 corresponding to the pain phenotype in the affected persons under investigation (Figure 1A).
Haplotype analysis was carried out to identify the recombination events in the JLTH family (Figure 1A). The affected individuals III:3 and IV:3 showed a recombination event between markers D3S1277 and D3S3623. The affected individual V:6 displayed a recombination event between markers D3S3517 and D3S3685. Taken together, these results suggested that the gene responsible for the pain phenotype located between D3S1277 and D3S3685 on chromosome 3p22.3–p21.32, a genomic region of 7.81 Mb (Figures 1A and 2A), which contains 55 RefSeq genes.
Figure 2.
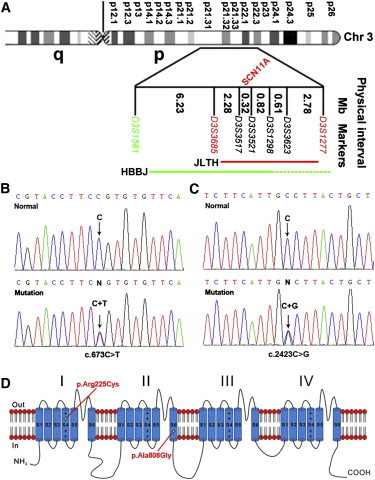
Diagram of Chromosome 3 Showing the Critical Region and Identification of SCN11A Mutations from the JLTH and HBBJ Families
(A) The linkage intervals and flanking markers of the critical region are indicated.
(B and C) DNA sequence chromatograms showing the different heterozygous mutations in SCN11A identified in our study families.
(D) Schematic structure diagram of the Nav1.9 protein with a summary of pain-associated mutations.
Our linkage analysis and haplotyping in the HBBJ family also suggested that the disease locus was positioned in the same region as in the JLTH family (Figures 1C and 2A). Two-point LOD scores for all three markers D3S1298, D3S3521, and D3S3685 were positive (Table S1).
To identify the pathogenic mutations, genomic DNA (3 μg) from the proband of the JLTH family was sent to BGI for whole-exome sequencing. The genomic DNA was randomly fragmented by the Covaris E10 system and the size of the library fragments was distributed mainly between 250 and 300 bp. The protein-coding regions were captured with a Nimblegen SeqCap EZ Human Exome Library v.2.0. High-throughput sequencing was performed on Hiseq2000 platform. The sequences of each individual were generated as 90 bp pair-end reads. Clean reads were mapped onto the human reference genome (UCSC Genome Browser hg19) with software SOAP and BWA for SNP detection and indel detection, respectively. Mean depth of target region was obtained as 81.13 × and coverage of target region as 99.54%. The called SNPs and indels were annotated by CCDS, RefSeq, Ensembl, and Encode databases. dbSNP (v.132) was used to filter known variants. Protein functions of missense SNP were predicted with SIFT. A variant (c.673C>T) in SCN11A (RefSeq accession number NM_014139.2), which is within the linked interval, was found. This variant is predicted to result in an Arg to Cys substitution at amino acid residue 225 (p.Arg225Cys) in the transmembrane segment S4 at DI region of the Nav1.9 channel protein (Figures 2B and 2D). It is worth noting that there is a variation (COSM108712/rs138607170) for the mutation (c.673C>T [p.Arg225Cys]) of SCN11A in Ensembl databases. Although further analysis showed that such a variation arises because the gene was derived from skin tumor rather than normal tissue, it nevertheless draws our attention to the importance of using public gene databases carefully, especially for researchers in genetics.
Further Sanger sequencing of SCN11A identified a different missense mutation (c.2423C>G) in the proband of the HBBJ family (the primers used are shown in Table S4). This mutation is predicted to result in an Ala to Gly substitution (p.Ala808Gly) in the transmembrane segment S6 at DII region of the Nav1.9 channel protein (Figures 2C and 2D). Because neither c.673C>T nor c.2423C>G would result in a gain or a loss of a restriction site, we designed mismatched primers to create a restriction site and used PCR restriction fragment length polymorphism (RFLP) assay to determine whether the two mutant alleles cosegregated with the pain phenotype. The primers and restrictive endonucleases used are listed in Table S2.
The results of the examination of all available members of the JLTH and HBBJ families via restriction analysis showed that the two missense mutations fully segregated with the pain phenotype in each of the families (Figures 1B and 1D). We also performed a screening for the mutations of c.673C>T and c.2423C>G in 1,021 unrelated Han populations by restriction analysis, but no mutations were found (data not shown). In addition, neither of our identified mutations is present in either the 1000 Genomes Project database or the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) database.
The protein sequence alignment showed that both Arg225 and Ala808 amino acid residues of human Nav1.9 are highly conserved from chicken to human (Figure S2A). Meanwhile, these two residues are also highly conserved in different Na+ channel types, including Nav1.1 through Nav1.9 and Nax in humans (Figure S2B).
