

Abstract
Copy-number variants (CNVs) represent a significant interpretative challenge, given that each CNV typically affects the dosage of multiple genes. Here we report on five individuals with coloboma, microcephaly, developmental delay, short stature, and craniofacial, cardiac, and renal defects who harbor overlapping microdeletions on 8q24.3. Fine mapping localized a commonly deleted 78 kb region that contains three genes: SCRIB, NRBP2, and PUF60. In vivo dissection of the CNV showed discrete contributions of the planar cell polarity effector SCRIB and the splicing factor PUF60 to the syndromic phenotype, and the combinatorial suppression of both genes exacerbated some, but not all, phenotypic components. Consistent with these findings, we identified an individual with microcephaly, short stature, intellectual disability, and heart defects with a de novo c.505C>T variant leading to a p.His169Tyr change in PUF60. Functional testing of this allele in vivo and in vitro showed that the mutation perturbs the relative dosage of two PUF60 isoforms and, subsequently, the splicing efficiency of downstream PUF60 targets. These data inform the functions of two genes not associated previously with human genetic disease and demonstrate how CNVs can exhibit complex genetic architecture, with the phenotype being the amalgam of both discrete dosage dysfunction of single transcripts and also of binary genetic interactions.
Introduction
Copy-number variants (CNV) are a potential source both of genetic diversity1,2 and of deleterious lesions that can drive both rare and common human disorders.3 The now-common use of array-comparative genomic hybridization (aCGH) has increased the discovery of CNVs and has highlighted the fact that the mechanisms that lead to genomic rearrangements are, collectively, ∼1,000 times more prevalent per meiosis than the point mutation rate.4 In the last decade, CNVs have been associated with a variety of human phenotypes, including respiratory, cardiac, renal, and craniofacial defects.5–11 Notably, CNVs are found commonly to drive neurodevelopmental and cognitive disorders.5,12–19
CNVs typically affect the dosage balance of multiple transcripts, rendering the determination of the contribution of individual genes within each CNV to the clinical phenotype challenging. Previous studies of recurrent, common CNVs that used a variety of genetic tools and model organisms have made progress in identifying specific transcripts whose dosage imbalance can explain CNV-associated phenotypes or discrete endophenotypes.20–25 By contrast, nonrecurrent CNVs are significantly more challenging to dissect owing to the presence of variable boundaries and thus a dearth of individuals with the same lesion.
Genomic rearrangements of the distal bands of the long arm of chromosome 8 have been associated previously with several clinical entities; e.g., 8q23.3 deletions in colorectal cancer (MIM 114500)26 and Lynch syndrome I (MIM 120435),27 8q23.3–q24.1 deletions in myoclonic epilepsy (FAME [MIM 601068]),28 8q23.2–q24.12 deletions in microcephaly-thin corpus callosum syndrome,29 and 8q23.3–q24.13 deletions in tricho-rhino-phalangeal syndrome type I (Langer-Giedion syndrome, TRPS1 [MIM 190350]).30 Interstitial 8q23–q24 duplications have also been reported, in which the affected children have craniofacial features, visual anomalies, and developmental delay.31–33 Deletions are equally as rare as duplications; 8q23–q24 deletions have been described in individuals with Langer-Giedion syndrome and were often transmitted from a mosaic mother.31,34–36 Further, two individuals with coloboma, intellectual disability, cardiac defects, and craniofacial features were also reported to harbor an 8.5 Mb nonmosaic deletion that includes the Langer-Giedion syndrome region.37 Similar to other CNVs, transitioning from detection of these events to identifying their underlying genetic drivers is hampered by the paucity of individuals with recurrent events and by the variability in phenotypic expressivity. In the present study, through the detailed analysis of a cohort with overlapping CNVs, we narrowed the minimal critical region to a small segment of the genome containing three genes. Systematic functional evaluation of all three transcripts allowed us to identify discrete contributors to the clinical phenotype and to establish intricate genetic interactions between two of the transcripts to modulate the expressivity of specific endophenotypes. Further, we identified an individual with a de novo mutation in one of the two genes and, once again through the combinatorial use of clinical assessment and functional studies in vitro and in vivo, were able to show that the pathology of this individual is consistent with the predicted contribution of one of the transcripts to the complex 8q CNV phenotype.
Subjects and Methods
Subjects’ Ascertainment
Human genetic studies were approved by the institutional review board of Boston Children’s Hospital and Centre Hospitalier Universitaire Vaudois, Lausanne. Written informed consent was obtained from the subjects or their legal guardians for exome sequencing, RNA studies, and use of photographs.
Knockdown, Rescue Experiments, Alcian Blue Staining, Filtration Assay, and Embryo Manipulations
The splice-blocking morpholinos against scrib and puf60a were designed and obtained from Gene Tools: scrib MO (5′-GTGGCACAAAAGTTTGCATACCATT-3′) targeting the second exon-intron junction and puf60a MO (5′-TGCAGCAGCCGCCCCTCACCTATAA-3′) targeting the fifth exon-intron junction. Control morpholino was the scrambled nucleotide sequence from Gene Tools (5′-CCTCTTACCTCAGTTACAATTTATA-3′). We injected 0.5 nl of diluted morpholino (4 and 4.5 ng for scrib and puf60a MO, respectively) and/or RNA (100 pg) into wild-type zebrafish embryos at the 1- to 2-cell stage. Injected embryos were scored and classified into two groups, normal and mutant, on the basis of the phenotype compared with age-matched controls from the same clutch. For RNA rescue and overexpression experiments, the human wild-type mRNAs were cloned into the pCS2 vector and transcribed in vitro with the SP6 Message Machine kit (Ambion). All the experiments were repeated three times and we ran a t test or a Pearson’s chi-square test (χ2) to determine the significant differences of the morphant phenotype. Suppression of endogenous message was shown by PCR amplification of cDNA reverse transcribed from extracted total mRNA (puf60a and scrib primers are listed in Table S1 available online). Alcian blue staining of cartilaginous structures was performed for investigating the morphology of the head. Zebrafish embryos were fixed with 4% paraformaldehyde (PFA), and the cartilage structures were visualized by Alcian blue staining as previously described.38 The glomerular filtration assay was performed as follows: at 2 days postfertilization (dpf), FITC-labeled 50 kDa dextran (Molecular Probes) was injected into the cardiac venous sinus. For this injection, zebrafish were anesthetized in a 1:20 to 1:100 dilution of 4 mg/ml Tricaine (MESAB: ethyl-m-aminobenzoate methanesulfonate, 1% Na2HPO4 [pH 7.0]) (Sigma-Aldrich) and positioned ventral side up in a 1% agarose injection mold. After the injection, fish were returned to egg water for recovery.
Plasmids
To make pTT3-PUF60L-His169Tyr, the plasmid with the PUF60 His169Tyr mutation, pTT3-HisPUFL, was used as a template in a QuickChange Lightning Site-directed mutagenesis kit (Agilent) with primers PUF60-His169Tyr-F and PUF60-His169Tyr-R (Table S1). The plasmid, pTT3-PUF60Lrm, and pTT3-PUF60Lrm-His169Tyr were engineered with pTT3-PUF60L or pTT3-PUF60L-His169Tyr as a template in overlap-extension PCR reactions identical to the method used to make pMarx-PUF60rm.39
Cell Culture
HeLa cells were transfected with pTT3 empty vector, pTT3-PUF60Lrm, or pTT3-PUF60Lrm-His169Tyr and 20 nM of siRNA via Lipofectamine 2000 (Invitrogen). The siRNAs used were PUF60 r(GCAGAUGAACUCGGUGAUG)dTdT (sense strand, Dharmacon) and Allstars negative control siRNA (QIAGEN; cat. #1027281). After 72 hr cells were harvested for RNA by TRIzol reagent (Life Technologies) and protein by Laemmli buffer.
RT-PCR
RNA from affected individuals was collected with PAXgene tubes from peripheral blood. RNA from cell culture experiments was collected with TRIzol reagent (Invitrogen). Reverse transcription was performed with GoScript (Promega) with oligo dT primers. PCR with GoTaq polymerase (Promega) was carried out in reactions containing α-[32P]dCTP. Primers sequences are provided in Table S1. Products were separated on 6% native polyacrylamide gels. Quantitation is based on phosphorimage analysis (Typhoon 9400; GE Healthcare).
