

Abstract
Intraflagellar transport (IFT) depends on two evolutionarily conserved modules, subcomplexes A (IFT-A) and B (IFT-B), to drive ciliary assembly and maintenance. All six IFT-A components and their motor protein, DYNC2H1, have been linked to human skeletal ciliopathies, including asphyxiating thoracic dystrophy (ATD; also known as Jeune syndrome), Sensenbrenner syndrome, and Mainzer-Saldino syndrome (MZSDS). Conversely, the 14 subunits in the IFT-B module, with the exception of IFT80, have unknown roles in human disease. To identify additional IFT-B components defective in ciliopathies, we independently performed different mutation analyses: candidate-based sequencing of all IFT-B-encoding genes in 1,467 individuals with a nephronophthisis-related ciliopathy or whole-exome resequencing in 63 individuals with ATD. We thereby detected biallelic mutations in the IFT-B-encoding gene IFT172 in 12 families. All affected individuals displayed abnormalities of the thorax and/or long bones, as well as renal, hepatic, or retinal involvement, consistent with the diagnosis of ATD or MZSDS. Additionally, cerebellar aplasia or hypoplasia characteristic of Joubert syndrome was present in 2 out of 12 families. Fibroblasts from affected individuals showed disturbed ciliary composition, suggesting alteration of ciliary transport and signaling. Knockdown of ift172 in zebrafish recapitulated the human phenotype and demonstrated a genetic interaction between ift172 and ift80. In summary, we have identified defects in IFT172 as a cause of complex ATD and MZSDS. Our findings link the group of skeletal ciliopathies to an additional IFT-B component, IFT172, similar to what has been shown for IFT-A.
Main Text
Cilia are hair-like structures that project from the surface of most mammalian cells and are involved in diverse signaling pathways. Mutations in genes encoding ciliary proteins lead to “ciliopathies,” a collection of complex developmental disorders of multiple organ systems.1–3 Although there is broad clinical overlap, ciliopathies have been divided into subgroups on the basis of their predominant clinical phenotype and major organ involvement; nephronophthisis-related ciliopathies (NPHP-RCs) and skeletal ciliopathies are such examples. Whereas by definition NPHP-RCs show cystic renal degeneration,2 skeletal ciliopathies primarily manifest with a bone-related phenotype, such as polydactyly (e.g., in short-rib-polydactyly syndromes [SRPSs; MIM 263510] and Ellis-van-Creveld syndrome [EVC; MIM 225500]), thoracic dystrophy (e.g., in SRPSs and asphyxiating thoracic dystrophy [ATD], also known as Jeune syndrome [MIM 208500]), phalangeal cone-shaped epiphysis (e.g., in Mainzer-Saldino syndrome [MZSDS; MIM 266920]), or dolichocephaly and hypodontia and/or microdontia (e.g., in cranioectodermal dysplasia [CED], also known as Sensenbrenner syndrome [MIM 218330]).4 Whereas proteins associated with NPHP-RCs mainly function at the ciliary transition zone,5 most proteins associated with skeletal ciliopathies have been shown to participate in intraflagellar transport (IFT). IFT is an evolutionarily conserved kinesin- and dynein-mediated bidirectional trafficking system essential for cilium assembly and maintenance and is facilitated by two major subcomplexes, A (IFT-A) and B (IFT-B). Ciliary proteins found to be defective in skeletal disorders currently encompass the following four main subgroups: (1) all six subunits of IFT-A6–11 and its motor protein, DYNC2H1,12 whose defects have been shown to disrupt retrograde transport and cause IFT protein accumulation at the ciliary tip; (2) NEK1, a serine-threonine kinase involved in cell-cycle control and ciliogenesis;13 (3) EVC and EVC2, both located at the basal body as positive regulators of sonic hedgehog signaling;14,15 and (4) IFT80 (intraflagellar transport 80 homolog [Chlamydomonas]), one of 14 subunits of IFT-B, which is involved in anterograde IFT. Although all six IFT-A components are implicated in skeletal ciliopathies, none of the IFT-B components have been shown to play a role in human disease to date; the only exception is IFT80, encoded by IFT80 (MIM 611263), the first gene identified as causing ATD when mutated.16 Given the fact that IFT-B is critical for ciliogenesis in mice,17,18 we sought to elucidate whether additional IFT-B proteins are defective in individuals with ciliopathies, particularly skeletal ciliopathies.