To determine whether SCN11A mutations influence the channel function, we assessed the biophysical properties of human Nav1.9 (hNav1.9) mutant channels. The full-length cDNA of the wild-type SCN11A was subcloned into a pcDNA 3.1 (+) expression vector, and the c.673C>T or c.2423C>G mutant was introduced into the pcDNA 3.1-SCN11A plasmid by site-directed mutagenesis. The primers carrying the desired mutations are shown in Table S3. All recombinant constructs were confirmed by restriction enzyme digestion and verified by DNA sequencing.
The plasmids encoding wild-type, p.Arg225Cys, or p.Ala808Gly channels were transfected into mouse dorsal root ganglia (DRG) neurons by electroporation. The animal protocol used in this study was approved by the Institutional Animal Care and Use Committee of Peking University. Primary culture of DRG neurons were prepared as described previously with minor modification.21 In brief, 4- to 8-week-old adult C57 male mice (∼20 g) were killed by decapitation, and DRGs together with dorsal and ventral roots and attached spinal nerves were taken out from the spinal column. After removing the attached nerves and surrounding connective tissues, about 10–14 DRGs from thoracic and lumbar segments of spinal cords were immediately dissected and cleaned in Dulbecco’s modified Eagle’s medium type F12 (DMEM-F12, Gibco). Ganglia were dissociated by enzymatic treatment with collagenase (Type IA, 1 mg/ml) and trypsin (type I, 0.3 mg/ml) at 37°C for 40 min. Gentle mechanical trituration was performed every 10 min through fire-polished glass pipettes until solution became cloudy. The resulting suspension of single cells was centrifuged, and the cell pellet was resuspended in 100 μl R buffer (Life Technologies). hNav1.9 (WT) or mutant constructs and EGFP were mixed at a ratio of 10:1, and the mixture was electroporated into DRGs by using Neon transfection system for 20 ms at 1,200 mV twice (Life Technologies). After electroporation, cells were seeded onto poly-L-lysine-coated coverslips and maintained in DMEM (Gibco) containing 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin at 37°C in a humidified incubator with 5% CO2.
Conventional whole-cell patch-clamp recording was used to examine the biophysical properties of mutant hNav1.9. Patch pipettes were fabricated from borosilicate glass with a resistance below 5 MΩ when filled with the standard pipette solution. Currents were amplified with an integrated EPC10 amplifier with Pulse software (HEKA Elektronik, Germany). The liquid junction potential between the pipette and bath solutions was zeroed before seal formation. The compensation of pipette series resistance and capacitance were taken by using the built-in circuitry of the amplifier (>80%) to reduce voltage errors. All experiments were conducted at room temperature (22°C–25°C).
Patch-clamp recordings were performed 36–48 hr after electroporation of DRG neurons. The control bath solution contained (in mM) 140 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, 0.1 CdCl2, 20 TEA-Cl, 0.001 TTX, and 10–30 glucose (pH 7.4) (adjusted with NaOH). 20 mM tetraethylammonium chloride (TEA-Cl), 0.1 mM CdCl2, and 1 μM tetrodotoxin (TTX) were added into the bath solution to block endogenous voltage-gated potassium currents, calcium currents, and TTX-sensitive sodium currents, respectively. The standard pipette solution consisted of (in mM) 135 CsF, 10 NaCl, 2.5 MgCl2, 10 HEPES, 1 EGTA, 5 TEA-Cl, and 4 Mg-ATP (pH 7.4) (adjusted with CsOH). Fluoride anion was used to separate the activation and inactivation ranges of Nav1.9 and Nav1.8.22 For current-clamp recordings, the pipette solution contained (in mM) 140 KCl, 0.5 EGTA, 5 HEPES, and 2 Mg-ATP (pH 7.3) with KOH (adjusted to 315 mOsm with dextrose). The extracellular solution contained (in mM) 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES (pH 7.3) (adjusted with NaOH).
Data were analyzed with Igor software (Wavemetrics), Clampfit (Molecular Devices), SigmaPlot (SPSS), and Origin (OriginLab). Activation curves were fitted to a Boltzmann function as: G/Gmax = 1/(1 + exp[(V1/2 − Vm)/k]), in which Gmax is the maximal conductance, V1/2 is the half-maximum voltage, and k is the slope factor. Steady-state inactivation data were fitted by means of a function of the form I/Imax = 1/(1 + exp[(V1/2 − Vm)/k]). The activation time constants were measured by fitting the activation currents to a single exponential equation, I = A·exp(−t/τa) + C, where I is the current, A is the initial amplitude, t is the time for activation, τa is the time constant for activation, and C is the baseline. Data were presented as mean ± SEM. The statistical significance of difference for raw data was determined, when appropriate, by either Student’s t test or one-way ANOVA followed by Dunnett multiple comparisons versus control group (SigmaPlot). p < 0.05 was considered statistically significant.