Immunoblot
Protein samples were separated by SDS-PAGE and transferred to Immobilon-FL membrane (Millipore). Blots were probed with mouse monoclonal antibodies specific for PUF60 (generated by M.L.H.) or β-actin (Sigma), followed by Alexa Fluor 594-conjugated antimouse secondary antibody (Invitrogen) or horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody. Detection and quantitation of the signal was performed with a Typhoon 9400 Variable Mode Imager (GE Healthcare) and ImageQuant T software for fluorescently detected blots or with Lumi-Light Western Blotting Substrate (Roche Diagnostics) for HRP-labeled blots.
Exome Sequencing
Whole-exome sequencing of subject 6 and her parents was performed at the Broad Institute. Hybrid selection was performed with Agilent’s SureSelect Human All Exon Kit v.2 (Agilent Technologies). We sequenced the samples with the Illumina HiSeq 2000 platform (Illumina) and aligned the resulting reads to the UCSC Genome Browser (hg19) reference genome with Burrows-Wheeler Aligner. We then applied the Genome Analysis Toolkit, base quality score recalibration, and indel realignment and performed single-nucleotide polymorphism and indel discovery and genotyping across all samples simultaneously by using variant quality score recalibration. Variants were annotated for functional effect by SnpEff 2.0.5. Variants were filtered as previously described.40
Results
Clinical Assessment and Fine Mapping
We studied five individuals (individuals 1–5, Figure 1, Table 1) with a de novo interstitial 8q24.3 deletion ranging from 78 kb to 1 Mb, all of which contained the previously reported deletion.37
Figure 1.
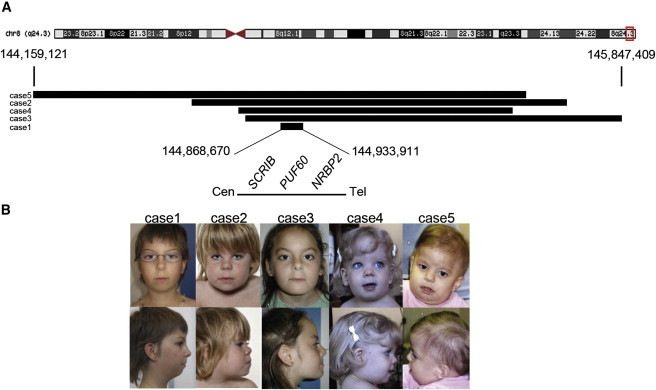
Positions of the 8q24.3 Deletions and the Corresponding Photographs of Affected Individuals
(A) Schematic representation of chromosome 8 showing the breakpoints of the 8q24.3 deletions. The minimal deletion (individual 1) includes SCRIB, PUF60, and NRBP2.
(B) Photographs of individuals 1–5 capture the core facial gestalt, including microcephaly, bitemporal narrowing, wide nasal bridge, anteverted nares, long and flat philtrum, and thin upper lip.
Table 1.
Phenotypes of the Five Individuals with 8q24.3 Deletions
| Clinical Features | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | Verheij, 1 |
|---|---|---|---|---|---|---|---|
| Coloboma | + | + | no | no | no | no | + |
| Renal | + | no | + | + | + | no | no |
| Low birth weight | + | no | + | + | no | + | + |
| Growth retardation | + | + | + | + | + | + | + |
| Microcephaly | + | + | no | no | + | + | no |
| Vertebral anomalya | + | no | + | no | + | + | no |
| Developmental delay | no | + | + | + | + | + | + |
| Hand anomaliesb | no | no | no | + | + | + | + |
| Feeding problems | no | no | + | + | + | no | + |
| Joint laxity and/or dislocationc | no | + | + | + | + | + | + |
| Long philtrum | + | + | + | + | + | + | + |
| Micro/retrognatism | + | no | no | no | no | + | + |
| Cardiac defects | no | no | + | no | + | + | + |
| Other signs | conductive hearing loss | short neck, hiatus hernia | high palate | none | short neck, cranial asymmetry, seizures | bronchopulmonary dysplasia, cleft palate, seizures | plagiocephaly, high palate, branchial arch remnant+fistula, hypoplastic corpus callosum |
| Size of deletion (MB) | 0.078 | 1 | 1 | 0.69 | 0.55 | p.His169Tyr | 15.1 |
| Break points proxd | 144,868,670 | 144,614,025 | 144,768,060 | 144,747,447 | 144,159,121 | none | 130,370,818 |
| Break points distd | 144,933,911 | 145,690,359 | 145,847,409 | 145,535,208 | 145,573,790 | none | 145,569,192 |
Sacral dysplasia, coccyx agenesis, fusion of L5-S1, hemivertebrae.
Short fifth fingers, clinodactyly, ectrodactyly (right thumb), postaxial polydactyly.
Hip dislocation unilateral/bilateral, left elbow subluxation.
UCAC Genome Browser hg19 coordinates.
Individual 1
This 10-year-old girl was the second child of healthy nonconsanguineous parents. The family history revealed no stillbirths, intellectual disability, or birth defects. Abdominal ultrasound showed a left renal agenesis. On physical examination at the age of 10 years, growth retardation and microcephaly were noted (weight: 22 kg, <3rd percentile; height: 125 cm, <3rd percentile; head circumference: 49 cm, <3rd percentile). Facial features included bilateral iris coloboma noted at birth, retrognatism, bilateral preauricular pits, long philtrum, thin upper lip, anteverted nares, and coarse hair (Figure 1). The association of middle ear malformations and renal agenesis prompted the molecular analysis of EYA1, which showed an intronic variant (c.1698−3C>T) that was considered nonpathogenic. Analysis of PAX2 was also performed because of the association of renal agenesis and bilateral coloboma and was normal. The karyotype was normal (46,XX). Array CGH analysis (Agilent 244 K) revealed a deletion of 65 to 78 kb on chromosome 8q24.3 (chr8: 144,868,670–144,933,911) (UCSC Genome Browser hg19). This deletion was absent in both parents.
Individual 2
This 6-year-old boy was the second child of nonconsanguineous parents. The family history revealed no stillbirths, intellectual disability, or birth defects. His growth was regular but below the 3rd percentile. He showed a moderate motor and speech delay. He had recurrent pneumonia, upper respiratory tract infections, and febrile seizures. He also had apnoeic episodes possibly resulting from esophageal reflux associated with a small hiatus hernia. At birth, bilateral coloboma of the iris, choroid, and retina without involvement of the optic nerves was noted. The vision was normal. The CT scan of the brain at 9 months and the MRI of the brain at 19 months showed slight temporal and frontal cerebral atrophy. On physical examination at the age of 2 years, growth retardation was noted (weight, –1 SD; height, −2 SD; head circumference, −1 SD). Facial features included bitemporal narrowing, long eyelashes, a flame nevus of the eyelids, short nose, anteverted nares, long philtrum, thin upper lip, full cheeks, and short neck with loose skin (Figure 1). He also had a single palmar crease and proximal placement of thumbs. Karyotype was normal (46,XY). Array CGH analysis (Agilent 244 K) revealed a 0.98 Mb (minimal size) deletion on chromosome 8q24.3 (chr8: 144,614,025–145,690,359) (UCSC Genome Browser hg19). The deletion was confirmed by real-time PCR and investigations of parental samples showed that the deletion was de novo.
Individual 3
This 17-year-old girl was the second child of nonconsanguineous parents. The family history revealed no stillbirths, intellectual disability, or birth defects. A truncus arteriosus type I associated with ventricular septal defect was diagnosed at birth. Her growth was regular but below the 3rd percentile. She smiled at 3 months, sat at 14 months, and walked at 28 months. She started to speak at 8 years of age and was still not reading nor writing at the age of 15 years. She had left polycystic kidney, fusion of L5-S1, and left elbow subluxation with malformation of the great sigmoid cavity. The brain MRI at the age of 15 years was normal. At 15 years of age, height was at −3.5 SD, BMI at +2.5 SD, and head circumference at 0 SD. Facial features included a prominent forehead, slight synophris, high and narrow palate, slight prognatism, bulbous nose tip, anteverted nares, and thin lips (Figure 1). Karyotype was normal (46,XX). Array CGH analysis (Agilent 105 K) revealed a 1 Mb deletion on chromosome 8q24.3 (chr8: 144,768,060–145,847,409) (UCSC Genome Browser hg19). Investigations by FISH of parental samples showed that the deletion was de novo.