To identify additional genes mutated in ciliopathies, we applied targeted candidate-gene sequencing and whole-exome capture with next-generation sequencing (also known as whole-exome resequencing [WER]) to a large multicenter cohort of 1,530 individuals with ciliopathies. Written informed consent was obtained from all individuals enrolled in this study and approved by the institutional review boards at the University of Michigan, the University College London Institute of Child Health (in partnership with the Great Ormond Street NHS Hospital Trust), Paris Descartes University, University of British Columbia, University of Queensland, University of Birmingham, and Duke University Medical Center. The diagnosis of NPHP-RCs and/or ATD was based on published clinical criteria.19 Mutation analysis was performed by three different approaches in five independent cohorts of individuals with NPHP-RCs or skeletal ciliopathies. In 12 families, we identified a total of 14 individuals who had biallelic mutations in IFT172 (intraflagellar transport 172 homolog [Chlamydomonas], also known as SLB, selective LIM binding factor homolog [RefSeq accession number NM_015662.1, MIM 607386]) and who shared a phenotype including skeletal abnormalities, nephronophthisis (NPHP), and liver and eye involvement, consistent with the diagnosis of complex ATD or MZSDS (Table 1). Whenever available, we obtained parental DNA to show segregation of a recessive trait by Sanger sequencing (Figure S1, available online, and Table 1).
Table 1.
IFT172 Mutations in 14 Individuals from 12 Families Affected by Skeletal Ciliopathies, ATD and MZSDS
| Individuala | Ethnic Origin | Nucleotide Mutationb | Deduced Protein Alteration | Exon or Intron (Zygosity, Segregation) | Amino Acid Evolutionary Conservation | PolyPhen-2 (HumVar) | MutationTaster | Parental Consanguinity | Clinical Diagnosis | Skeletal Features | Renal Disease (ESRD) | Other Clinical Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NPH2218 | Hungarian | c.432delA | p.Lys144Asnfs∗15 | 6 (het, p) | - | - | - | no | ATD, JBTS | TD, SS, SLB | NPHP (6 years) | RD, LF, OMA, CVH, ID, obesity |
| c.4161G>Ac | p.Arg1387Serfs∗7 | 38 (het, m) | - | - | - | |||||||
| A3189-21 | Pakistani | c.886C>Td | p.Arg296Trp | 9 (hom) | D. melanogaster | 0.967 | DC | yes | MZSDS | SS | NPHP (9 years) | RD, ID, died at 12 years |
| UCL-87 | Turkish | c.1232T>A | p.Ile411Asn | 13 (hom) (het, p/m) | D. melanogaster | 0.890 | DC | yes | ATD | TD, TA, PD (feet) | none (−) | LF, died at 18 months |
| UCL-107 | Turkish | c.1232T>A | p.Ile411Asn | 13 (hom) | D. melanogaster | 0.890 | DC | yes | ATD | TD, TA | none (−) | LF, died at 3 months |
| NPH2161 | French | c.1390_1395delGATATT | p.Asp464_Ile465 del | 14 (het) | D. melanogaster and D. rerio | - | - | ND | MZSDS | BD | NPHP (34 years) | RD, cholestasis |
| c.5179T>Ce | p.Cys1727Arg | 48 (het) | D. rerio | 0.648 | DC | |||||||