Nav1.9 channel conducts prominent persistent Na+ currents and displays slow inactivation kinetics at relatively depolarized potentials. Previous studies indicated that inclusion of fluoride instead of chloride in the pipette solution would cause a prominent negative shift in the activation threshold of Nav1.9 but not Nav1.8, and therefore separated the activation ranges from each other.22 After such a performance, we examined the activation characteristics of Na+ currents in DRG neurons expressed with hNav1.9 (WT), hNav1.9 (p.Arg225Cys), or hNav1.9 (p.Ala808Gly) channels in the presence of CdCl2 (0.1 mM), TEA-Cl (20 mM), and TTX (1 μM), in bath solutions, and with CsF (135 mM)-based solutions as pipette solutions. Under these conditions, Nav1.9 currents can be recorded individually. The whole-cell currents were recorded from small-sized DRG neurons (<30 pF) expression of wild-type, p.Arg225Cys, and p.Ala808Gly channels (Figure 3A). Please note that we performed the experiments only in the cells that displayed green fluorescence. Relatively low threshold inward Na+ currents, at about −70 mV, with slow inactivation were evoked by 200 ms test pulses ranging from −90 to −40 mV with 5 mV increments. Otherwise at more positive voltages (i.e., >−40 mV), much larger Na+ currents with fast inactivation kinetics arose, probably attributed to the activation of another type of Na+ channels, i.e., Nav1.8 channel. As such, we mainly analyzed the currents elicited from −90 to −50 mV. Wild-type channels and two alterations of Nav1.9 channels produced large persistent currents, and the voltage dependences were consistent with previous reports.12,22 The relationship between current densities at peak and voltages for wild-type, p.Arg225Cys, or p.Ala808Gly channels were compared (Figure 3B). In order to reduce the variation of current density arising from the channel protein expression level, we took the following experimental procedure as a strategy. In brief, after harvesting the DRG neurons, we divided the cells into three portions averagely and then transfected with the same amount of plasmid either for wild-type or mutant channels under the same electroporation conditions. In comparison to the difference in current density for different constructs, we conducted the electrophysiological experiments in parallel after establishing expression and analyzed the data collected in the same group. As illustrated in Figure 3B, the peak current densities for p.Arg225Cys and p.Ala808Gly channels at −55 mV were 23% (p = 0.31) and 30% (p = 0.16) larger in comparison to those of wild-type channels, respectively. The changes of peak current densities indicated that mutant channels, p.Arg225Cys and p.Ala808Gly, had a higher electrical activity than that of wild-type channels. In contrast, control cells (n = 6) transfected with empty vectors in our preparations showed very low responses displayed in hexagons. Parameters relating to activation and inactivation for WT, p.Arg225Cys, and p.Ala808Gly were also examined. For evaluation of inactivation changes, we measured the relative current for which persistent sodium currents (measured at the end of stimulation) were normalized by the respective maximum activation currents (the peak amplitude for activation). Figures 3C and 3D show that the averaged activation time constant (fit the activation currents by a single exponential equation) and the relative currents at different tested voltages were not significantly different from each other. For instance, at −50 mV, τa = 6.46 ± 0.51 ms (WT, n = 18); τa = 6.24 ± 0.53 ms (p.Arg225Cys, n = 16); and τa = 6.29 ± 0.57 ms (p.Ala808Gly, n = 9). Furthermore, the conductance-voltage curve and steady-state inactivation relationship for wild-type, p.Arg225Cys, and p.Ala808Gly channels were summarized in Figures 3E and 3F. As shown in Figure 3E, solid lines fitted to a single Boltzmann equation giving values of V1/2 = −59 ± 0.7 mV and k = 5.1 ± 0.5 mV for wild-type (n = 16); V1/2 = −57.7 ± 1.4 mV, k = 5.7 ± 0.7 mV for p.Arg225Cys (n = 15); and V1/2 = −60.4 ± 0.9 mV, k = 4.8 ± 0.6 mV for p.Ala808Gly (n = 12), respectively. In addition, Boltzmann fitting of steady-state inactivation curve in Figure 3F corresponded to V1/2 of −57.3 ± 0.7 mV, −60.6 ± 0.9 mV, and −56.5 ± 0.9 mV for wild-type, p.Arg225Cys, and p.Ala808Gly, respectively. Data were normalized with respect to fitted values for Gmax and Imax. No significant differences were observed from the conductance-voltage curve and steady-state inactivation among wild-type and p.Arg225Cys or p.Ala808Gly channels under our experiment conditions. Because p.Arg225Cys is positioned in the S4 segment of domain I where the voltage sensor is located, a substitution of this positively charged residue is expected to change the voltage-dependent gating of ion channels. Extensive studies of sodium channels have shown that substitution of the arginine residues in the S4 segments reduces the steepness of voltage-dependent gating. In addition, previous studies in rat brain IIA and II sodium channels showed that the positively charged residues in all four S4 segments may have an unequal contribution to the voltage-dependent properties of the sodium channel.23,24 However, the use of fluorid-based pipette solution may mask some differences of voltage dependence brought in by p.Arg225Cys because of the strong negative shift of voltage-dependent gating of the Nav1.9 channel by F− in this study. To investigate whether the alteration of p.Arg225Cys modifies voltage-dependent gating of Nav1.9 channel, we also conducted similar voltage activation experiments with CsCl-based internal solution. As illustrated in Figure S3, there was no significant change observed from the normalized conductance-voltage relationships at the voltage ranging from −90 to −25 mV (n = 7–12). Thus, the detailed mechanism underlying the higher current densities carried by substitutions (i.e., alterations of number of channels, open probability, unitary current, or combinations) still need further investigation.