Individual 4
She was the only child of nonconsanguineous and healthy parents. The family history revealed no stillbirths, intellectual disability, or birth defects. Her growth was regular but below the 3rd percentile. She sat at 6–7 months and walked at 18 months. She had fewer than 10 words at 20 months and started to speak in sentences at 36 months. Investigations for short stature showed a normal growth hormone stimulation test, normal thyroid hormone status, and a delayed bone age (at the chronological age of 9 years and 1 month, the bone age was 6 years and 10 months). She had feeding problems. At the age of 4 months she was found to have ectopic fused kidneys after investigations for urinary tract infection. The MRI of the brain at the age of 2 years and 10 months showed small areas of encephalomalacia over the anterior part of the left cerebellar hemisphere and inferior vermis originating from an old ischemic event within the area served by the posterior inferior cerebellar artery. On physical examination at the age of 20 months, growth retardation was noted (weight, −3.5 SD; height, 2nd percentile; head circumference, 50th percentile). Facial features included bitemporal narrowing, tall and narrow forehead, wide nasal bridge, anteverted nares, long philtrum, thin upper lip, wide palpebral fissure, bilateral preauricular pits, and a capillary hemangioma in the middle of her forehead (Figure 1). Karyotype was normal (46,XX). Array CGH analysis (GenomeDx microarray v.3.1) of the affected individual and parents revealed a de novo 688 kb deletion on chromosome 8q24.3 (chr8: 144,747,447–145,535,208) (UCSC Genome Browser hg19).
Individual 5
This 11-year-old girl was the second child of healthy nonconsanguineous parents. The family history revealed no stillbirths, intellectual disability, or birth defects. Dysmorphic features were noted at birth: bitemporal narrowing with trigonocephaly, bilateral epicanthal folds, broad nasal root, depressed nasal tip, long philtrum, short neck with excess neck skin posteriorly, clinodactyly, bilateral transverse palmar creases, sacral dimple, and bilateral dislocated hips. Between 1 and 2 months, she experienced respiratory distress. Hemivertebrae were diagnosed on chest X-ray and ventricular septal defect associated with aorta coarctation on echocardiography. She suffered reflux and tracheomalacia until 7 months old and received nasogastric feeding for 6 months. She suffers from severe cyclic vomiting since the age of 3. At 1 year, she was diagnosed with right kidney hypoplasia, solitary renal cyst in this kidney, scoliosis, and joint hypermobility. She showed moderate motor and speech delay. She was diagnosed with hyperopia at the age of 3 and seizures at the age of 9. The MRI of the brain at the age of 6 and 7 years noted volume loss in the left temporal lobe. At 10 years of age, her growth remained regular below the 3rd percentile for all parameters (microcephaly). Facial features included cranial asymmetry with right occipital flattening, broad nasal root, long and flat philtrum, thin upper lip, short broad neck with low posterior hairline, bilateral epicanthal folds, bilateral transverse palmar creases, and shortening of the 5th digit with 5th finger clinodactyly (Figure 1). Karyotype was normal (46,XX). Array CGH analysis (GenomeDx microarray v.3.1) revealed a 0.55 Mb (minimal size) deletion on chromosome 8q24.3 (chr8:144,159,121–145,573,790) (UCSC Genome Browser hg19). This deletion was not present in either parent by FISH analysis.
The minimal common deletion (Figure 1; Table 1) encompassed three genes: SCRIB (scribbled homolog [MIM 607733]), NRBP2 (nuclear receptor binding protein 2), and PUF60 (poly-U binding splicing factor 60 kDa [MIM 604819]), raising the possibility that haploinsufficiency of some or all these three genes might give rise to the observed phenotypes.
In Vivo Testing of Minimal CNV Genes
Manipulation of zebrafish has enhanced our understanding of the function of point mutations in human genetic disorders38,41–46 (also see review47). More recently, this model has also been used to investigate human dosage-sensitive genes involved in CNV-induced phenotypes, wherein suppression and/or overexpression of each gene within the CNV, followed by binary epistatic tests, can facilitate the dissection of CNV-associated defects.20,48 We therefore asked whether we could examine systematically the potential contribution of the three genes within the minimal 8q24.3 CNV for anatomic traits by assaying the organ systems relevant to the human pathology with objective measurements. We first queried the zebrafish genome for each of the three genes; we identified a single ortholog each for SCRIB and PUF60 (60% identity for scrib [NM_001007175.1] and 82% identity for puf60a [NM_001105588.1]) and two orthologs for NRBP2 (62% and 61% identity for nrbp2, [XP_001920672.2] and NRBP2 (2 of 2) [XP_005163579.1], respectively). Next, we generated splice-blocking morpholinos (MOs) against each of scrib, nrbp2 (both copies), and puf60 and injected embryos at the two-cell stage, followed by phenotyping of key anatomical hallmarks of the CNV. Suppression of either copy of nrbp2 did not give any appreciable phenotypes, whereas a concomitant double suppression led to either phenotypically normal embryos at low MO doses or global delay and embryonic lethality with no organ-specific phenotypes at higher doses (n = 80 embryos, 3 independent experiments, scoring at 3 dpf). As such, although a definitive statement about this gene cannot be made with regard to its contribution to the phenotypes of the individuals studied here, we consider this transcript unlikely to be a primary driver of the discrete pathology of this CNV.
In contrast, injection of 4 ng of scrib MO or 4.5 ng of puf60 MO recapitulated multiple 8q24.3 CNV-associated phenotypes. Injection of MO followed by RT-PCR testing of whole embryo lysates showed the inclusion of intronic sequences for both scrib and puf60a messages at 4.5 dpf, thus confirming the efficiency of the splice-blocking MO (Figure S2). First, given that short stature has been observed in our individuals with 8q24.3 deletions (Table 1), we measured the body length of 3 dpf embryos injected with either scrib or puf60 MOs. We observed a modest but significant and reproducible reduction of body length of each of scrib and puf60 morphants compared to sham-injected embryos (5% reduction, p = 0.003; and 12% reduction, p < 0.0001, respectively) (Figure 2A).
Figure 2.
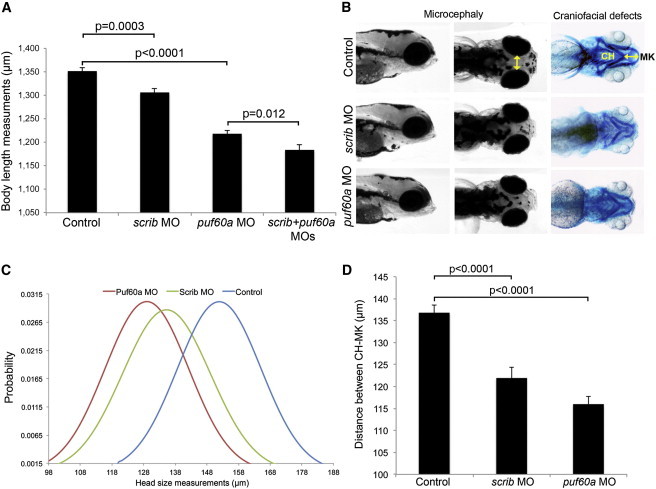
Suppression of scrib and puf60 in Zebrafish Leads to Short Stature, Small Head Size, and Craniofacial Defects
(A) Quantification of total body length was performed in embryo batches injected with sham (control), scrib MO, puf60a MO, and double MOs. Bars represent the mean length of 80 embryos at 3 dpf per condition, which were scored blind to injection cocktail.
(B) Lateral and dorsal views of representative control embryos and embryos injected with scrib or puf60a MO at 5 dpf; yellow line indicates the distance across the convex tips of the eye cups. Right panel, ventral views of corresponding embryos stained with Alcian blue at 5 dpf to visualize cartilage structures; yellow line indicates distance between ceratohyal (CH) and Meckel’s cartilages (MK).
