

Abstract
“Nagashima-type” palmoplantar keratosis (NPPK) is an autosomal recessive nonsyndromic diffuse palmoplantar keratosis characterized by well-demarcated diffuse hyperkeratosis with redness, expanding on to the dorsal surfaces of the palms and feet and the Achilles tendon area. Hyperkeratosis in NPPK is mild and nonprogressive, differentiating NPPK clinically from Mal de Meleda. We performed whole-exome and/or Sanger sequencing analyses of 13 unrelated NPPK individuals and identified biallelic putative loss-of-function mutations in SERPINB7, which encodes a cytoplasmic member of the serine protease inhibitor superfamily. We identified a major causative mutation of c.796C>T (p.Arg266∗) as a founder mutation in Japanese and Chinese populations. SERPINB7 was specifically present in the cytoplasm of the stratum granulosum and the stratum corneum (SC) of the epidermis. All of the identified mutants are predicted to cause premature termination upstream of the reactive site, which inhibits the proteases, suggesting a complete loss of the protease inhibitory activity of SERPINB7 in NPPK skin. On exposure of NPPK lesional skin to water, we observed a whitish spongy change in the SC, suggesting enhanced water permeation into the SC due to overactivation of proteases and a resultant loss of integrity of the SC structure. These findings provide an important framework for developing pathogenesis-based therapies for NPPK.
Main Text
The congenital palmoplantar keratoses (PPKs) are a heterogeneous group of diseases. Phenotypic classification of hereditary PPKs is based mainly on the specific morphology and distribution of the hyperkeratosis, the presence or absence of associated features, and the inheritance pattern and is assisted by additional criteria such as the presence of skin lesions in areas other than the palms and soles, the age at onset of the hyperkeratosis, the severity of the disease process, and histopathological findings.1
“Keratosis palmoplantaris Nagashima”2 or “Nagashima-type” PPK (NPPK)3 has been proposed as a clinical entity included within the diffuse hereditary PPKs without associated features.1 A familial case of two siblings was first reported as a distinct clinical type of PPK in 1989.2,4 Because Nagashima briefly described this type of hereditary PPK in the Japanese literature in 1977,5 the name “keratosis palmoplantaris Nagashima” was proposed.2 Although about 20 cases of Japanese individuals with NPPK have been reported in the Japanese literature since then, this clinical entity was not described in detail in the English language literature until 2008.3
An autosomal recessive trait has been suggested in NPPK.2,3 The clinical features of NPPK are characterized by well-demarcated reddish and diffuse palmoplantar hyperkeratosis that extends to the dorsal surfaces of the hands, feet, inner wrists, ankles, and the Achilles tendon area.2–6 Involvement of the elbows and knees and high frequencies of hyperhidrosis on palms and soles have been noted.3 Clinical observations revealed no differences between males and females, no seasonal change, and no association with squamous cell carcinoma or any other malignancy. Although mild T cell infiltration in the affected skin area has been reported,7 the pathophysiology of the skin redness and hyperkeratosis are still uncharacterized.
An autosomal recessive trait, transgressive diffuse hyperkeratosis, and the absence of associated features are also characteristic of Mal de Meleda (MDM [MIM 248300]),8 PPK Gamborg Nielsen (Norrbotten recessive type PPK [MIM 244850]),9,10 and acral keratoderma.11 These other diffuse PPKs show more severe and more progressive features than does NPPK, such as thick hyperkeratosis, leading to flexion contractures (MDM) and constricting bands surrounding the digits (MDM, PPK Gamborg Nielsen, and acral keratoderma), occasionally resulting in spontaneous amputation (MDM and acral keratoderma).1 NPPK shows only mild and nonprogressive hyperkeratosis and does not show flexion contractures or constricting bands. Thus, NPPK is distinguishable clinically from these other PPKs. Mutations in the coding region of the SLURP1 (MIM 606119) have been identified in MDM but not in NPPK, suggesting that MDM and NPPK are genetically distinct diseases.3,12
To identify gene mutations responsible for NPPK, we performed whole-exome sequencing in three unrelated Japanese NPPK individuals (KDex8 [II-1 of family 1 in Figure 1A], KDex14 [II-2 of family 2], and KDex20 [II-3 of family 3]) who showed the characteristic symptoms of NPPK; Figures 1B and 1C; see Figure S1 available online. Clinical features are summarized in Table 1. The major clinical differentiating points by which we diagnosed these individuals with NPPK among the diverse hereditary PPKs without associated features are summarized in Table 2. The study was conducted after obtaining written informed consent according to the guidelines of the Institutional Review Board of Keio University School of Medicine, National Center for Child Health and Development, Kyoto University, and Tokyo Medical University in accordance with the Helsinki guidelines.
Figure 1.
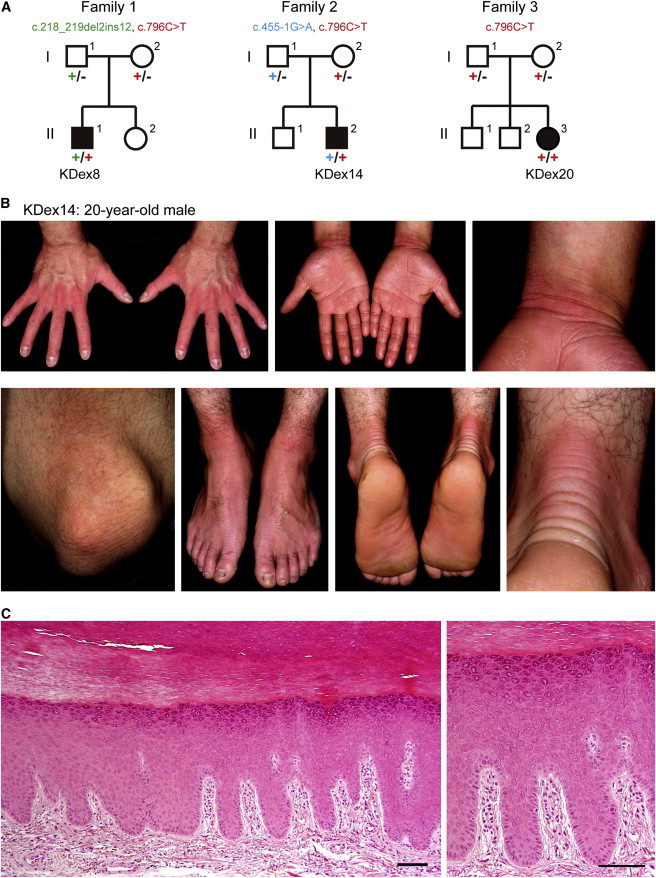
Family Pedigrees and Skin Manifestations of the Probands with NPPK
(A) Pedigrees for the families in which exome sequencing and analyses were performed on the probands (KDex8, KDex14, and KDex20). Segregation of the mutations identified in each pedigree is shown.