Figure 3.
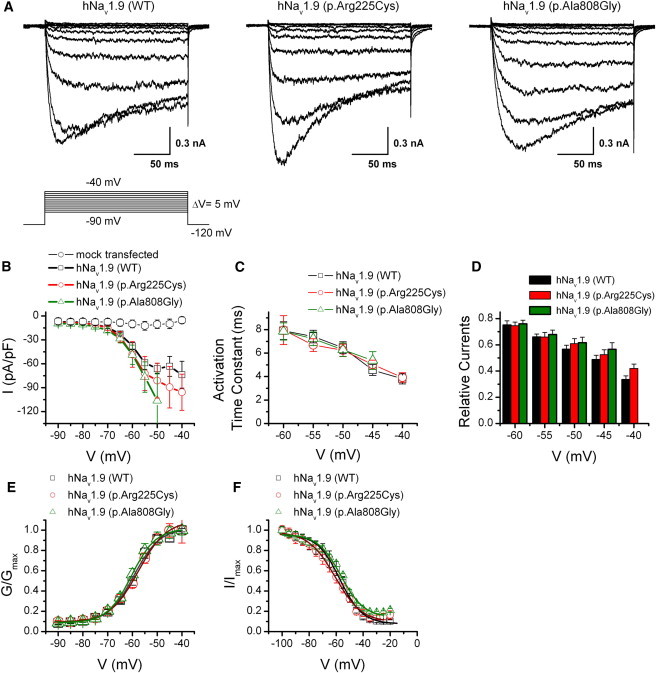
Alterations p.Arg225Cys and p.Ala808Gly Increase Current Densities of hNav1.9 Channels and Leave Voltage Dependence for Activation Unchanged when Overexpressed in Mouse DRG Neurons through Electroporation
(A) Representative whole-cell recordings evoked by voltage for wild-type channels (left) and mutant channels p.Arg225Cys (middle) and p.Ala808Gly (right), respectively. Currents were elicited by 200 ms test pulses stepped from −90 to −40 mV with 5 mV increments. Note the pipette solutions contained 135 mM fluoride. Vh = −120 mV. The recording was performed at least 10 min after achieving whole-cell recording with fluoride to reduce the time-dependent shift of the parameters of current properties.
(B) Current-voltage relationships of p.Arg225Cys and p.Ala808Gly versus the wild-type for data in (A). The peak current density (normalized by membrane capacitance) is plotted (n = 8–15, p > 0.05). The endogenous responses from mouse DRG neurons activated by the same voltage protocol are displayed in hexagons (6 of 16 cells). Data are presented as mean ± SEM.
(C) Mean activation time constant fit by a single exponential equation is plotted against voltage (n = 8–15). There was no significant change for the time-to-peak values from each other (p > 0.05).
(D) Summary plot of relative current at the voltage ranging from −60 mV to −40 mV for WT and two substitutions. For the relative currents, persistent sodium currents (measured at the end of stimulation) were normalized by the respective maximum activation currents (the peak amplitude for activation).
(E) Normalized conductance-voltage relationships. Boltzmann fits correspond to V1/2 = −59 ± 0.7 mV and k = 5.1 ± 0.5 mV for wild-type (n = 16), to V1/2 = −57.7 ± 1.4 mV, k = 5.7 ± 0.7 mV for p.Arg225Cys (n = 20), and to V1/2 = −60.4 ± 0.9 mV, k = 4.8 ± 0.6 mV for p.Ala808Gly (n = 18), respectively.