| B1 | Belgian | c.1671_1672dupAG | p.Val558Glufs∗12 | 16 (het, p) | - | - | - | no | ATD | TD, TA, PSCE, BD, PD | none (−) | RD, ID |
| c.5179T>Ce | p.Cys1727Arg | 48 (het, m) | D. rerio | 0.648 | DC | |||||||
| SKDP-44.3 | British | c.2158delC | p.Arg720Valfs∗28 | 21 (het, m) | - | - | - | no | ATD | TD, TA, SS, BD | mild structural abnormalities | RD, cholestasis, OMA, ID, obesity |
| c.5179T>Ce | p.Cys1727Arg | 48 (het, p) | D. rerio | 0.648 | DC | |||||||
| A3215-21 | South American | c.2716C>T | p.Gln906∗ | 25 (het, m) | - | - | - | no | ATD | TD, SS, GV | NPHP (12 years), RTX (13 years) | ID |
| c.4607T>C | p.Leu1536Pro | 42 (het, p) | C. reinhardtii | 0.807 | DC | |||||||
| F108-21 | German | c.3228+1G>A | 5′ splice site | 29 (het, m) | - | - | - | no | MZSDS | PCSE, BD | NPHP (11 years) | RD, LF, IGT, obesity |
| c.4607T>C | p.Leu1536Pro | 42 (het, p) | C. reinhardtii | 0.807 | DC | |||||||
| SKDP-165.3 | Singaporean and Malaysian | c.3907C>T | p.Arg1303∗ | 35 (het, p) | - | - | - | no | ATD | TD, TA, PD, SLB | early cystic dysplasia | LF, VSD, hydrocephalus, died by induced abortion |
| c.4630C>T | p.Arg1544Cys | 42 (het, m) | D. melanogaster | 0.991 | DC | |||||||
| A2052-21 and A2052-22 | Filipino | c.4630C>T | p.Arg1544Cys | 42 (hom) (het, p/m) | D. melanogaster | 0.991 | DC | yes | ATD, MZSDS, JBTS | TD, TA, PCSE, BD | NPHP (2 years), RTX (4 years) | RD, LF, OMA, CVH, ID |
| A3037-21 and A3037-22 | European American | c.4925_4928del GAGA | p.Arg1642Lysfs∗32 | 46 (het, p) | - | - | - | no | MZSDS | PCSE, BD | NPHP (20 years) | RD, LF, obesity |
| c.5179T>Ce | p.Cys1727Arg | 48 (het, m) | D. rerio | 0.648 | DC |
Abbreviations are as follows: BD, brachydactyly; ESRD, end-stage renal disease; CVH, cerebellar vermis hypoplasia; DC, predicted to be “disease causing”; GV, genu valgum; het, heterozygous; hom, homozygous; ID, intellectual disability; IGT, impaired glucose tolerance; ATD, asphyxiating thoracic dystrophy; JBTS, Joubert syndrome; LF, liver fibrosis; m, maternal; MZSDS, Mainzer-Saldino syndrome; ND, no data; NPHP, nephronophthisis; OMA, ocular motor apraxia; p, paternal; PD, polydactyly; PCSE, phalangeal cone-shaped epiphysis; RD, retinal degeneration; RTX, renal transplantation; SLB, short long bone; SS, short stature; TA, trident acetabulum; TD, thoracic dystrophy (small bell-shaped thorax); and VSD, ventriculoseptal defect.
In sibling cases, clinical information refers to the underlined individual.
cDNA mutations are numbered according to human cDNA RefSeq NM_015662.1 (IFT172); +1 corresponds to the A of the ATG start translation codon.
This variant abrogates the 3′ splice site (Figure S2). It is in 1000 Genomes (its minor allele frequency is not annotated), but not in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS).
NHLBI EVS (n = 6,503 control subjects): T/T = 0; T/C = 1; C/C = 6,502.
NHLBI EVS (n = 6,503 control subjects): C/C = 0; C/T = 1; T/T = 6,502.