(B) Skin manifestations of the proband KDex14.
(C) Hematoxylin and eosin staining of the plantar epidermis of the proband KDex8. Scale bars represent 100 μm.
Table 1.
SERPINB7 Mutations and Clinical Phenotypes in Individuals with NPPK
| Affected Individual | Gender / Age |
Allele 1 |
Allele 2 |
Onset | Other Involved Areas | Hyperhidrosis | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Base Change | Amino Acid Change | Segregation | Base Change | Amino Acid Change | Segregation | |||||
| Homozygous Mutations | ||||||||||
| KDex20a | F/10 | c.796C>T | p.Arg266∗ | Paternal | c.796C>T | p.Arg266∗ | Maternal | At birth | Knees | + |
| KDex55 | F/2 | c.796C>T | p.Arg266∗ | Paternal | c.796C>T | p.Arg266∗ | Maternal | Early infancy | − | − |
| KDex62 | M/31 | c.796C>T | p.Arg266∗ | NA | c.796C>T | p.Arg266∗ | NA | 1 week | Knees | + |
| KDex72 | F/5 | c.796C>T | p.Arg266∗ | Paternal | c.796C>T | p.Arg266∗ | Maternal | At birth | Knees and elbows | + |
| KDex79 | M/31 | c.796C>T | p.Arg266∗ | NA | c.796C>T | p.Arg266∗ | NA | At birth | Knees and elbows | + |
| KDex90 | M/14 | c.796C>T | p.Arg266∗ | Paternal | c.796C>T | p.Arg266∗ | Maternal | 9-10 years | Knees and elbows | + |
| Compound Heterozygous Mutations | ||||||||||
| KDex8a | M/38 | c.796C>T | p.Arg266∗ | Maternal | c.218_219del2ins12 | p.Gln73Leufs∗17b | Paternal | At birth | Knees and elbows | + |
| KDex59 | F/16 | c.796C>T | p.Arg266∗ | Paternal | c.218_219del2ins12 | p.Gln73Leufs∗17b | Maternal | At birth | − | + |
| KDex60 | F/30 | c.796C>T | p.Arg266∗ | Paternal | c.218_219del2ins12 | p.Gln73Leufs∗17b | Maternal | At birth | Knees | + |
| KDex64 | F/28 | c.796C>T | p.Arg266∗ | Paternal | c.218_219del2ins12 | p.Gln73Leufs∗17b | Maternal | 2 years | − | + |
| KDex66 | F/64 | c.796C>T | p.Arg266∗ | NA | c.218_219del2ins12 | p.Gln73Leufs∗17b | NA | Early infancy | − | − |
| KDex14a | M/20 | c.796C>T | p.Arg266∗ | Maternal | c.455-1G>A | p.Gly152Valfs∗21b | Paternal | At birth | Knees and elbows | + |
| KDex58 | M/51 | c.796C>T | p.Arg266∗ | NA | c.455-1G>A | p.Gly152Valfs∗21b | NA | 5–6 years | Knees and elbows | + |
Abbreviations: M, male; F, Female; NA, not available; c.218_219del2ins12, c.218_219delAGinsTAAACTTTACCT.
Whole-exome sequencing performed.
Predicted from genomic sequences.
Table 2.
Major Clinical Differentiating Points among Diffuse Hereditary Palmoplantar Keratoses without Associated Features
| Types | Vörner37 | Unna-Thost38,39 | Greither40 | Sybert41 | Bothnian31 | Mal de Meleda8 | Nagashima2,3 | Gamborg Nielsen9,10 | Acral Keratoderma11 |
|---|---|---|---|---|---|---|---|---|---|
| Other names | Diffuse Epidermolytic PPK | Diffuse Nonepidermolytic PPK | Progressive PPK | Keratosis Palmoplantaris Transgradiens of Siemens | |||||
| MIM number | 144200 | 600962 | 144200 | 600231 | 248300 | 244850 | |||
| Mode of inheritance | AD | AD | AD | AD | AD | AR | AR | AR | AR |
| Responsible gene | KRT1,42KRT943,44 | KRT145 | KRT146 | Unknown | AQP532,33 | SLURP112 | SERPINB7a | Unknown | Unknown |
| Prevalence rate | 4.4/100,000 populations in Northern Ireland47 | Clinical entity in doubt1,48,49 | Rare | Rare | Rare | Relatively common in the island of Meleda. 1/100,000 in general populations50 | 1.2/10,000 in Japana, 3.1/10,000 in Chinaa | Rare | Rare |
| Age of onset | Within the first year of life | Within the first 2 years of life | Ages 8 to 10 | Within the first year of life | During childhood, not as early as during the first year of life | Early infancy | Mostly within the first year of life | ||
| Pathologic findings | Epidermolytic hyperkeratosis | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic | Nonepidermolytic |
| Hyperkeratosis | Thick | Thick | Thick | Thick | Mild to thick | Severe | Mild | Thick | Thick |
| Transgrediens | − | − | + | + | + | + | + | + (1 of 4) | + |
| Hyperhidrosis | − | − | + | Not described | + | + | + | Not described | Not described |
| Whitish change upon water exposure | − | − | − | − | + | − | + | − | − |
| Development on other areas | − | − | Elbows, knees, flexural areas, and Achilles tendon | Natal cleft, groin, elbows, knees, posterior aspects of forearms, and anterior aspects of legs | − | Knees and elbows, perioral erythema, and periorbital erythema | Knees, elbows, and Achilles tendon area | Only knuckle pads on the dorsa of the fingers | Knees, elbows, ankles, Achilles tendon area |
| Constricting bands | − | − | + | + | − | + | − | + | + |
| Spontaneous amputation | − | − | + | + | − | Occasionally | − | Not described | + |
| Flexion contractures | − | − | − | − | − | + | − | − | − |
Abbreviations: AD, autosomal dominant inheritance; AR, autosomal recessive inheritance.
This study.