(F) Steady-state inactivation curves of wild-type, p.Arg225Cys, and p.Ala808Gly are plotted. Steady-state availability data were derived by measuring the peak current amplitude evoked by −50 mV after 500 ms pre-pulse to voltages ranging from −120 mV to 0 mV. The smooth lines are fits to a Boltzmann equation giving values of V1/2 of −57.3 ± 0.7 mV, −60.6 ± 0.9 mV, and −56.5 ± 0.9 mV for wild-type, p.Arg225Cys, and p.Ala808Gly, respectively (p > 0.05). All experiments in the figure were carried out in the presence of 135 mM fluoride in the pipette solutions and 1 μM TTX in the bath solutions.
To gain further insight into the effect of substitutions (p.Arg225Cys and p.Ala808Gly) on excitability, we utilized current-clamp recordings to investigate the action potential firing in DRG neurons. We injected current ranging from 0 to 225 pA with 25 pA increments lasting for 500 ms into DRG neurons expressed with wild-type, p.Arg225Cys, or p.Ala808Gly channels, and the representative current-clamp responses to 500 ms current pulse injection are shown in Figure 4A. We note that the action potential firing traces were mainly chosen to emphasize the action potential firing frequency changes and may not faithfully reflect the kinetics information. Statistics data indicated that both the resting membrane potential (wild-type: −46.5 ± 0.8 mV, n = 40; p.Arg225Cys: −45.6 ± 1.3 mV, n = 20; p.Ala808Gly: −42.8 ± 1.2 mV, n = 25; mock: −47.9 ± 2.0 mV, n = 7) and threshold voltage at which action potential take-off occurs (wild-type: −21.4 ± 0.5 mV, n = 40; p.Arg225Cys: −23.5 ± 0.6 mV, n = 20; p.Ala808Gly: −23.7 ± 0.6 mV, n = 25; mock: −22.3 ± 0.9 mV, n = 7) were not significantly different in DRG neurons expressed with wild-type or mutant channels (Figure 4B). Both alterations (p.Arg225Cys and p.Ala808Gly) produced an increase in the proportion of spontaneously firing DRG neurons as compared to that expressed with WT. Only ∼10% (5 of 51 cells) of DRG neurons expressed with wild-type Nav1.9 channels displayed spontaneous firing. In contrast, ∼25% (6 of 24 cells and 7 of 28 cells for p.Arg225Cys and p.Ala808Gly, respectively) of DRG neurons expressed with p.Arg225Cys or p.Ala808Gly displayed spontaneous firing, significantly more than that of WT (p < 0.05). Moreover, averaged number of action potential firing stimulated at 25 pA was 3.0 ± 0.5, 3.8 ± 0.8, and 5.6 ± 0.9 for WT, p.Arg225Cys, and p.Ala808Gly, respectively. We therefore did not determine the current threshold for action potential generation. Action potential firing increased along with elevation of input currents. In many but not all of the neurons studied no matter whether they were transfected with WT, p.Arg225Cys, or p.Ala808Gly, there was a progressive diminution in the amplitude of action potentials as well as the voltage of afterhyperpolarization; furthermore, such a phenomenon varied from different cells, even the cultured cells coming from the same cover glass. As illustrated in Figure 4C, DRG neurons overexpressed with hNav1.9 channels resulted in more action potential firings as compared to that of mock transfected cells (p < 0.05), and the averaged frequencies of action potential firing in DRG neurons expressed with p.Arg225Cys or p.Ala808Gly channels exhibited significant promotion compared to that of DRG neurons expressed with wild-type channels over a wide range of stimuli. More importantly, DRG neurons expressed with p.Ala808Gly fired even more action potentials than those neurons expressed with p.Arg225Cys under the same conditions (Figures 4A and 4C).
Figure 4.
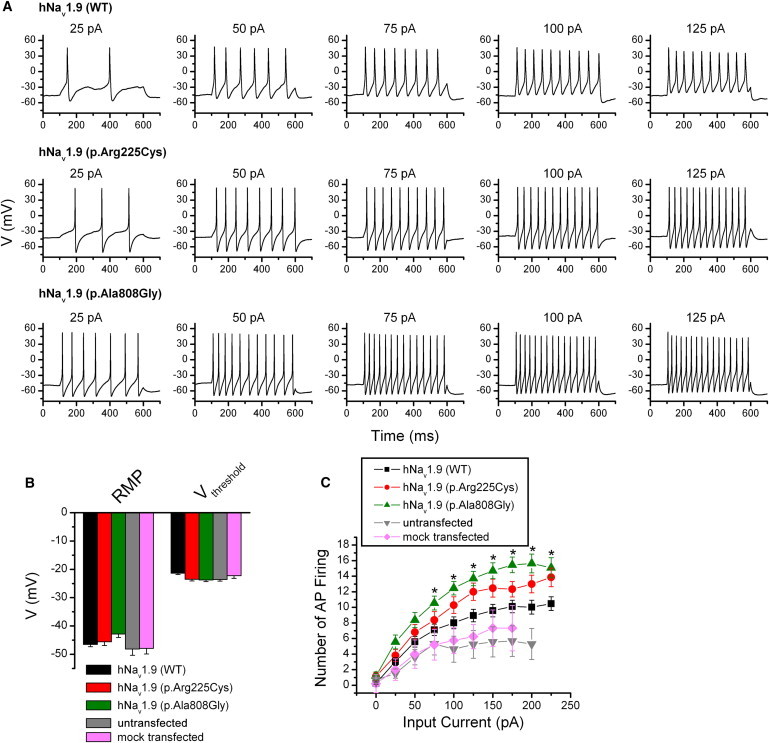
Both Alterations p.Arg225Cys and p.Ala808Gly Increase the Excitability of DRG Neurons
(A) Current-clamp responses of DRG neurons expressed with wild-type, p.Arg225Cys, and p.Ala808Gly channels to 500 ms current pulse injection, respectively. Amplitude of injected current is indicated on the top of each panel.