First, we performed a candidate-gene screening of all 14 genes encoding IFT-B complex proteins (Table S1) in 1,056 affected individuals with NPHP-RCs by applying a recently developed mutation-analysis method of microfluidic array-based multiplex PCR and consecutive barcoded next-generation sequencing (NGS).20 As a result, we detected seven individuals with recessive IFT172 mutations in five families. Three individuals from two families were homozygous for missense mutations (A3189-21, c.886C>T [p.Arg296Trp]; A2052-21 and A2052-22, c.4630C>T [p.Arg1544Cys]). Another four individuals from three families were compound heterozygous for a truncating and a missense mutation (A3215-21, c.2716C>T [p.Gln906∗] and c.4607T>C [p.Leu1536Pro]; F108-21, c.3228+1G>A and c.4607T>C [p.Leu1536Pro]; A3037-21 and A3037-22, c.4925_4928delGAGA [p.Arg1642Lysfs∗32] and c.5179T>C [p.Cys1727Arg]). All detected missense residues were highly conserved throughout evolution (Table 1). Most affected individuals exhibited NPHP with progressive renal insufficiency in childhood and reached end-stage renal disease (ESRD) by 20 years of age. Three subjects (A3215-21, A2052-21, and A2052-22) showed thoracic dystrophy with chronic respiratory distress, necessitating intermittent mechanical ventilation (Figure 1A). All three presented with the clinical characteristics of ATD: thoracic dystrophy with a trident acetabular roof and shortening of the long bones (Figure 1 and Table 1). The affected siblings from family A2052, as well as three other individuals (A3037-21, A3037-22, and F108-21), displayed phalangeal cone-shaped epiphysis, a hallmark of MZSDS (Figure 1G), in addition to liver fibrosis and retinal dystrophy. Interestingly, both siblings from family A2052 also exhibited cerebellar vermis hypoplasia, representing an exceedingly rare co-occurrence of ATD, MZSDS, and Joubert syndrome (JBTS [MIM 213300]).21 Three of the individuals with MZSDS (A3037-21, A3037-22, and F108-21) also presented with obesity and impaired glucose tolerance, suggesting a phenotypic overlap with Bardet-Biedl syndrome (BBS [MIM 209900]) (Table 1). Mutations in genes most frequently associated with NPHP (NPHP1–NPHP13) had been previously excluded in all affected individuals.22
Figure 1.
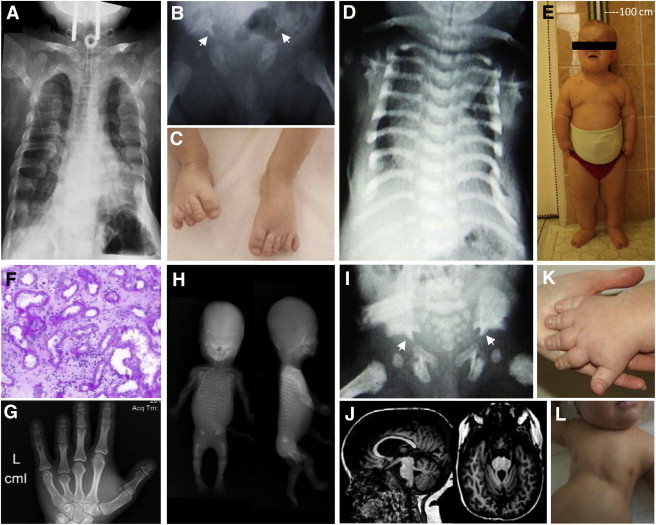
Clinical Characteristics of Individuals with Recessive Mutations in IFT172
(A) A chest X-ray of individual A3215-21 shows a narrowed, bell-shaped thorax and short ribs. Note the tracheostomy for ventilation.
(B) A hip X-ray of individual UCL-87 demonstrates a trident acetabular roof with spurs (white arrowheads).
(C) Postaxial polydactyly of the feet in individual UCL-87.
(D) A chest X-ray of individual UCL-107 shows a narrowed, bell-shaped thorax.
(E) Obesity and short stature of individual NPH2218 at 10 years of age.
(F) Renal histology of individual NPH2218 exhibits dilated tubules, disruption of the tubular basement membrane, and extensive interstitial fibrosis.
(G) A hand X-ray of individual A3037-21 shows brachydactyly with cone-shaped epiphysis of the middle phalanges.
(H) A babygram of individual SKDP-165.3 shows a turricephaly-like skull shape, absent nasal bone, postaxial tetramelic hexadactyly, shortened and curved long bones, short ribs, mild platyspondyly, and spur-like projections of the acetabular roof.
(I) A trident acetabular roof with spurs (white arrowheads) in individual UCL-107.
(J) Cranial MRI depicts partial agenesis of the cerebellar vermis in individual NPH2218.
(K) Brachydactyly of individual NPH2218.
(L) Narrow thorax of individual UCL-107.