Whole-exome sequencing and data analyses were performed as described previously.13 Whole-exome sequencing produced approximately 100,000,000 paired reads per sample, approximately 80% of which were mapped to the hs37d5 exon region of the human genome sequence assembly.14 The average coverage of the exonic region was 87.5×, with more than 93.2% of targeted bases covered at 10× reads. No SLURP1 mutation (Refseq: NM_020427.2) was identified in any of the three NPPK individuals. A genome informatics study found 693, 677, and 747 allelic variants in three NPPK individuals (KDex8, 14, and 20, respectively), showing a minor allele frequency of less than 1% in the 1092 individuals from the 1000 Genomes Project.14 Because NPPK is possibly inherited in an autosomal recessive manner,2,3 the causative mutation was expected to be a homozygous or compound heterozygous variant shared by the affected individuals but absent or found only in a heterozygous manner in the control cohort. Among the identified variants, only mutations in the SERPINB7 (MIM 603357) fulfilled these requirements, suggesting a causative role in NPPK (Table 1).
SERPINB7 consists of eight exons, with three distinct transcription start sites (exons 1a–c; Figure 2A). The start codon is located within exon 2, and the termination codon within exon 8 (Figure 2A). The SERPINB7 transcript (RefSeq: NM_001040147.2) encodes a 380 amino-acid protein. Mutations identified by whole-exome sequencing were confirmed by Sanger sequencing by using the primers in Table S1 (Figure 2B). A nonsense mutation encoding a c.796C>T alteration (p.Arg266∗) in the last exon of the SERPINB7 was found in all three NPPK individuals. KDex20 was homozygous for the c.796C>T nonsense mutation. KDex8 was a compound heterozygote of a maternal c.796C>T mutation and a paternal small indel mutation of c.218_219delAGinsTAAACTTTACCT (c.218_219del2ins12) at the end of exon 3, predicted to lead to a premature stop codon (p.Gln73Leufs∗17). KDex14 was a compound heterozygote of a maternal c.796C>T mutation and a paternal mutation of c.455-1G>A in the splice acceptor site upstream of exon 6 of SERPINB7, which was also predicted to lead to a premature stop codon (p.Gly152Valfs∗21) at chromosome 18: 61465837 in the hs37d5 human genome sequence.14
Figure 2.
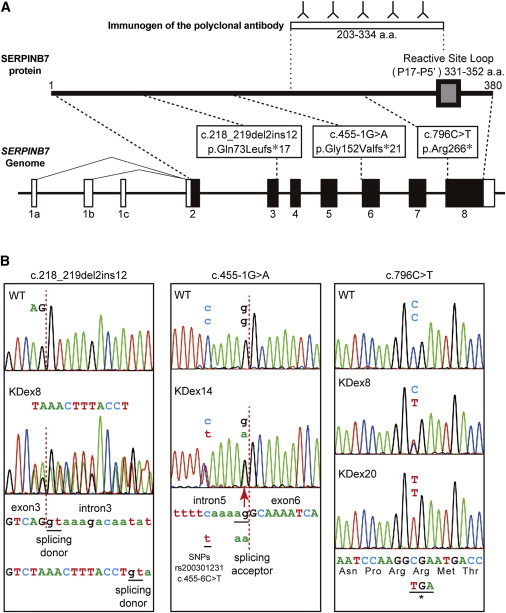
Genomic Organization of SERPINB7, Reactive Site Loop for Protease Inhibitory Activity of the SERPINB7 Protein, and Location of NPPK-Causing Mutations
(A) Schematic presentation of the genomic structure of SERPINB7 (lower) and its encoded protein (middle), SERPINB7. The gray box indicates the reactive site loop indispensable for protease inhibitory activity of SERPINB7. Open and filled boxes indicate exons of untranslated regions and coding regions, respectively. The positions of SERPINB7 mutations identified in this study are indicated. The immunogen of the anti-SERPINB7 polyclonal antibody is shown at the top.
(B) Heterozygous or homozygous mutated sequences of affected individuals (KDex8, KDex14, and KDex20) compared with the corresponding wild-type sequences. The base and amino acid sequences are shown. The intron-exon junctions are shown with red dotted lines. Intron and exon sequences are shown in lower case and upper case, respectively.
Abbreviations are as follows: c.218_219 del2ins12, c.218_219delAGinsTAAACTTTACCT.
To confirm mutations in SERPINB7 as a cause of NPPK, we analyzed ten additional unrelated NPPK individuals. The clinical manifestations of these individuals are presented in Table 1. Sanger sequencing failed to detect a mutation in SLURP1 in any of the ten individuals by using methods described previously.3 When the entire coding region of SERPINB7 was analyzed by Sanger sequencing with the primers in Table S1, five of the ten individuals were homozygous for the c.796C>T mutation, four were compound heterozygotes of the c.796C>T and c.218_219 del2ins12 mutations, and one was a compound heterozygote of the c.796C>T and c.455-1G>A mutations (Table 1). These results confirmed that mutations in SERPINB7 are a major cause of NPPK and that c.796C>T and c.218_219 del2ins12 are major mutations for NPPK in a Japanese population.
From our clinical experience, NPPK is much more common than other types of hereditary PPKs in Japan, although no statistical analysis has been reported. NPPK has not been recognized as a clinical entity within PPKs in Western populations, probably because it is rare. Next, we evaluated the variant databases of the cohort of 1,092 individuals in the 1000 Genomes Project14 to estimate the frequency of SERPINB7 mutations classified by ethnicity. The nonsense mutation of c.796C>T was identified as an SNP (rs142859678) with a minor allele frequency of 0.4% and found in a heterozygous manner in two of 89 Japanese individuals, four of 97 Han Chinese individuals from Beijing, and two of 100 Han Chinese individuals from southern China. On the other hand, the c.796C>T mutation was not found in any of 806 non-Asian individuals, suggesting that the c.796C>T mutation is a founder mutation causing NPPK in Asian populations. We also found another putative causative mutation, c.336+2T>G (an SNP of rs201433665), in one of 97 Han Chinese individuals from Beijing in a heterozygous manner. Other mutations found in this study were not identified in the 1,092 individuals.
From these results, the prevalence rate of NPPK was estimated as 1.2/10,000 in Japanese populations and 3.1/10,000 in Chinese populations. In contrast, no putative causative mutation (nonsense, missense, insertion, deletion, or exon-intron boundary mutation) was identified in 806 individuals of non-Asian origin in the 1000 Genomes Project.14 We further searched causative mutations in European-American and African-American populations by using the NHBLI Exome Variant Server and found only one putative causative mutation, c.309delT in the exon 4 (1 of 12,517 alleles), predicted to lead to a premature stop codon (p.Phe103Leufs∗33). Thus, the prevalence rate of NPPK in non-Asian populations was ∼0.5/100,000,000. These results well explain why NPPK is so common in PPKs in Japanese populations but has not been reported from non-Asian countries.