(C and D) Quantification of head size (C) and craniofacial defects (D) was performed in control and embryo batches injected with scrib or puf60a MO by measuring distances as shown by the yellow lines in (B). Head size measurements are represented as a normal probability distribution curve in which the y axis represents the probability that the values of x fall within a certain interval.
(C) Significant differences were observed for the microcephaly phenotype; p < 0.0001 between control and puf60a morphants and p < 0.0001 between control and scrib morphants (three independent experiments; two-tailed t test comparisons).
(A and D) Data are shown as the standard error of the mean, SEM from three independent experiments, n = 80 embryos. The corresponding p values are denoted on the bar graphs (two-tailed t test comparisons).
Second, because three out of five of the individuals studied here are microcephalic, we asked whether SCRIB and/or PUF60 might be involved in the regulation of head size. By using previously established methods of masked scoring of embryos at 5 dpf,20,38 we observed significant microcephaly in each of scrib and puf60 morphants compared to controls (p < 0.0001) (Figures 2B and 2C). Neither the body length nor the head size phenotypes were likely driven by gross developmental delay; morphants were normal with regard to the development of the swim bladder and pigmentation. Moreover, both the body length and microcephaly phenotypes were specific; they could each be rescued with coinjection of wild-type human full-length mRNAs (Figure S3).
Third, individuals with 8q24.3 deletions share craniofacial defects, with retrognathia being common across the cohort (Table 1). To test for this phenotype, we stained 5 dpf MO-injected embryos with Alcian blue and performed quantitative morphometric analyses of the cartilage of the jaw as described.38 We observed a significant reduction of the distance between the Meckel’s and ceratohyal cartilages for each of scrib and puf60 morphants, which indicated a reduced jaw size compared to sham-injected embryos (p < 0.0001) (Figures 2B and 2D).
Other phenotypes were unique to either scrib or puf60 morphants. Suppression of scrib resulted in coloboma, a phenotype observed in two out of six of the affected individuals, consisting of an absence of closure of the iris in one or both eyes in 53% of the embryos at 3 dpf; none of the embryos injected with puf60 MO showed this phenotype (Figure 3A). Further, four out of five of the individuals studied here have reported renal abnormalities, prompting us to perform a glomerular filtration assay, in which we injected 50 kDa FITC-labeled dextran into the cardiac venous sinus at 2 dpf followed by imaging at 5 dpf as described.49,50 Diffusion of the dextran throughout the body and gross edema often reflect the loss of the embryo’s ability to maintain fluid homeostasis.50 We observed gross edema and abnormal diffusion of dextran in 32% of scrib morphants; by contrast, all puf60 morphants were indistinguishable from controls (Figure 3B). We observed similarly restricted phenotypes for PUF60 as well; assessment of heart edema as an established surrogate of cardiac structural defects,51–54 a phenotype present in two out of six of the affected individuals, showed scrib embryos to be indistinguishable from controls, and 34% of puf60 MO-injected embryos exhibited heart edema (Figure 3C).
Figure 3.
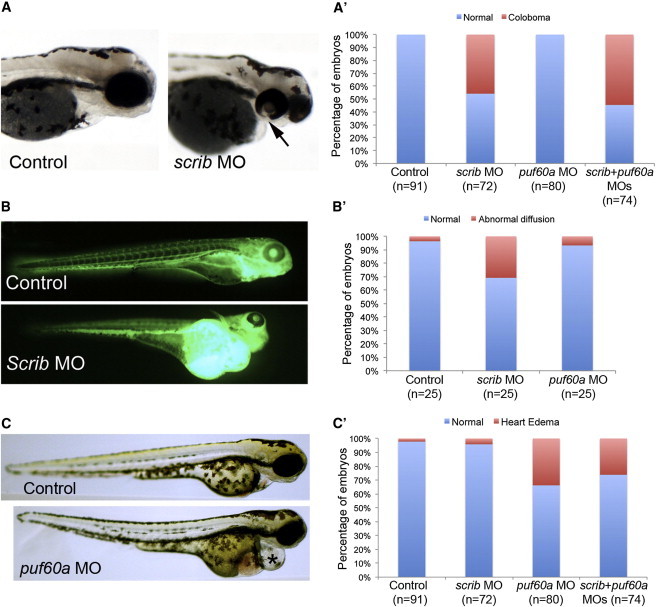
Suppression of scrib or puf60 in Zebrafish Lead to Unique Phenotypes
(A) Lateral views of representative control (sham-injected) embryos and those injected with scrib MO at 3 dpf. Coloboma was detected in scrib morphants (black arrow) but was absent in puf60a morphants.
(A′) Corresponding quantification of the coloboma phenotype in control, scrib, and puf60a morphants. There is no statistical difference between scrib morphants and the double morphants; ∼50% of the scrib and double morphants have coloboma.
(B) Lateral views of representative control embryos and those injected with scrib MO at 5 dpf. 50 kDa FITC-labeled dextran was injected in the cardiac venous sinus at 2 dpf followed by imaging. At 5 dpf Scrib morphants have pericardiac/yolk edema and absence of dextran in the intersegmental vessels compared to control.
(B′) Qualitative scoring of the glomerular filtration was performed in control and embryo batches injected with scrib MO or puf60a MO (n = 25, Dextran-injected embryos at 2 dpf followed by scoring at 5 dpf).
(C) Lateral views of representative control embryos and those injected with puf60a MO at 3 dpf.
(C′) Qualitative scoring of heart edema was performed in embryo batches injected with scrib, puf60a, and double MOs (80 embryos per injection cocktail).
Combinatorial Effect of Gene Suppression
Taken together, our data suggest that suppression of scrib or puf60 can induce phenotypes likely relevant to the components of the 8q24.3 syndromic presentation. Some phenotypes were found upon suppression of either gene (head size, craniofacial defects, short stature), whereas others were specific for one or the other transcript (scrib for coloboma and renal defects; puf60 for cardiac anomalies). We therefore asked whether combinatorial suppression of scrib and puf60 might have an additive or multiplicative effect. To test this possibility, we coinjected scrib and puf60 MOs into zebrafish embryos and asked whether we could observe genetic interaction. We observed no changes in penetrance or expressivity for coloboma, cardiac, head size, retrognathic, or renal defects compared to zebrafish receiving a single MO injection (Figures 3A, 3C, and 4). However, coinjection of scrib and puf60 MOs increased the severity of the short stature phenotype from 1,300 μm body length for scrib MO and 1,220 μm for puf60 MO alone to 1,175 μm for the double MOs (p = 0.012) (Figure 2A). Taken together, our data suggest a complex genetic architecture for the clinical phenotype associated with the minimal 8q24.3 deletion, wherein several phenotypes could be induced by the loss of either SCRIB or PUF60, other phenotypes could be driven exclusively by a single transcript, and one phenotype could be exacerbated from the genetic interaction of both genes.
Figure 4.
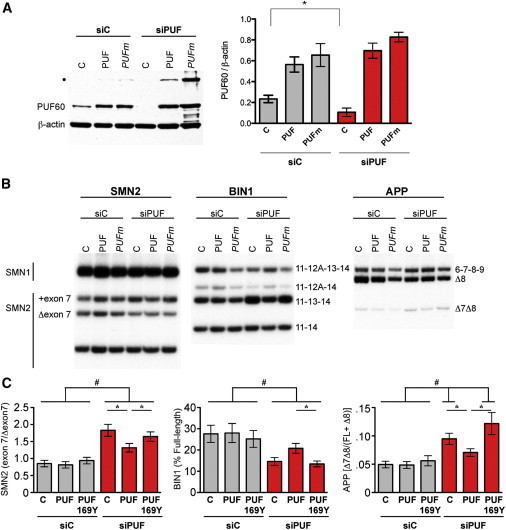
Puf60 p.His169Tyr Is Nonfunctional in Splicing
(A) Immunoblot analysis of PUF60 in lysates from cells transfected with a nonspecific control siRNA (siC) or a siRNA specific for PUF60. Cells were cotransfected with an empty expression plasmid (C) or a plasmid expressing wild-type PUF60 (PUF) or PUF60 p.His169Tyr mutant (PUFm). Both PUF60 cDNA expression plasmids have silent mutations introduced into the siPUF target region to render them insensitive to siRNA silencing. Blots were probed with an Alexa 594-tagged secondary antibody and quantitated with a Typhoon phosphorimager (right). Error bars represent SEM (n = 4); ∗p < 0.05 (two-tailed, paired Student’s t test). Blots were subsequently probed with an HRP-labeled secondary antibody (left). β-actin is included as a loading control. Asterisk indicates an SDS-resistant PUF60 dimer.