Similarly, we conducted bidirectional Sanger sequencing of the coding exons and intron-exon boundaries of all 14 IFT-B-encoding genes in another cohort of 296 individuals with ciliopathies. We thereby identified compound-heterozygous changes in IFT172: the previously identified missense mutation c.5179T>C (p.Cys1727Arg) and a frameshifting 2 bp deletion, c.1671_1672dupAG (p.Val558Glufs∗12), in a Belgian female with ATD (B1, Table 1 and Figure S1). Notably, her initial symptoms were bilateral postaxial polydactyly of the hands at birth and night blindness at 5 years of age. Subsequent clinical evaluation revealed a mildly hypoplastic left thoracic cage, rhizomelic shortening of the limbs with brachydactyly, short phalanges, and a trident acetabulum. Ultrasonography at the time of ascertainment revealed no significant abnormalities in the kidneys, liver, or pancreas.23
Second, by applying exon-enriched NGS of 1,209 ciliary candidate genes, including those encoding all 14 IFT-B components (“ciliome sequencing”),11 in another 115 individuals with NPHP-RCs, we found two individuals with compound-heterozygous IFT172 mutations. Individual NPH2218 carried two truncating mutations, a frameshift mutation in exon 6 (c.432delA [p.Lys144Asnfs∗15]), and a nucleotide change that affected the first base of exon 38 and thus abrogated the acceptor splice site and led to a truncated protein (c.4161G>A [p.Arg1387Serfs∗7]) (Figure S2). This individual exhibited a severe phenotype with shortened long bones, resulting in severe dwarfism, obesity, brachydactyly, and NPHP with early-onset ESRD (Figures 1E, 1F, and 1K). Additionally, he also presented with liver failure, retinal degeneration, severe intellectual disability, oculomotor apraxia, and partial agenesis of the cerebellar vermis, consistent with JBTS (Figure 1J). In contrast, individual NPH2161 displayed a milder phenotype evoking MZSDS as a result of late-onset retinitis pigmentosa, NPHP with adult-onset ESRD (at 34 years), cholestasis, and short hands. This individual carried a missense allele (c.5179T>C [p.Cys1727Arg], conserved to D. rerio) and an in-frame deletion (c.1390_1395delGATATT [p.Asp464_Ile465del], conserved to D. rerio and D. melanogaster).
Third, WER was independently performed in two separate ATD cohorts, one from the United Kingdom and one from Australia. In the United Kingdom cohort, we performed WER in 56 individuals with the clinical diagnosis of ATD. We thereby identified a homozygous missense mutation in IFT172 (c.1232T>A [p.Ile411Asn], conserved to D. melanogaster) in an individual of consanguineous Turkish descent (UCL-87). Parallel sequencing of 60 more ATD cases with the use of a NGS gene-panel approach revealed the same mutation in a second individual of consanguineous Turkish descent (UCL-107). In addition to showing characteristic ATD features, such as a bell-shaped narrow thorax with short ribs, handlebar clavicles, and a trident acetabulum (Figures 1B, 1D, 1I, and 1L), both individuals displayed hepatosplenomegaly, dilated intrahepatic bile ducts, and liver failure similarly to the previously detected individuals (F108-21, A3037-21, and A2052-21). In contrast to most of the described subjects, UCL-87 additionally presented with postaxial polydactyly of the feet (Figure 1C). Renal disease was not reported in either of them. However, because both individuals died within the first 18 months of life as a result of respiratory (UCL-107)24 or liver (UCL-87) failure, renal involvement could not be completely excluded or might have developed later in life. WER variant analysis was performed as previously described.25 In UCL-87, the above mutation was one out of three remaining homozygous missense variants found in three different genes. Only two variants, the one in IFT172 and one in ERCC6, were located on a long homozygosity stretch corresponding to parental consanguinity.26 ERCC6 is known to cause Cockayne syndrome type B (MIM 133540), a recessive UV-sensitive nucleotide-excision-repair disorder characterized by neurological and sensory impairment, cachectic dwarfism, and photosensitivity.27 Therefore, considering the individual’s phenotype and taking the evolutionary conservation of both missense variants into account, IFT172 remained the most likely disease-associated candidate (Table S2).