Serpins were originally identified as serine protease inhibitors. Serpin molecules are evolutionarily old because even bacteria and Archaea possess them.15–17 Most serpins identified to date possess protease inhibitory activity, although their protease targets are now known not to be restricted to serine proteases.16,17 Serpins form covalent complexes with target proteases to inhibit protease activity irreversibly. Human serpins have been divided into nine clades (A–I) by phylogenetic analyses.15,18 Several congenital diseases have been reported to be caused by deficiencies in the protease inhibitory function of serpins—for example, plasminogen activator inhibitor-1 deficiency (MIM 613329) with mutations in SERPINE1 (MIM 173360)19—or to be caused by polymerization and accumulation of mutated serpins, for example, familial encephalopathy with neuroserpin inclusion bodies (MIM 604218) with mutations in SERPINI1 (MIM 602445).20
SERPINB7 is located on chromosome 18q21.3, forming a cluster of clade-B serpin genes.18 Clade-B serpins are intracellular serpins, possibly protecting cells from exogenous and endogenous protease-mediated injury.18 The protease-inhibitory activity of serpins is dependent on the reactive site loop to form a covalent bond with target proteases.16 The center of the reactive site loop (P1–P1’) is located at amino acids 347–348 of SERPINB7,16 and the entire region of the reactive site loop (P17–P5′, corresponding to the amino acid region 331–352 of SERPINB7) is predicted to be absent in all of the mutant proteins (Figure 2A). Thus, all of the mutations identified in this study presumably result in a complete loss of the protease inhibitory activity of SERPINB7.
SERPINB7 was originally described as being expressed in kidney mesangial cells and was named MEGSIN.21 However, no renal manifestation has been identified in NPPK individuals. A recent report using a bacterial artificial chromosome transgene expressing Cre in mice under the control of Serpinb7 regulatory elements showed specific expression of Cre in cornified stratified epithelial cells, but not in kidney mesangial cells,22 suggesting that Serpinb7 might be specifically expressed in epidermal keratinocytes in mice. Thus, we next analyzed the expression of SERPINB7 in human skin. We used a commercial polyclonal antibody (HPA024200; Sigma-Aldrich) raised against a peptide corresponding to the amino acid 203–334 region of human SERPINB7 (Figure 2A). To characterize the antibody, we performed immunoblotting against GST-fused full-length human SERPINB7 and GST-fused p.Arg266∗ mutant that were produced in Escherichia coli BL21(DE3) by using the pGEX 5X-1 vector (GE Healthcare) in inclusion bodies and purified by washing with 1% Triton X-100 and 4 M urea. The purified proteins showed molecular weights of ∼62 kDa and ∼50 kDa in SDS-PAGE analysis, respectively (Figure 3A). In immunoblotting analysis, the anti-SERPINB7 antibody recognized both the GST-fused full-length SERPINB7 and GST-fused p.Arg266∗ mutant (Figure 3A). The immunosignals for the full-length SERPINB7 were stronger than those for the truncated p.Arg266∗ mutant, suggesting that this polyclonal antibody includes antibodies against peptides corresponding to both the amino acid 203–265 region and the 266–334 region of human SERPINB7 (Figure 2A).
Figure 3.
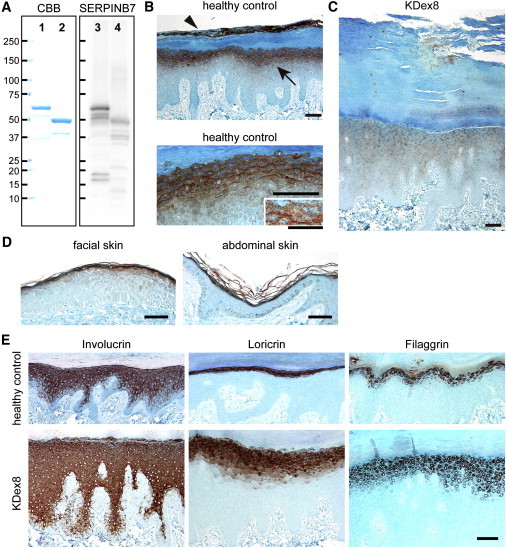
SERPINB7 Localization in the Epidermis and Immunohistochemical Analysis of the Affected Skin
(A) Investigation of the anti-SERPINB7 antibody. GST-fused recombinant full-length human SERPINB7 (lanes 1 and 3) and GST-fused recombinant p.Arg266∗ mutant (lanes 2 and 4) were analyzed by electrophoresis with Coomassie Brilliant Blue staining (lanes 1 and 2) or with immunoblotting with the anti-SERPINB7 rabbit polyclonal antibody (lanes 3 and 4). Scale bars indicate molecular weights (kDa).
(B) Immunohistochemistry of SERPINB7 in plantar skin of a healthy control. Upper panel shows SERPINB7 in the stratum granulosum (arrow) and in the upper part of the stratum corneum (arrowhead). Lower panel shows the intracellular distribution of SERPINB7 in the stratum granulosum. Scale bars represent 100 μm. Inset in the lower panel shows stratum granulosum cells and intercellular spaces at higher magnification (scale bar represents 50 μm).
(C) Immunohistochemistry of SERPINB7 in the hyperkeratotic plantar skin of NPPK individual of KDex8. Scale bar represents 100 μm.
(D) Immunohistochemistry of SERPINB7 in facial and abdominal skin sections of a healthy control. Scale bars represent 100 μm.
(E) Immunohistochemistry of epidermal differentiation-related proteins in plantar skin of a healthy control (upper panels) and a NPPK individual (KDex8; lower panels). Scale bar represents 100 μm.