(B) Statistics plot showing no significant changes for RMP (resting membrane potential) and Vthreshold (the threshold at which action potential take-off occurs) from DRG neurons expressed with wild-type channels. RMP of neurons expressed with WT (−46.5 ± 0.8 mV, n = 40), p.Arg225Cys (−45.6 ± 1.3 mV, n = 20; p > 0.05), and p.Ala808Gly (−42.8 ± 1.2 mV, n = 25; p > 0.05) was not significantly different. Voltage threshold of DRG neurons expressed with WT (−21.4 ± 0.5 mV, n = 40), p.Arg225Cys (−23.5 ± 0.6 mV, n = 20; p > 0.05), and p.Ala808Gly (−23.7 ± 0.6 mV, n = 25; p > 0.05) was not significantly altered, either.
(C) Comparison of averaged frequency of action potential (AP) firing (numbers of action potential firing) evoked by 500 ms current injection ranging from 0 to 225 pA in DRG neurons untransfected or overexpressed with empty pcDNA3.1 vector, wild-type, p.Arg225Cys, and p.Ala808Gly channels (∗p < 0.05 as compared to wild-type).
In this study, we identified two gain-of-function point mutations in SCN11A (c.673C>T [p.Arg225Cys] and c.2423C>G [p.Ala808Gly]) as the cause of familial episodic pain. These substitutions are highly penetrant, similar to TRPA1 mutation (c.2564A>G [p.Asn855Ser]), giving rise to stereotyped episodes of severe pain.15 The difference is that TRPA1 mutation resulted in familial episodic pain affecting principally the upper body triggered by cold and fasting, whereas the pain caused by SCN11A mutations mainly localized to the distal lower extremities and usually was aggravated by fatigue. Electrophysiology experiments with isolated DRG neurons showed that p.Arg225Cys and p.Ala808Gly alterations did increase peak current densities and produced higher electrical activities than that of wild-type channels. Higher electrical activities promoted action potential firing in DRG neurons and might be the cause of episodic pain.
Nav1.7, Nav1.8, and Nav1.9 are preferentially expressed in peripheral somatosensory neurons and have been implicated in injury-induced neuronal hyperexcitability.25 Nav1.7 and Nav1.8 have been linked to human pain in heredity.8,12 Nevertheless, a direct link of Nav1.9 to human pain has not been reported. Previous studies in Scn11a knockout mice have confirmed that Nav1.9 is an important molecule in generating hyperalgesia in infammatory pain states,18,19 suggesting that Nav1.9 channels may play an important role in nociception.26,27 We herein provided the genetic evidence that mutations in SCN11A were directly involved in human episodic pain. In addition, Lolignier et al. reported that Nav1.9 channel plays an important role in the generation of heat and mechanical pain hypersensitivity in both subacute and chronic inflammatory models.28
Compared to other Nav channels, Nav1.9 generates a persistent TTX-r current with a more hyperpolarized voltage dependence and ultraslow recovery from inactivation in DRG neurons.19,29–31 From DRG neurons in Nav1.9 knockout mice, the persistent TTX-r current is missing and can be restored by expression of recombinant Nav1.9 channels.18,19,29 It has been suggested that the Nav1.9 channel is involved in the modulation of nociceptor membrane potential and makes important contributions to establish the membrane voltage that may in turn influence gating of other Nav channels present in DRG neurons and overall neuronal excitability.31,32 In contrast, Nav1.8 channels evoked by strong depolarizations have been thought to mediate the rapid upstroke of action potentials of sensory neurons.32–34 Although the Nav1.9 channel is not directly responsible for action potential generation,30,31 it may enhance various types of firing behavior such as burst firing and rebound firing.27,29
In the present study, we found that both mutations of Nav1.9 channels had higher electrical activities in DRG neurons. DRG neurons overexpressed with mutant channels also exhibited significantly augmented neuronal action potential firing rate. Taken together, our results suggested that the higher electrical activities of mutant Nav1.9 channels might remarkably alter the membrane potentials and induce the opening of other Nav channels (such as Nav1.8 channel), and thus cause DRG neurons to be hyperexcitable, which contributed to the familial episodic pain disorder.