In the Australian cohort, we performed WER in seven individuals with ATD and identified two individuals with compound-heterozygous mutations in IFT172 (SKDP-165.3 and SKDP-44.3). SKDP-165.3 carried a truncating mutation (c.3907C>T [p.Arg1303∗]) and a missense mutation (c.4630C>T [p.Arg1544Cys], conserved to D. melanogaster). SKDP-44.3 carried a single-base frameshift deletion (c.2158delC [p.Arg720Valfs∗28]) and a missense mutation (c.5179T>C [p.Cys1727Arg], conserved to D. rerio). Internal WER data of 993 unrelated control individuals did not demonstrate any other person with compound-heterozygous mutations in IFT172. An ultrasound scan of SKDP-165.3 at 16 weeks of gestation demonstrated a facial cleft with an absent nasal bone, hydrocephalus, cardiac malformation, tetramelic polydactyly, short long bones, and echogenic kidneys; the pregnancy was terminated at this stage. Postmortem examination demonstrated intrauterine growth restriction, a turricephaly-like skull shape, upper-lip paramedian cleft extending into the palate, hypoplasia of the nasal bridge and nose, postaxial hexadactyly of all four limbs, hydrocephalus, possible brain heterotopia, a ventriculoseptal defect, bilateral adrenal hypoplasia, prominent hepatic ductal plates, and early renal cystic dysplasia (Table 1). Other skeletal changes included shortened and curved long bones, relatively short ribs, mild platyspondyly, and spur-like projections of the acetabular roof (Figure 1H). ATD was considered the most likely diagnosis. WER was performed as previously described.25 Given parental nonconsanguinity, compound heterozygosity was thought the most likely form of inheritance. Seven genes carried two or more novel (absent from public databases) or rare (minor allele frequency < 0.001) nonsynonymous SNPs, small insertions, or exon or splice-site deletions predicted to be damaging.
Two of these genes (IFT172 and STXBP5L) were represented in the cilia proteome and had appropriate familial segregation of variants; however, only IFT172 had two variants that were in highly conserved regions and had a deleterious effect predicted by PolyPhen-2, SIFT, and MutationTaster. Thus, IFT172 was considered the most likely candidate (Tables S3 and S4).
SKDP-44.3, the last individual of the Australian cohort, had neonatal respiratory distress. Poor vision was noted from the age of 6 weeks, leading to the diagnosis of retinal dystrophy. Speech was delayed, and brain MRI revealed mild ventriculomegaly. Frequent and severe chest infections led to a skeletal survey, which showed narrowing of the thorax and resulted in a diagnosis of ATD. This individual was also noted to have mild renal structural abnormalities, obesity, marked rhizomelic shortening, and brachydactyly. WER data for SKDP-44.3 was similarly filtered; after Sanger sequencing for appropriate segregation within the family, only IFT172 remained as a possible candidate gene (Tables S3 and S4).
IFT172 encodes IFT172, a 1,749-residue protein (the largest of all known IFT proteins) containing 9 N-terminal WD-40, 1 LIM, and 14 C-terminal TPR (tetratricopeptide repeat) domains. The detected mutations lead to protein changes in both principal domain structures and have a slight predominance toward the C-terminal end, which neighbors the loci of two extensively studied animal mutants, the wimple mouse (wim)17 and the thermosensitive Chlamydomonas fla1128 (Figures 2A and 2B). Interestingly, although both mutants represent missense changes (p.Leu1564Pro for wim and p.Leu1615Pro for fla11), they result in severe phenotypes and, in the case of wim, embryonic lethality.17
Figure 2.
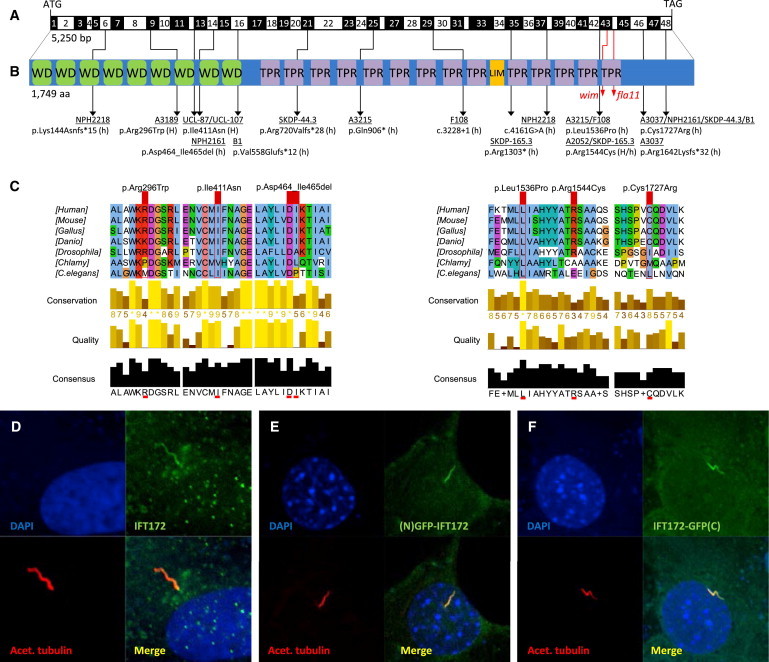
Biallelic IFT172 Mutations, Deduced Impact at Protein Level, and Subcellular Localization of WT IFT172
(A) Exon structure of human IFT172 cDNA. The positions of the start codon (ATG) and stop codon (TGA) are indicated.