Using this antibody, we performed an immunohistochemical analysis of paraffin wax-embedded sections of healthy human skin and NPPK skin, with antigen retrieval with 15 min boiling in a microwave oven in 100 mM Tris-HCl and 1 mM EDTA buffer (pH 9.0), immunosignal detection with ImmPRESS kit and NovaRed substrates (Vector Laboratories), and counterstaining with methyl green (Wako Pure Chemical). The immunosignals of the antibody were specifically detected from the stratum granulosum and from the upper part of the SC in healthy control plantar skin (Figure 3B). No signal was detected from the lower part of the SC, probably because the tightly packed intracorneocyte proteinaceous structure prevents access of the antibody to the antigen. When the stratum granulosum was observed at higher magnification, signals were observed in the cytoplasm, with a mild concentration to the apical side of the stratum granulosum cells (Figure 3B). In NPPK individuals, the immunosignals of the stratum granulosum and the SC were markedly diminished (KDex8, a compound heterozygote of the c.796C>T and c.218_219del2ins12 mutations; Figure 3C. For other affected individuals, data are not shown or skin biopsies were not performed). Thus, the immunosignals observed in healthy control plantar skin were considered to represent the distribution of SERPINB7. Some nuclear staining was observed in both the healthy control skin and the NPPK skin, which was considered to be nonspecific background (Figures 3B and 3C). Weak cytoplasmic immunosignals were observed in the NPPK skin, which were considered to be due to the p.Arg266∗ mutant of SERPINB7 or nonspecific background (Figure 3C).
To clarify whether SERPINB7 expression was limited to the palmoplantar area of the skin, we immunostained facial and abdominal skin sections of healthy controls. SERPINB7 immunosignals were specifically detected from the stratum granulosum and the SC in facial and abdominal epidermis (Figure 3D), suggesting that SERPINB7 is expressed in the epidermis of the whole body.
Next, we investigated whether loss of functional SERPINB7 affected epidermal differentiation by using NPPK skin. In NPPK plantar skin, hematoxylin and eosin staining showed acanthosis and orthohyperkeratosis (Figure 1C), as described previously.3 The localization of epidermal differentiation markers, loricrin, involucrin, and filaggrin, which were detected with anti-loricrin (ab24722; Abcam), anti-involucrin (clone SY5; Sigma Aldrich), and anti-filaggrin (clone FLG01; Thermo Scientific) antibodies, respectively, showed no major keratinocyte differentiation defect in NPPK skin (Figure 3E). Transmission electron microscopic studies of NPPK skin failed to show any major defect in the stratum granulosum or the SC (data not shown).
Loss of functional SERPINB7 might induce overactivation of target proteases in the stratum granulosum and the SC. Because no apparent change was observed in the stratum granulosum except for thickening, we reinvestigated the skin phenotype of NPPK, looking especially for any finding of changes in the SC. We found that the NPPK skin showed a whitish spongy appearance within 10 min of water exposure specifically in the reddish hyperkeratotic area (Figure 4A). The wrinkling of palms that is observed after water exposure in cystic fibrosis (MIM 219700)23,24 was not apparent, even after 30 min of water exposure (Figure 4A). These phenotypes suggested enhanced water permeation into the surface of the SC in NPPK lesional skin.
Figure 4.
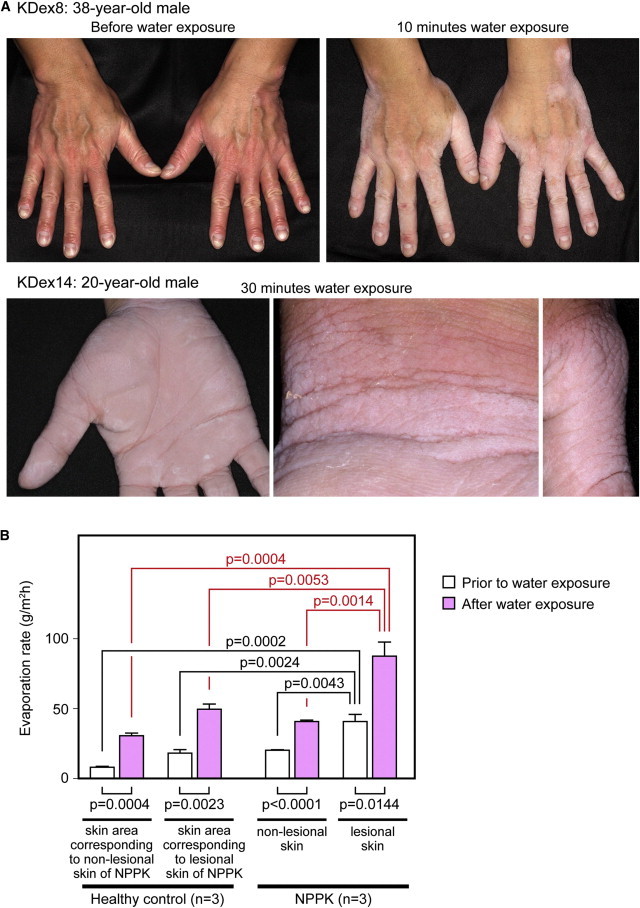
Changes upon Water Exposure in NPPK Lesional Skin
(A) Clinical phenotype of the hands of the proband (KDex8) prior to water exposure (upper left panel) and after 10 min water exposure (upper right panel), and the clinical phenotype of the hands of the proband (KDex14) after 30 min water exposure: the palm (lower left panel), inner wrist (lower middle panel), and dorsa of the thumb (lower right panel).
(B) Means of TEWL values prior to water exposure and after 30 min of water exposure in the lesional skin and nonlesional skin of NPPK individuals (n = 3; KDex8, KDex14, and KDex79) and in the corresponding skin area of healthy controls (n = 3). In each skin condition, the means of TEWL were compared upon water exposure (lower lines). The means of TEWL were compared between lesional and nonlesional skin of NPPK individuals and the corresponding skin area of healthy controls before water exposure (upper black lines) and after water exposure (upper red lines).
Thus, we next performed a transepidermal water loss (TEWL) analysis prior to and after water exposure in three NPPK individuals and three healthy controls. TEWL was measured at the lesional and nonlesional skin of dorsal hands and inner wrists in each NPPK individual and at the corresponding skin area in each healthy control with a Vapo Scan AS-VT100RS (Asahi Biomed) at room temperature (20°C–22°C) and 40%–60% humidity to avoid the effects of hyperhidrosis. The mean TEWL value was calculated from measurements of at least eight different points under each skin condition. Before water exposure, the mean TEWL values were higher in the lesional skin of NPPK individuals than in the nonlesional skin of NPPK individuals or the corresponding skin area of normal healthy controls (Figure 4B), when analyzed by using the Tukey-Kramer multiple-comparisons test with the Prism software (ver. 6; GraphPad Software). Next, the hands of NPPK individuals and healthy controls were immersed in water at 37°C for 30 min. After water exposure, TEWL values were significantly elevated in all skin conditions in all NPPK skin and in all healthy control skin (data not shown), and the mean TEWL values were significantly elevated on water exposure in any skin condition (Figure 4B) when analyzed with Student’s t test with the Prism software.