(B) Representative images of radiolabelled RT-PCR analysis of SMN2, BIN1, and APP alternatively spliced RNA isolated from cells treated as described in (A).
(C) Quantitation of alternative splicing shown in (B) by a Typhoon phosphorimager. Error bars represent standard error (n = 6). Asterisk (∗) indicates statistically different from siPUF rescued with PUF60, and hatch sign (#) indicates statistically different from equivalently treated siC (ANOVA with Tukey post-test).
A De Novo Mutation in PUF60 Causes 8q CNV Endophenotypes
We reasoned that if this model is correct, individuals with heterozygous loss-of-function mutations in either SCRIB or PUF60 should manifest the above-defined phenotypic components. In parallel to and independent of the CNV dissection, we exome sequenced an individual with developmental delay, microcephaly, and craniofacial and cardiac defects but no coloboma or renal defects (individual 6, Table 1) and we identified a de novo c.505C>T mutation leading to a p.His169Tyr change in PUF60 (RefSeq accession number NM_078480.2; GI, 402794025). This 21-year-old girl was treated in her early years for severe growth retardation. Because of her slow interval growth, she was given a course of growth hormone treatment, which resulted in increased growth velocity. She was noted to have several orthopedic anomalies: she was born with bilateral hip dislocation corrected with Pavlik Harness. Because of a lumbar hemivertebra and tethered cord-inducing severe scoliosis, she underwent L4-S1 laminotomies and removal of tethered cord at 8 years of age and resection of fatty filum and fusion of T4-L5 to treat the scoliosis at 13 years of age. Her other dysmorphic features included hemifacial microsomia, retrognathia, submucosal cleft palate, short neck, right arm and hand shorter/smaller than the left ones, pes planus, an absent right thumb, hypoplastic metacarpal and scaphoid bones, and malar hypoplasia (Figure S1). On her last physical examination at the age of 20 years, she demonstrated severe short stature with a height of 128.7 cm (−5.31 SD), weight 38.2 kg (−3.34 SD), and BMI 23.1 kg/m2 (0.5 SD). She had marked microcephaly with a head circumference of 50 cm (−4.1 SD). Her global developmental delay affected cognitive, behavior, and physical growth. She was able to sit at 3 years and walk at 5 years of age. She is nonverbal but is able to communicate with rudimentary sign language. She developed seizures at 9 years of age, controlled with antiepileptic therapy. She was noted to have a ventricular septal defect that closed spontaneously along with a bicuspid aortic valve. The MRI of the brain at the age of 12 years noted slight enlargement of the third and lateral ventricles with a thin corpus callosum and microcephaly. She had no large CNVs but did have a 13.946 kb intronic deletion (COL5A2; chr2: 189,993,689–190,007,634). This lesion is unlikely to be relevant to the phenotype.
The allele affects a highly conserved residue (Figure S5) and was absent from all control data sets, including 1000 Genomes, ESP, and internal controls, and is predicted to be damaging with a Polyphen2 score of 0.999.
Our CNV dissection data predict that, based on the phenotype, the p.His169Tyr allele would have a loss-of-function effect on the protein. To test this possibility, we first knocked down PUF60 expression in HeLa cells by using siRNA and assayed quantitatively the splicing of BIN1, APP, and SMN2, all of which are known to be differentially spliced based on PUF60 function.39 We then asked whether plasmids expressing 169His or 169Tyr, with additional silent mutations engineered to render them insensitive to RNAi, could rescue that phenotype. Knockdown of PUF60 led to significant changes in these genes’ mRNA splicing profiles. Specifically, upon knockdown of the endogenous PUF60, we observed an increase of the SMN2 exon 7 splicing (ratio of exon 7 included/Δexon 7) and more skipping of APP exons 7 and 8 (ratio Δ7Δ8/FL+Δ8) compared to control cells transfected with the sham siRNA (siC) (Figures 4A–4C). We also observed a decrease of the BIN1 full-length transcript in the absence of PUF60 (Figures 4B and 4C). These splicing changes could be rescued by wild-type but not mutant PUF60 (p < 0.05) (Figures 4B and 4C).
To explore this effect directly in our cohort, we extracted whole-blood RNA from the individual with the p.His169Tyr change (individual 6), from three individuals with 8q24.3 deletion (individuals 1, 2, and 4), and from six unaffected family members. We found similar patterns of alternative splicing affecting SMN2 and APP, but not BIN1, in individuals with the deletion compared to controls as were seen in the in vitro knockdown experiments (p = 0.0002 and p = 0.054 for SMN2 and APP, respectively) (Figure 5A). Strikingly, the splicing of PUF60 itself was affected in all three individuals with 8q24.3 deletion, as well as in individual 6, in whom exon 5 was skipped more frequently compared to unaffected family members, with a net result of the individual producing less of the long isoform of PUF60 (p = 0.04) (Figure 5A).
Figure 5.
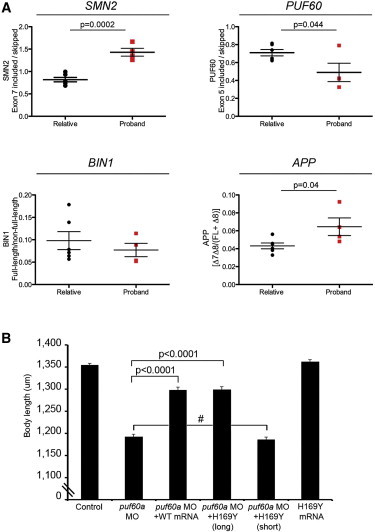
In Vivo Data: Individuals’ RNA and Zebrafish
(A) Deregulation of PUF60-dependent alternative splicing in affected individuals. Quantitation of RT-PCR analysis of alternatively spliced mRNA isoforms from SMN2, PUF60, BIN1, and APP from relatives (n = 6) or proband (n = 4) with a mutation or deletion of PUF60.
(B) Quantification of the total body length was performed in embryo batches injected with puf60a MO alone or puf60a MO along with either human wild-type (WT) or mutant PUF60 (p.His169Tyr) messages. The p.His169Tyr mutation was introduced in the long and short isoforms. Bars represent the average length of 80 embryos at 3 dpf, which were scored blind to injection cocktail. Injection of long WT and mutant mRNAs rescued the body length phenotype observed in puf60a morphants (p < 0.0001; two-tailed t test comparisons). The short mutant PUF60 mRNA failed to rescue the phenotype and showed no significant difference with the embryos injected with the puf60a MO alone. Data are shown as the mean ± SEM (three independent experiments). Hatch sign (#) indicates nonsignificant.
As a final test, we sought to evaluate the relative functionality of the long and short isoforms of PUF60 in vivo with regard to the phenotypes seen to be driven by the PUF60 mutation or deletion. We therefore engineered the two PUF60 isoforms and introduced the 169Tyr allele in both of them. We then coinjected zebrafish embryos with puf60 MO and either wild-type (WT) or mutant mRNAs and measured the body length at 3 dpf (80 embryos/injection, masked to the injection cocktail). We observed a significant rescue of the body length phenotype in WT coinjections and in coinjections with the 169Tyr mutant on the long PUF60 isoform (p < 0.0001). In contrast, the short mutant isoform of PUF60 failed to rescue the phenotype (Figure 5B). Together with the in vitro data, these findings suggest that the p.His169Tyr change is a loss-of-function allele that perturbs the splicing patterns of PUF60 itself, biasing the locus toward production of a short splice isoform. Critically, this bias is probably relevant to the pathology, given that the human phenotypes approximated in puf60 morphants could be rescued only by the long human PUF60 isoform.