Affected individuals in the HBBJ family had many more recurrent episodic pains than did those of the JLTH family according to the affected persons’ description, which is coincident with the actual fact that DRG neurons expressed with p.Ala808Gly provoked even more action potential firings than those expressed with p.Arg225Cys under the same conditions.
Several studies have suggested that Nav1.9 channel currents were upregulated by inflammatory mediators and that the excitability of nociceptors was increased as a result.35 Therefore, the Nav1.9 channel is thought of as an effector of peripheral inflammatory pain hypersensitivity.27,32 Intriguingly, the pain is relieved in patients from both Chinese families after oral administration of anti-inflammatory medicines (pediatric paracetamol tablets and Fenbid capsules). It is of great interest to investigate whether the Nav1.9 channel protein is the direct target of these two anti-inflammatory drugs, or whether drug-regulated inflammatory factors regulate the gating of Nav1.9 channels. Our above analysis provides evidence that the Nav1.9 channel would be an appealing target for the development of specific drugs to relieve pain.
Acknowledgments
We thank the affected individuals and their families’ members who participated in the study for their continuous support. This study was mainly supported by the National Natural Science Foundation of China grants 31230045 and 81271252 (J.Y.L.), 31271209 (J.Y.), and 31100597 (L.H.Z.), by the Program for Changjiang Scholars and Innovative Research Team in University (LRT1006 to X.Z.), by the National Science & Technology Pillar Program during the 12th Five-year Plan Period (2012BAI09B04) (J.Y.L.), and by Specialized Research Fund for the Doctoral Program of Higher Education of Ministry of Education of China (No. 20120142120062 to J.Y.), and the National Basic Research Program of China 2014CB910304/02 (J.Y.) and 2012CB518006 (Z.Z.).
Contributor Information
Jing Yao, Email: jyao@whu.edu.cn.
Jing Yu Liu, Email: liujy@mail.hust.edu.cn.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Marshfield Clinic Medical Genetics database, http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Nimblegen SeqCap EZ Human Exome Library v.2.0, http://www.nimblegen.com/products/seqcap/ez/v2/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Raouf R., Quick K., Wood J.N. Pain as a channelopathy. J. Clin. Invest. 2010;120:3745–3752. doi: 10.1172/JCI43158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lampert A., O’Reilly A.O., Reeh P., Leffler A. Sodium channelopathies and pain. Pflugers Arch. 2010;460:249–263. doi: 10.1007/s00424-009-0779-3. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg Y.P., Pimstone S.N., Namdari R., Price N., Cohen C., Sherrington R.P., Hayden M.R. Human Mendelian pain disorders: a key to discovery and validation of novel analgesics. Clin. Genet. 2012;82:367–373. doi: 10.1111/j.1399-0004.2012.01942.x. [DOI] [PubMed] [Google Scholar]
- 4.Hoeijmakers J.G., Merkies I.S., Gerrits M.M., Waxman S.G., Faber C.G. Genetic aspects of sodium channelopathy in small fiber neuropathy. Clin. Genet. 2012;82:351–358. doi: 10.1111/j.1399-0004.2012.01937.x. [DOI] [PubMed] [Google Scholar]
- 5.Ogata N., Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 2002;88:365–377. doi: 10.1254/jjp.88.365. [DOI] [PubMed] [Google Scholar]
- 6.Isom L.L., De Jongh K.S., Patton D.E., Reber B.F., Offord J., Charbonneau H., Walsh K., Goldin A.L., Catterall W.A. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 7.Goldin A.L., Barchi R.L., Caldwell J.H., Hofmann F., Howe J.R., Hunter J.C., Kallen R.G., Mandel G., Meisler M.H., Netter Y.B. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y., Wang Y., Li S., Xu Z., Li H., Ma L., Fan J., Bu D., Liu B., Fan Z. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dib-Hajj S.D., Cummins T.R., Black J.A., Waxman S.G. From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Fertleman C.R., Baker M.D., Parker K.A., Moffatt S., Elmslie F.V., Abrahamsen B., Ostman J., Klugbauer N., Wood J.N., Gardiner R.M., Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Dabby R., Sadeh M., Gilad R., Lampl Y., Cohen S., Inbar S., Leshinsky-Silver E. Chronic non-paroxysmal neuropathic pain - Novel phenotype of mutation in the sodium channel SCN9A gene. J. Neurol. Sci. 2011;301:90–92. doi: 10.1016/j.jns.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Faber C.G., Lauria G., Merkies I.S., Cheng X., Han C., Ahn H.S., Persson A.K., Hoeijmakers J.G., Gerrits M.M., Pierro T. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA. 2012;109:19444–19449. doi: 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox J.J., Reimann F., Nicholas A.K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss J., Pyrski M., Jacobi E., Bufe B., Willnecker V., Schick B., Zizzari P., Gossage S.J., Greer C.A., Leinders-Zufall T. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–190. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kremeyer B., Lopera F., Cox J.J., Momin A., Rugiero F., Marsh S., Woods C.G., Jones N.G., Paterson K.J., Fricker F.R. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dib-Hajj S.D., Black J.A., Cummins T.R., Waxman S.G. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 2002;25:253–259. doi: 10.1016/s0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- 17.Dib-Hajj S.D., Tyrrell L., Waxman S.G. Structure of the sodium channel gene SCN11A: evidence for intron-to-exon conversion model and implications for gene evolution. Mol. Neurobiol. 2002;26:235–250. doi: 10.1385/MN:26:2-3:235. [DOI] [PubMed] [Google Scholar]
- 18.Priest B.T., Murphy B.A., Lindia J.A., Diaz C., Abbadie C., Ritter A.M., Liberator P., Iyer L.M., Kash S.F., Kohler M.G. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. USA. 2005;102:9382–9387. doi: 10.1073/pnas.0501549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amaya F., Wang H., Costigan M., Allchorne A.J., Hatcher J.P., Egerton J., Stean T., Morisset V., Grose D., Gunthorpe M.J. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 2006;26:12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C., Li Y., Shi L., Ren J., Patti M., Wang T., de Oliveira J.R., Sobrido M.J., Quintáns B., Baquero M. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 2012;44:254–256. doi: 10.1038/ng.1077. [DOI] [PubMed] [Google Scholar]
- 21.Zhang C., Xiong W., Zheng H., Wang L., Lu B., Zhou Z. Calcium- and dynamin-independent endocytosis in dorsal root ganglion neurons. Neuron. 2004;42:225–236. doi: 10.1016/s0896-6273(04)00189-8. [DOI] [PubMed] [Google Scholar]
- 22.Coste B., Osorio N., Padilla F., Crest M., Delmas P. Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol. Cell. Neurosci. 2004;26:123–134. doi: 10.1016/j.mcn.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 23.Kontis K.J., Rounaghi A., Golgin A.L. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physion. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stühmer W., Conti F., Suzuki H., Wang X.D., Noda M., Yahagi N., Kubo H., Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 25.Shields S.D., Cheng X., Uçeyler N., Sommer C., Dib-Hajj S.D., Waxman S.G. Sodium channel Na(v)1.7 is essential for lowering heat pain threshold after burn injury. J. Neurosci. 2012;32:10819–10832. doi: 10.1523/JNEUROSCI.0304-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keh S.M., Facer P., Simpson K.D., Sandhu G., Saleh H.A., Anand P. Increased nerve fiber expression of sensory sodium channels Nav1.7, Nav1.8, And Nav1.9 in rhinitis. Laryngoscope. 2008;118:573–579. doi: 10.1097/MLG.0b013e3181625d5a. [DOI] [PubMed] [Google Scholar]
- 27.Scroggs R.S. The distribution of low-threshold TTX-resistant Na+ currents in rat trigeminal ganglion cells. Neuroscience. 2012;222:205–214. doi: 10.1016/j.neuroscience.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Lolignier S., Amsalem M., Maingret F., Padilla F., Gabriac M., Chapuy E., Eschalier A., Delmas P., Busserolles J. Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE. 2011;6:e23083. doi: 10.1371/journal.pone.0023083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostman J.A., Nassar M.A., Wood J.N., Baker M.D. GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J. Physiol. 2008;586:1077–1087. doi: 10.1113/jphysiol.2007.147942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosmans F., Puopolo M., Martin-Eauclaire M.F., Bean B.P., Swartz K.J. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 2011;138:59–72. doi: 10.1085/jgp.201110614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herzog R.I., Cummins T.R., Waxman S.G. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 2001;86:1351–1364. doi: 10.1152/jn.2001.86.3.1351. [DOI] [PubMed] [Google Scholar]
- 32.Dib-Hajj S.D., Cummins T.R., Black J.A., Waxman S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 33.Scroggs R.S. Serotonin upregulates low-and high-thresthold etrodotoxin-resistant sodium channels in the same subpopulation of rat nociceptors. Neuroscience. 2010;165:1293–1300. doi: 10.1016/j.neuroscience.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 34.Blair N.T., Bean B.P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maingret F., Coste B., Padilla F., Clerc N., Crest M., Korogod S.M., Delmas P. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 2008;131:211–225. doi: 10.1085/jgp.200709935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.