After water exposure, the mean TEWL values were higher in the lesional skin of NPPK individuals than in the nonlesional skin of NPPK individuals or the corresponding skin areas of healthy controls (Figure 4B) when analyzed with the Tukey–Kramer multiple-comparisons test. Because the TEWL instrument measures water evaporation from the skin surface, the TEWL values after water exposure might correspond mostly to water evaporation from water-swollen SC. Thus, these results suggest that water permeation into the SC is specifically facilitated in NPPK lesional skin.
Here, we identified that loss-of-function mutations in SERPINB7 cause NPPK and established NPPK genetically as a distinct clinical entity within hereditary diffuse PPKs without associated features. While SERPINB7 was considered to be expressed in the epidermis of the whole body, the affected skin area of NPPK is limited to hands, feet, knees, and elbows, the reason for which remains unknown. Such limitations in the affected skin area with a deficiency of gene products that are ubiquitously expressed in the epidermis have been observed in several other types of PPK: Vohwinkel syndrome (MIM 124500), caused by mutations in GJB2 (MIM 121011),25 and type I striate PPK (MIM 148700), caused by mutations in DSG1 (MIM 125670).26 The effects on the knees and elbows in NPPK suggest that chronic exposure to mechanical stress might have a role in the development of NPPK skin lesions, and the lesions in NPPK are limited to chronic mechanical stress-exposed areas of the skin. Thus, SERPINB7 might inhibit mechanical stress-induced proteases and protect keratinocytes or corneocytes from protease-mediated cellular damage.
Our findings suggest that NPPK is a genetic dermatosis caused by a deficiency of an intracellular protease inhibitor. Deficiencies of the protease inhibitors, LEKTI, encoded by SPINK5 (MIM 605010), and cystatin A, encoded by CSTA (MIM 184600), have been reported in Netherton syndrome (MIM 256500)27 and exfoliative ichthyosis (MIM 607936),28 respectively. In Netherton syndrome, overactivation of secreted extracellular proteases, kallikreins, has been suggested to induce overdesquamation via excessive degradation of cell adhesion molecules in the SC29 and skin inflammatory responses through thymic stromal lymphopoietin expression, mediated by unregulated activation of protease-activated receptor-2.30 In exfoliative ichthyosis, defects in desmosome-mediated cell-cell adhesion in the lower levels of the epidermis have been suggested to cause coarse peeling of skin on the palms and soles.28 However, the precise pathophysiology or protease overactivation induced by the loss of cystatin A has not yet been characterized.
As corneocytes lose the cell membrane on cornification, it is unclear whether SERPINB7 is held within corneocytes at the SC. But the phenotype of NPPK differs completely from that of Netherton syndrome because desquamation is rather prolonged in the erythematous hyperkeratotic area in NPPK, suggesting that the target proteases of SERPINB7 are unlikely to be associated with the desquamation process. Here, we observed a whitish spongy change in the SC on exposure to water in the lesional skin of NPPK. This change is caused by a loss of integrity in the SC structure, probably due to overactivation of target proteases of SERPINB7. Such a whitish change in the skin upon water exposure has been reported in an autosomal-dominant Bothnian-type PPK (MIM 600231) with mutations in AQP5 (MIM 600442),31–33 and in the aquagenic keratoderma associated with cystic fibrosis with mutations in CFTR (MIM 602421),23,24 but the pathophysiology of the whitish changes might differ among these diseases.
Together with the strong immunosignals of SERPINB7 in the SC, we propose that loss of functional SERPINB7 induces overactivation of intracorneocyte proteases specifically in the affected skin area, which induces degradation of the integrated proteinaceous structure of the corneocytes and facilitates water permeation into the SC. Additional functional assays and molecular biological analyses are required to investigate the changes in the water repellant properties of the SC surface in NPPK skin.
Various proteases are present in the stratum granulosum and the SC34–36. Additionally, the epidermis is attacked by various exogenous proteases—originating from bacteria, fungi, virus, pollen, and house dust mites—and endogenous proteases, originating from infiltrating cells.35 Appropriate control of the activity of these proteases by endogenous protease inhibitors is likely important in maintaining skin homeostasis. Our discovery of loss-of-function mutations in SERPINB7 in NPPK should provide insights into the functions and regulatory mechanisms of proteases and protease inhibitors in the epidermis. Future studies will aim to identify the target proteases of SERPINB7 in the steady state and in mechanically stressed states. It is also important to understand the pathophysiology of the putative protease overactivation in NPPK skin; that is, how the proteinaceous structure of the SC and integrity of the SC barrier are affected and whether the reddish hyperkeratosis and inflammatory cell infiltrations are secondary changes via augmented external stimuli through protease-mediated damage to the SC or direct effects of intraepidermal overactivation of proteases. The development of specific protease inhibitors mimicking SERPINB7 might allow pathogenesis-based therapies for NPPK.
Acknowledgments
We thank our clinical colleagues, and family members who contributed the samples used in this study. We also thank Nobuyo Nishimura, Hiromi Sakuragi, Asami Hirakiyama, Kazunari Shibata, and the Collaborative Research Resources of Keio University School of Medicine for technical support. This work was supported in parts by Health and Labour Sciences Research Grants for Research on Rare and Intractable Diseases, for Research on Allergic Disease and Immunology, and for Research on Measures for Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan, The Grant of National Center for Child Health and Development (24-5), and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This manuscript is dedicated to the memory of Professor Masaji Nagashima. Potential conflict of interest: M.A. has consultancy arrangements with Daiichi Sankyo Co. A.K., A.S., and T.S. have been supported by one or more grants from MSD K.K. and Maruho Co., Ltd. This work was supported in part by a grant from Maruho Co., Ltd.