Discussion
In contrast to recurrent CNVs that are mediated by nonallelic homologous recombination and thus have typically fixed boundaries, rare, nonrecurrent CNVs present a vexing interpretative problem. Here we have shown that by combining clinical assessment, genomic and genetic analyses, and systematic functional studies of all genes within the minimal CNV region, it is possible to determine the contribution of dosage imbalance of each transcript to the phenotype. Our studies suggest that haploinsufficiency of SCRIB and PUF60 drive the majority of the syndromic phenotypes found in our individuals with the 8q24.3 CNV.
Papillorenal syndrome (RCS [MIM 120330]) is characterized by optic nerve dysplasia (coloboma) and renal malformations. To date, PAX2 mutations (MIM 167409) have been the only identified genetic cause associated with RCS, but mutations remain unidentified in ∼50% of RCS-positive individuals. Notably, individual 1, harboring the minimal 8q24.3 deletion including SCRIB, exhibits coloboma and renal malformations. PAX2 and SCRIB share a similar major role in planar cell polarity (PCP), so SCRIB thus appears as a potential candidate gene for RCS considering the phenotypic overlap between RCS and the 8q24.3 deletion-associated phenotypes. The scribble complex, containing scribble (the Drosophila ortholog of the human SCRIB), lethal giant larvae (Lgl), and discs large (Dig), localizes to the lateral surface in polarized epithelia.55 Scribble also regulates cell-cell interaction56 and endothelial cell migration.57 Requirement of scribble has been reported in the formation of the pronephros by controlling the renal epithelial polarity via the procadherin Fat1 and Hippo signaling.58 The presence of scribble is required in PCP establishment during the development of the Drosophila eye and wing.59 Defective PCP signaling has been associated previously with microphthalmia and retinal coloboma exemplified by the presence of coloboma in Frizzled560–62 and Lrp663 knockout mice (with 50% and full penetrance, respectively). Finally, scribble plays a role in synaptic plasticity and synaptic vesicle dynamics64 and promotes cell polarization during astrocyte migration.65
A retrospective evaluation of the clinical phenotype of individual 6, as well as of the PUF60-driven phenotypes of the CNV carriers, suggested a striking overlap with Nager syndrome (MIM 154400) and mandibulofacial dysostosis with microcephaly (MFDM [MIM 610536]). Nager syndrome and MFDM are due to haploinsufficiency in SF3B4 (MIM 605593) and EFTUD2 (MIM 603892), both encoding proteins with a key role in splicing.66–69 SF3B4 is a component of U2 snRNP and EFTUD2 is a U5 snRNP protein.70 PUF60 encodes an additional splicing factor, which interacts directly with SF3B439 and plays a role in the recognition of the 3′ splice site during splicing and recruitment of U2 snRNP and subsequently U5 snRNP to the intron for splicing. Taken together, our data suggest that Nager syndrome, MFDM, and now PUF60 deficiency all lead to a spectrum of disorders resulting from partial ablation of spliceosome function.
Experimental animal models, including the zebrafish experiments described here, support the notion that individual cell types and developmental events may require differential PUF60 expression, possibly because of a role for PUF60 in the alternative splicing of specific gene transcripts. The Drosophila ortholog of PUF60, pUf68 (HFP), is required for normal development.71 Hypomorphic females produce short eggs with defects in germline mitosis, nuclear morphology, and dorsal ventral patterning possibly resulting from observed defects in developmentally regulated alternative splicing.71 PUF60 may have a partially redundant function in constitutive splicing, as previous studies have shown,39 but may be specifically required for the splicing of a subset of tissue-specific splicing events that, when deregulated in the absence of PUF60, affect the development of organs such as the brain, heart, kidney, and the eye.
Our studies also contribute to emerging theme of complex genetic architecture of CNVs with regard to the contribution of genes to the phenotype. Here, haploinsufficiency of each of SCRIB or PUF60 contribute uniquely to specific endophenotypes (e.g., coloboma, heart defects), and binary interaction potentially exacerbates other aspects of the clinical pathology of individuals with 8q24.3 deletion. We speculate that this genetic architecture will be common in both recurrent and nonrecurrent CNVs. Although the number of CNVs dissected to date is modest, KCTD13 (MIM 608947) has been shown to be the major driver of neuroanatomical defects in the recurrent 16p11.2 deletion and reciprocal duplication, where it can interact genetically with MVP and MAPK3 (MIM 601795), and PRRT2 (MIM 614386), which also maps in that CNV, probably contributes uniquely to the CNV-associated epilepsy.20,72 Similarly, two genes have been implicated in the etiology of the Williams-Beuren syndrome (MIM 194050), with each gene responsible for a different phenotypic component in individuals with the 7q11.23 CNV. Haploinsufficiency of elastin (ELN [MIM 130160]) is responsible for the arteriopathies and LIMK-1 (MIM 601329) contributes to the cognitive defects.73–75
Finally, we note that our approach is limited to the assessment of anatomical phenotypes that have reproducible, credible surrogates in zebrafish embryos; it is not currently possible to determine the contribution of specific genes to cognitive or behavioral defects. Further, we caution that the phenotypes driven by CNVs are not necessarily restricted to dosage imbalance for genes within the CNV; deletions or duplications can have significant effects on the regulation of neighboring genes that are not possible to predict and dissect functionally at present. Nonetheless, given that a substantial fraction of CNVs do present with modelable anatomical defects,76 we suggest that unbiased, systematic modeling of all genes within each discovered CNV will provide insight into both causality and genetic architecture, while also providing tools for the initial dissection of the biological underpinnings of the phenotype. Further insights will be gained through integration of this CNV modeling data with genomic sequence data from humans with novel potentially pathogenic point mutations in the involved genes.