Supplemental Data
Web Resources
The URLs for data presented here are as follows:
1000 Genomes, http://browser.1000genomes.org
NCBI dbSNPs, http://www.ncbi.nlm.nih.gov/projects/SNP/
NCBI RefSeq, http://www.ncbi.nlm.nih.gov/refseq/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Lucker G.P., Van de Kerkhof P.C., Steijlen P.M. The hereditary palmoplantar keratoses: an updated review and classification. Br. J. Dermatol. 1994;131:1–14. doi: 10.1111/j.1365-2133.1994.tb08450.x. [DOI] [PubMed] [Google Scholar]
- 2.Mitsuhashi Y., Hashimato I. Keratosis palmoplantaris Nagashima. Dermatologica. 1989;179:231. [Google Scholar]
- 3.Kabashima K., Sakabe J., Yamada Y., Tokura Y. “Nagashima-type” keratosis as a novel entity in the palmoplantar keratoderma category. Arch. Dermatol. 2008;144:375–379. doi: 10.1001/archderm.144.3.375. [DOI] [PubMed] [Google Scholar]
- 4.Mitsuhashi Y., Hashimoto I., Takahashi M. Meleda type keratosis palmoplantaris (Nagashima) Practical Dermatol. 1989;11:297–300. (in Japanese) [Google Scholar]
- 5.Nagashima M. Palmoplantar keratoses. In: Miura O., Ochiai K., editors. Volume 9. Igaku Shoin; Tokyo: 1977. pp. 23–27. (Handbook of Human Genetics). (in Japanese) [Google Scholar]
- 6.Isoda H., Kabashima K., Tokura Y. ‘Nagashima-type’ keratosis palmoplantaris in two siblings. J. Eur. Acad. Dermatol. Venereol. 2009;23:737–738. doi: 10.1111/j.1468-3083.2009.03206.x. [DOI] [PubMed] [Google Scholar]
- 7.Sakabe J., Kabashima K., Sugita K., Tokura Y. Possible involvement of T lymphocytes in the pathogenesis of Nagashima-type keratosis palmoplantaris. Clin. Exp. Dermatol. 2009;34:e282–e284. doi: 10.1111/j.1365-2230.2008.03202.x. [DOI] [PubMed] [Google Scholar]
- 8.Hovorka O., Ehlers E. Mal de Meleda. Arch. Dermatol. Res. 1897;40:251–256. [Google Scholar]
- 9.Gamborg Nielsen P. Two different clinical and genetic forms of hereditary palmoplantar keratoderma in the northernmost county of Sweden. Clin. Genet. 1985;28:361–366. doi: 10.1111/j.1399-0004.1985.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 10.Kastl I., Anton-Lamprecht I., Gamborg Nielsen P. Hereditary palmoplantar keratosis of the Gamborg Nielsen type. Clinical and ultrastructural characteristics of a new type of autosomal recessive palmoplantar keratosis. Arch. Dermatol. Res. 1990;282:363–370. doi: 10.1007/BF00372085. [DOI] [PubMed] [Google Scholar]
- 11.Nesbitt L.T., Jr., Rothschild H., Ichinose H., Stein W., 3rd, Levy L. Acral keratoderma. Arch. Dermatol. 1975;111:763–768. [PubMed] [Google Scholar]
- 12.Fischer J., Bouadjar B., Heilig R., Huber M., Lefèvre C., Jobard F., Macari F., Bakija-Konsuo A., Ait-Belkacem F., Weissenbach J. Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum. Mol. Genet. 2001;10:875–880. doi: 10.1093/hmg/10.8.875. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki T., Niizeki H., Shimizu A., Shiohama A., Hirakiyama A., Okuyama T., Seki A., Kabashima K., Otsuka A., Ishiko A. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J. Dermatol. Sci. 2012;68:36–44. doi: 10.1016/j.jdermsci.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 14.The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irving J.A., Pike R.N., Lesk A.M., Whisstock J.C. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10:1845–1864. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 16.Gettins P.G.W. Serpin structure, mechanism, and function. Chem. Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 17.Law R.H.P., Zhang Q., McGowan S., Buckle A.M., Silverman G.A., Wong W., Rosado C.J., Langendorf C.G., Pike R.N., Bird P.I., Whisstock J.C. An overview of the serpin superfamily. Genome Biol. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silverman G.A., Whisstock J.C., Askew D.J., Pak S.C., Luke C.J., Cataltepe S., Irving J.A., Bird P.I. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 2004;61:301–325. doi: 10.1007/s00018-003-3240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fay W.P., Shapiro A.D., Shih J.L., Schleef R.R., Ginsburg D. Brief report: complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. N. Engl. J. Med. 1992;327:1729–1733. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 20.Davis R.L., Shrimpton A.E., Carrell R.W., Lomas D.A., Gerhard L., Baumann B., Lawrence D.A., Yepes M., Kim T.S., Ghetti B. Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet. 2002;359:2242–2247. doi: 10.1016/S0140-6736(02)09293-0. [DOI] [PubMed] [Google Scholar]
- 21.Miyata T., Nangaku M., Suzuki D., Inagi R., Uragami K., Sakai H., Okubo K., Kurokawa K. A mesangium-predominant gene, megsin, is a new serpin upregulated in IgA nephropathy. J. Clin. Invest. 1998;102:828–836. doi: 10.1172/JCI2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Guo Q., Casey A., Lin C., Chen F. A new tool for conditional gene manipulation in a subset of keratin-expressing epithelia. Genesis. 2012;50:899–907. doi: 10.1002/dvg.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garçon-Michel N., Roguedas-Contios A.-M., Rault G., Le Bihan J., Ramel S., Revert K., Dirou A., Misery L. Frequency of aquagenic palmoplantar keratoderma in cystic fibrosis: a new sign of cystic fibrosis? Br. J. Dermatol. 2010;163:162–166. doi: 10.1111/j.1365-2133.2010.09764.x. [DOI] [PubMed] [Google Scholar]
- 24.Weibel L., Spinas R. Images in clinical medicine. Aquagenic wrinkling of palms in cystic fibrosis. N. Engl. J. Med. 2012;366:e32. doi: 10.1056/NEJMicm1103833. [DOI] [PubMed] [Google Scholar]
- 25.Maestrini E., Korge B.P., Ocaña-Sierra J., Calzolari E., Cambiaghi S., Scudder P.M., Hovnanian A., Monaco A.P., Munro C.S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- 26.Rickman L., Simrak D., Stevens H.P., Hunt D.M., King I.A., Bryant S.P., Eady R.A., Leigh I.M., Arnemann J., Magee A.I. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum. Mol. Genet. 1999;8:971–976. doi: 10.1093/hmg/8.6.971. [DOI] [PubMed] [Google Scholar]