Acknowledgments
We thank Anthony Hinrich and Yangfan Phoebe Liu for technical assistance. This work was conducted with support from Harvard Catalyst, The Harvard Clinical and Translational Science Center (National Institutes of Health Award #UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic health care centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University, and its affiliated academic health care centers, the National Center for Research Resources, or the National Institutes of Health. This work was also supported by the grant from the Simons Foundation SFARI # 239983 (N.K.) and the Eunice Kennedy Shriver National Institute of Child Health and Development grant K23HD073351 (A.D.). S.J. is supported by the Swiss National Fund professorship grant (PP00P3_144902). N.K. is a Distinguished Brumley Professor.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
DECIPHER, https://decipher.sanger.ac.uk/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Primer3, http://frodo.wi.mit.edu/primer3/
SnpEff, http://snpeff.sourceforge.net/
ZFIN, http://zfin.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 3.McCarroll S.A., Altshuler D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007;39(Suppl):S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 4.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fakhro K.A., Choi M., Ware S.M., Belmont J.W., Towbin J.A., Lifton R.P., Khokha M.K., Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. USA. 2011;108:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osoegawa K., Vessere G.M., Utami K.H., Mansilla M.A., Johnson M.K., Riley B.M., L’Heureux J., Pfundt R., Staaf J., van der Vliet W.A. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J. Med. Genet. 2008;45:81–86. doi: 10.1136/jmg.2007.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanna-Cherchi S., Kiryluk K., Burgess K.E., Bodria M., Sampson M.G., Hadley D., Nees S.N., Verbitsky M., Perry B.J., Sterken R. Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 2012;91:987–997. doi: 10.1016/j.ajhg.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott D.A., Klaassens M., Holder A.M., Lally K.P., Fernandes C.J., Galjaard R.J., Tibboel D., de Klein A., Lee B. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia. Hum. Mol. Genet. 2007;16:424–430. doi: 10.1093/hmg/ddl475. [DOI] [PubMed] [Google Scholar]
- 11.Serra-Juhé C., Rodríguez-Santiago B., Cuscó I., Vendrell T., Camats N., Torán N., Pérez-Jurado L.A. Contribution of rare copy number variants to isolated human malformations. PLoS ONE. 2012;7:e45530. doi: 10.1371/journal.pone.0045530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coe B.P., Girirajan S., Eichler E.E. The genetic variability and commonality of neurodevelopmental disease. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:118–129. doi: 10.1002/ajmg.c.31327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar R.A., KaraMohamed S., Sudi J., Conrad D.F., Brune C., Badner J.A., Gilliam T.C., Nowak N.J., Cook E.H., Jr., Dobyns W.B., Christian S.L. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 15.McCarthy S.E., Makarov V., Kirov G., Addington A.M., McClellan J., Yoon S., Perkins D.O., Dickel D.E., Kusenda M., Krastoshevsky O., Wellcome Trust Case Control Consortium Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., Autism Consortium Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 20.Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horev G., Ellegood J., Lerch J.P., Son Y.E., Muthuswamy L., Vogel H., Krieger A.M., Buja A., Henkelman R.M., Wigler M., Mills A.A. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacaria M., Saha P., Potocki L., Bi W., Yan J., Girirajan S., Burns B., Elsea S., Walz K., Chan L. A duplication CNV that conveys traits reciprocal to metabolic syndrome and protects against diet-induced obesity in mice and men. PLoS Genet. 2012;8:e1002713. doi: 10.1371/journal.pgen.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao J., Kochilas L., Nowotschin S., Arnold J.S., Aggarwal V.S., Epstein J.A., Brown M.C., Adams J., Morrow B.E. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- 24.Walz K., Caratini-Rivera S., Bi W., Fonseca P., Mansouri D.L., Lynch J., Vogel H., Noebels J.L., Bradley A., Lupski J.R. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Mol. Cell. Biol. 2003;23:3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan J., Keener V.W., Bi W., Walz K., Bradley A., Justice M.J., Lupski J.R. Reduced penetrance of craniofacial anomalies as a function of deletion size and genetic background in a chromosome engineered partial mouse model for Smith-Magenis syndrome. Hum. Mol. Genet. 2004;13:2613–2624. doi: 10.1093/hmg/ddh288. [DOI] [PubMed] [Google Scholar]
- 26.Tomlinson I.P., Webb E., Carvajal-Carmona L., Broderick P., Howarth K., Pittman A.M., Spain S., Lubbe S., Walther A., Sullivan K., CORGI Consortium. EPICOLON Consortium A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 2008;40:623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 27.Talseth-Palmer B.A., Scott R.J., Vasen H.F., Wijnen J.T. 8q23.3 and 11q23.1 as modifying loci influencing the risk for CRC in Lynch syndrome. Eur. J. Hum. Genet. 2012;20:487–488. doi: 10.1038/ejhg.2011.232. author reply 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikami M., Yasuda T., Terao A., Nakamura M., Ueno S., Tanabe H., Tanaka T., Onuma T., Goto Y., Kaneko S., Sano A. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am. J. Hum. Genet. 1999;65:745–751. doi: 10.1086/302535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halevy A., Basel-Vanagaite L., Shuper A., Helman S., Har-Zahav A., Birk E., Maya I., Kornreich L., Inbar D., Nürnberg G. Microcephaly-thin corpus callosum syndrome maps to 8q23.2-q24.12. Pediatr. Neurol. 2012;46:363–368. doi: 10.1016/j.pediatrneurol.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto Y., Oguro N., Miyao M., Yanagisawa M. Tricho-rhino-phalangeal syndrome type I with severe mental retardation due to interstitial deletion of 8q23.3-24.13. Am. J. Med. Genet. 1989;32:133–135. doi: 10.1002/ajmg.1320320128. [DOI] [PubMed] [Google Scholar]
- 31.Naritomi K., Hirayama K. Partial trisomy of distal 8q derived from mother with mosaic 8q23.3----24.13 deletion, and relatively mild expression of trichorhinophalangeal syndrome I. Hum. Genet. 1989;82:199–201. doi: 10.1007/BF00284059. [DOI] [PubMed] [Google Scholar]
- 32.Stengel-Rutkowski S., Lohse K., Herzog C., Apacik C., Couturier J., Albert A., Belohradsky B. Partial trisomy 8q. Two case reports with maternal translocation and inverted insertion: phenotype analyses and reflections on the risk. Clin. Genet. 1992;42:178–185. doi: 10.1111/j.1399-0004.1992.tb03234.x. [DOI] [PubMed] [Google Scholar]
- 33.Wheeler P.G. 8q23-q24 duplication—further delineation of a rare chromosomal abnormality. Am. J. Med. Genet. A. 2010;152A:459–463. doi: 10.1002/ajmg.a.33237. [DOI] [PubMed] [Google Scholar]
- 34.Bühler E.M., Bühler U.K., Beutler C., Fessler R. A final word on the tricho-rhino-phalangeal syndromes. Clin. Genet. 1987;31:273–275. doi: 10.1111/j.1399-0004.1987.tb02806.x. [DOI] [PubMed] [Google Scholar]
- 35.Shanske A.L., Patel A., Saukam S., Levy B., Lüdecke H.J. Clinical and molecular characterization of a patient with Langer-Giedion syndrome and mosaic del(8)(q22.3q24.13) Am. J. Med. Genet. A. 2008;146A:3211–3216. doi: 10.1002/ajmg.a.32615. [DOI] [PubMed] [Google Scholar]
- 36.Wuyts W., Roland D., Lüdecke H.J., Wauters J., Foulon M., Van Hul W., Van Maldergem L. Multiple exostoses, mental retardation, hypertrichosis, and brain abnormalities in a boy with a de novo 8q24 submicroscopic interstitial deletion. Am. J. Med. Genet. 2002;113:326–332. doi: 10.1002/ajmg.10845. [DOI] [PubMed] [Google Scholar]
- 37.Verheij J.B., de Munnik S.A., Dijkhuizen T., de Leeuw N., Olde Weghuis D., van den Hoek G.J., Rijlaarsdam R.S., Thomasse Y.E., Dikkers F.G., Marcelis C.L. An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur. J. Hum. Genet. 2009;52:353–357. doi: 10.1016/j.ejmg.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Beunders G., Voorhoeve E., Golzio C., Pardo L.M., Rosenfeld J.A., Talkowski M.E., Simonic I., Lionel A.C., Vergult S., Pyatt R.E. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013;92:210–220. doi: 10.1016/j.ajhg.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hastings M.L., Allemand E., Duelli D.M., Myers M.P., Krainer A.R. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65) PLoS ONE. 2007;2:e538. doi: 10.1371/journal.pone.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dauber A., Stoler J., Hechter E., Safer J., Hirschhorn J.N. Whole exome sequencing reveals a novel mutation in CUL7 in a patient with an undiagnosed growth disorder. J. Pediatr. 2013;162:202–204. doi: 10.1016/j.jpeds.2012.07.055. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bicknell L.S., Walker S., Klingseisen A., Stiff T., Leitch A., Kerzendorfer C., Martin C.A., Yeyati P., Al Sanna N., Bober M. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat. Genet. 2011;43:350–355. doi: 10.1038/ng.776. [DOI] [PubMed] [Google Scholar]
- 42.Kuo A.J., Song J., Cheung P., Ishibe-Murakami S., Yamazoe S., Chen J.K., Patel D.J., Gozani O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–119. doi: 10.1038/nature10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Margolin D.H., Kousi M., Chan Y.M., Lim E.T., Schmahmann J.D., Hadjivassiliou M., Hall J.E., Adam I., Dwyer A., Plummer L. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 45.Sarparanta J., Jonson P.H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J.M., Mahjneh I., Vihola A. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012;44:450–455. doi: 10.1038/ng.1103. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santoriello C., Zon L.I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 2012;122:2337–2343. doi: 10.1172/JCI60434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doelken S.C., Köhler S., Mungall C.J., Gkoutos G.V., Ruef B.J., Smith C., Smedley D., Bauer S., Klopocki E., Schofield P.N. Phenotypic overlap in the contribution of individual genes to CNV pathogenicity revealed by cross-species computational analysis of single-gene mutations in humans, mice and zebrafish. Dis. Model. Mech. 2013;6:358–372. doi: 10.1242/dmm.010322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hentschel D.M., Mengel M., Boehme L., Liebsch F., Albertin C., Bonventre J.V., Haller H., Schiffer M. Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. Am. J. Physiol. Renal Physiol. 2007;293:F1746–F1750. doi: 10.1152/ajprenal.00009.2007. [DOI] [PubMed] [Google Scholar]
- 50.Hentschel D.M., Park K.M., Cilenti L., Zervos A.S., Drummond I., Bonventre J.V. Acute renal failure in zebrafish: a novel system to study a complex disease. Am. J. Physiol. Renal Physiol. 2005;288:F923–F929. doi: 10.1152/ajprenal.00386.2004. [DOI] [PubMed] [Google Scholar]
- 51.Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011;91:279–288. doi: 10.1093/cvr/cvr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mitchell I.C., Brown T.S., Terada L.S., Amatruda J.F., Nwariaku F.E. Effect of vascular cadherin knockdown on zebrafish vasculature during development. PLoS ONE. 2010;5:e8807. doi: 10.1371/journal.pone.0008807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qu X., Jia H., Garrity D.M., Tompkins K., Batts L., Appel B., Zhong T.P., Baldwin H.S. Ndrg4 is required for normal myocyte proliferation during early cardiac development in zebrafish. Dev. Biol. 2008;317:486–496. doi: 10.1016/j.ydbio.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun X., Zhang R., Lin X., Xu X. Wnt3a regulates the development of cardiac neural crest cells by modulating expression of cysteine-rich intestinal protein 2 in rhombomere 6. Circ. Res. 2008;102:831–839. doi: 10.1161/CIRCRESAHA.107.166488. [DOI] [PubMed] [Google Scholar]
- 55.Pieczynski J., Margolis B. Protein complexes that control renal epithelial polarity. Am. J. Physiol. Renal Physiol. 2011;300:F589–F601. doi: 10.1152/ajprenal.00615.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yates L.L., Schnatwinkel C., Hazelwood L., Chessum L., Paudyal A., Hilton H., Romero M.R., Wilde J., Bogani D., Sanderson J. Scribble is required for normal epithelial cell-cell contacts and lumen morphogenesis in the mammalian lung. Dev. Biol. 2013;373:267–280. doi: 10.1016/j.ydbio.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michaelis U.R., Chavakis E., Kruse C., Jungblut B., Kaluza D., Wandzioch K., Manavski Y., Heide H., Santoni M.J., Potente M. The polarity protein Scrib is essential for directed endothelial cell migration. Circ. Res. 2013;112:924–934. doi: 10.1161/CIRCRESAHA.112.300592. [DOI] [PubMed] [Google Scholar]
- 58.Skouloudaki K., Puetz M., Simons M., Courbard J.R., Boehlke C., Hartleben B., Engel C., Moeller M.J., Englert C., Bollig F. Scribble participates in Hippo signaling and is required for normal zebrafish pronephros development. Proc. Natl. Acad. Sci. USA. 2009;106:8579–8584. doi: 10.1073/pnas.0811691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Courbard J.R., Djiane A., Wu J., Mlodzik M. The apical/basal-polarity determinant Scribble cooperates with the PCP core factor Stbm/Vang and functions as one of its effectors. Dev. Biol. 2009;333:67–77. doi: 10.1016/j.ydbio.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burns C.J., Zhang J., Brown E.C., Van Bibber A.M., Van Es J., Clevers H., Ishikawa T.O., Taketo M.M., Vetter M.L., Fuhrmann S. Investigation of Frizzled-5 during embryonic neural development in mouse. Dev. Dyn. 2008;237:1614–1626. doi: 10.1002/dvdy.21565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu C., Bakeri H., Li T., Swaroop A. Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hum. Mol. Genet. 2012;21:1848–1860. doi: 10.1093/hmg/ddr616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H., Xu S., Wang Y., Mazerolle C., Thurig S., Coles B.L., Ren J.C., Taketo M.M., van der Kooy D., Wallace V.A. Ciliary margin transdifferentiation from neural retina is controlled by canonical Wnt signaling. Dev. Biol. 2007;308:54–67. doi: 10.1016/j.ydbio.2007.04.052. [DOI] [PubMed] [Google Scholar]
- 63.Zhou C.J., Molotkov A., Song L., Li Y., Pleasure D.E., Pleasure S.J., Wang Y.Z. Ocular coloboma and dorsoventral neuroretinal patterning defects in Lrp6 mutant eyes. Dev. Dyn. 2008;237:3681–3689. doi: 10.1002/dvdy.21770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roche J.P., Packard M.C., Moeckel-Cole S., Budnik V. Regulation of synaptic plasticity and synaptic vesicle dynamics by the PDZ protein Scribble. J. Neurosci. 2002;22:6471–6479. doi: 10.1523/JNEUROSCI.22-15-06471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Osmani N., Vitale N., Borg J.P., Etienne-Manneville S. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr. Biol. 2006;16:2395–2405. doi: 10.1016/j.cub.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 66.Gordon C.T., Petit F., Oufadem M., Decaestecker C., Jourdain A.S., Andrieux J., Malan V., Alessandri J.L., Baujat G., Baumann C. EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J. Med. Genet. 2012;49:737–746. doi: 10.1136/jmedgenet-2012-101173. [DOI] [PubMed] [Google Scholar]
- 67.Lines M.A., Huang L., Schwartzentruber J., Douglas S.L., Lynch D.C., Beaulieu C., Guion-Almeida M.L., Zechi-Ceide R.M., Gener B., Gillessen-Kaesbach G., FORGE Canada Consortium Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 2012;90:369–377. doi: 10.1016/j.ajhg.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luquetti D.V., Hing A.V., Rieder M.J., Nickerson D.A., Turner E.H., Smith J., Park S., Cunningham M.L. “Mandibulofacial dysostosis with microcephaly” caused by EFTUD2 mutations: expanding the phenotype. Am. J. Med. Genet. A. 2013;161A:108–113. doi: 10.1002/ajmg.a.35696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernier F.P., Caluseriu O., Ng S., Schwartzentruber J., Buckingham K.J., Innes A.M., Jabs E.W., Innis J.W., Schuette J.L., Gorski J.L., FORGE Canada Consortium Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 2012;90:925–933. doi: 10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Champion-Arnaud P., Reed R. The prespliceosome components SAP 49 and SAP 145 interact in a complex implicated in tethering U2 snRNP to the branch site. Genes Dev. 1994;8:1974–1983. doi: 10.1101/gad.8.16.1974. [DOI] [PubMed] [Google Scholar]
- 71.Van Buskirk C., Schüpbach T. Half pint regulates alternative splice site selection in Drosophila. Dev. Cell. 2002;2:343–353. doi: 10.1016/s1534-5807(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 72.Labate A., Tarantino P., Palamara G., Gagliardi M., Cavalcanti F., Ferlazzo E., Sturniolo M., Incorpora G., Annesi G., Aguglia U., Gambardella A. Mutations in PRRT2 result in familial infantile seizures with heterogeneous phenotypes including febrile convulsions and probable SUDEP. Epilepsy Res. 2013;104:280–284. doi: 10.1016/j.eplepsyres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 73.Delio M., Pope K., Wang T., Samanich J., Haldeman-Englert C.R., Kaplan P., Shaikh T.H., Cai J., Marion R.W., Morrow B.E., Babcock M. Spectrum of elastin sequence variants and cardiovascular phenotypes in 49 patients with Williams-Beuren syndrome. Am. J. Med. Genet. A. 2013;161A:527–533. doi: 10.1002/ajmg.a.35784. [DOI] [PubMed] [Google Scholar]
- 74.Frangiskakis J.M., Ewart A.K., Morris C.A., Mervis C.B., Bertrand J., Robinson B.F., Klein B.P., Ensing G.J., Everett L.A., Green E.D. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 1996;86:59–69. doi: 10.1016/s0092-8674(00)80077-x. [DOI] [PubMed] [Google Scholar]
- 75.Micale L., Turturo M.G., Fusco C., Augello B., Jurado L.A., Izzi C., Digilio M.C., Milani D., Lapi E., Zelante L., Merla G. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur. J. Hum. Genet. 2010;18:317–323. doi: 10.1038/ejhg.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golzio C., Katsanis N. Genetic architecture of reciprocal CNVs. Curr. Opin. Genet. Dev. 2013;23:240–248. doi: 10.1016/j.gde.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.