- 27.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafé J.L., Wilkinson J., Taïeb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 28.Blaydon D.C., Nitoiu D., Eckl K.-M., Cabral R.M., Bland P., Hausser I., van Heel D.A., Rajpopat S., Fischer J., Oji V. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am. J. Hum. Genet. 2011;89:564–571. doi: 10.1016/j.ajhg.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Descargues P., Deraison C., Bonnart C., Kreft M., Kishibe M., Ishida-Yamamoto A., Elias P., Barrandon Y., Zambruno G., Sonnenberg A., Hovnanian A. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 2005;37:56–65. doi: 10.1038/ng1493. [DOI] [PubMed] [Google Scholar]
- 30.Briot A., Deraison C., Lacroix M., Bonnart C., Robin A., Besson C., Dubus P., Hovnanian A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009;206:1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lind L., Lundström A., Hofer P.A., Holmgren G. The gene for diffuse palmoplantar keratoderma of the type found in northern Sweden is localized to chromosome 12q11-q13. Hum. Mol. Genet. 1994;3:1789–1793. doi: 10.1093/hmg/3.10.1789. [DOI] [PubMed] [Google Scholar]
- 32.Blaydon D.C., Lind L.K., Plagnol V., Linton K.J., Smith F.J.D., Wilson N.J., McLean W.H.I., Munro C.S., South A.P., Leigh I.M. Mutations in AQP5, Encoding a Water-Channel Protein, Cause Autosomal-Dominant Diffuse Nonepidermolytic Palmoplantar Keratoderma. Am. J. Hum. Genet. 2013;93:330–335. doi: 10.1016/j.ajhg.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao X., Yin J., Wang H., Zhao J., Zhang J., Dai L., Zhang J., Jiang H., Lin Z., Yang Y. Mutation in AQP5, Encoding Aquaporin 5, Causes Palmoplantar Keratoderma Bothnia Type. J. Invest. Dermatol. 2013 doi: 10.1038/jid.2013.302. in press. [DOI] [PubMed] [Google Scholar]
- 34.Kamata Y., Taniguchi A., Yamamoto M., Nomura J., Ishihara K., Takahara H., Hibino T., Takeda A. Neutral cysteine protease bleomycin hydrolase is essential for the breakdown of deiminated filaggrin into amino acids. J. Biol. Chem. 2009;284:12829–12836. doi: 10.1074/jbc.M807908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer-Hoffert U. Reddish, scaly, and itchy: how proteases and their inhibitors contribute to inflammatory skin diseases. Arch. Immunol. Ther. Exp. (Warsz.) 2009;57:345–354. doi: 10.1007/s00005-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 36.Matsui T., Miyamoto K., Kubo A., Kawasaki H., Ebihara T., Hata K., Tanahashi S., Ichinose S., Imoto I., Inazawa J. SASPase regulates stratum corneum hydration through profilaggrin-to-filaggrin processing. EMBO Mol Med. 2011;3:320–333. doi: 10.1002/emmm.201100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vörner H. Zur Kenntnis des Keratoma hereditarium palmare et plantare. Arch. Dermatol. Syph. 1901;56:3–31. [Google Scholar]
- 38.Thost A. (1880). Über erbliche ichthyosis palmaris et plantaris cornea. (Med. Diss). Horning J., ed. Heidelberg.
- 39.Unna P. Über das Keratoma palmare et plantare hereditarium, eine Studie zur Kerato-Nosologie. Arch. Dermatol. Syph. 1883;15:231–270. [Google Scholar]
- 40.Greither A. Keratosis extremitatum hereditaria progrediens mit dominantem Erbgang. Hautarzt. 1952;3:198–203. [PubMed] [Google Scholar]
- 41.Sybert V.P., Dale B.A., Holbrook K.A. Palmar-plantar keratoderma. A clinical, ultrastructural, and biochemical study. J. Am. Acad. Dermatol. 1988;18:75–86. [PubMed] [Google Scholar]
- 42.Hatsell S.J., Eady R.A., Wennerstrand L., Dopping-Hepenstal P., Leigh I.M., Munro C., Kelsell D.P. Novel splice site mutation in keratin 1 underlies mild epidermolytic palmoplantar keratoderma in three kindreds. J. Invest. Dermatol. 2001;116:606–609. doi: 10.1046/j.1523-1747.2001.13041234.x. [DOI] [PubMed] [Google Scholar]
- 43.Reis A., Hennies H.C., Langbein L., Digweed M., Mischke D., Drechsler M., Schröck E., Royer-Pokora B., Franke W.W., Sperling K. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (EPPK) Nat. Genet. 1994;6:174–179. doi: 10.1038/ng0294-174. [DOI] [PubMed] [Google Scholar]
- 44.Küster W., Reis A., Hennies H.C. Epidermolytic palmoplantar keratoderma of Vörner: re-evaluation of Vörner’s original family and identification of a novel keratin 9 mutation. Arch. Dermatol. Res. 2002;294:268–272. doi: 10.1007/s00403-002-0328-9. [DOI] [PubMed] [Google Scholar]
- 45.Kimonis V., DiGiovanna J.J., Yang J.M., Doyle S.Z., Bale S.J., Compton J.G. A mutation in the V1 end domain of keratin 1 in non-epidermolytic palmar-plantar keratoderma. J. Invest. Dermatol. 1994;103:764–769. doi: 10.1111/1523-1747.ep12412771. [DOI] [PubMed] [Google Scholar]
- 46.Gach J.E., Munro C.S., Lane E.B., Wilson N.J., Moss C. Two families with Greither’s syndrome caused by a keratin 1 mutation. J. Am. Acad. Dermatol. 2005;53(Suppl 1):S225–S230. doi: 10.1016/j.jaad.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 47.Covello S.P., Irvine A.D., McKenna K.E., Munro C.S., Nevin N.C., Smith F.J., Uitto J., McLean W.H. Mutations in keratin K9 in kindreds with epidermolytic palmoplantar keratoderma and epidemiology in Northern Ireland. J. Invest. Dermatol. 1998;111:1207–1209. doi: 10.1046/j.1523-1747.1998.00445.x. [DOI] [PubMed] [Google Scholar]
- 48.Hamm H., Happle R., Butterfass T., Traupe H. Epidermolytic palmoplantar keratoderma of Vörner: is it the most frequent type of hereditary palmoplantar keratoderma? Dermatologica. 1988;177:138–145. doi: 10.1159/000248531. [DOI] [PubMed] [Google Scholar]
- 49.Küster W., Becker A. Indication for the identity of palmoplantar keratoderma type Unna-Thost with type Vörner. Thost’s family revisited 110 years later. Acta Derm. Venereol. 1992;72:120–122. [PubMed] [Google Scholar]
- 50.Bouadjar B., Benmazouzia S., Prud’homme J.F., Cure S., Fischer J. Clinical and genetic studies of 3 large, consanguineous, Algerian families with Mal de Meleda. Arch. Dermatol. 2000;136:1247–1252. doi: 10.1001/archderm.136.10.1247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.