(B) Domain structure of IFT172, which contains 9 WD-40 repeats (WD), located N-terminal to 14 tetratricopeptide repeats (TPR) and 1 LIM domain. For the mutations detected, black arrows indicate positions in relation to exons and protein domains. Family numbers are underlined. Abbreviations are as follows: H, homozygous; and h, heterozygous. IFT172 animal mutants wim (mouse, p.Leu1564Pro) and fla11 (C. reinhardtii, p.Leu1615Pro) are indicated by red arrows. Note the proximity of the detected missense changes p.Leu1536Pro and p.Arg1544Cys to the wim locus at position Leu1564.
(C) A partial protein alignment of IFT172 shows evolutionary conservation of the identified missense changes (p.Arg296Trp, p.Ile411Asn, p.Leu1536Pro, p.Arg1544Cys, and p.Cys1727Arg).
(D) Antibody staining (polyclonal rabbit antibody, Abcam, 1:100) of WT IFT172 in human control fibroblasts shows axonemal and pericentriolar localization in comparison to acetylated tubulin (anti-acetylated alpha tubulin, mouse monoclonal antibody, Abcam, 1:1000).
(E and F) Localization of human WT IFT172 constructs, once with an N-terminal GFP tag (E) and once with a C-terminal GFP tag (F), after transfection of a 48 hr serum-starved NIH 3T3 cell line. Immunofluorescence on a confocal microscope (Zeiss, LSM 720) confirmed axonemal localization with enrichment at the ciliary base upon overexpression.
In accordance with previously reported human mutations of genes encoding the IFT-A or IFT-B complex, most affected individuals carried one highly conserved missense allele in trans with a functional null nonsense or frameshift allele (Figures 2B and 2C). Accordingly, the observed phenotypes of the subjects in this study, especially the phenotype of SKDP-165.3, are reminiscent of the hypomorphic avc1 mouse, which displays shortening of the long bones, preaxial polydactyly, renal dysplasia, atrioventricular septal defect, and hydrocephalus.29 Only NPH2218 carried two truncating mutations, consequently associated with a severe phenotype of multiple organ involvement, notably malformations of the CNS. However, the homozygous c.1232T>A (p.Ile411Asn) mutation of UCL-87 and UCL-107 was also associated with a severe phenotype and early death. This might be due to the substitution’s N-terminal localization, given that the N terminus was recently shown to be necessary for anterograde transport in Tetrahymena.30 However, health-care standards in the individuals’ respective countries have to be taken into account before a genotype-phenotype correlation can be considered. On the other hand, SKDP-165.3 also demonstrated an extremely severe phenotype; whether this would have been compatible with postnatal life is unknown. This individual carried one earlier truncating mutation and the same C-terminally located missense mutation as did family A2052. In conclusion, even missense mutations can result in a severe phenotype if located within a specific domain structure of crucial protein function, as already demonstrated for wim (Figures 2A and 2B).17
To investigate the subcellular localization of the wild-type (WT) protein, we first performed immunofluorescence in primary human skin fibroblasts of healthy controls. WT IFT172 localized to the axoneme and around the base of the cilium (Figure 2D). Similar to endogenous IFT172, N-terminally and C-terminally tagged constructs localized to the ciliary axoneme upon overexpression (Figures 2E and 2F).
To evaluate the impact of the identified human mutations on ciliogenesis, ciliary morphology, and composition, we examined cultured human fibroblasts from affected individuals A2052-21, NPH2161, and NPH2218 and compared them to those of healthy controls. No significant difference in the number of ciliated cells was observed between mutants and controls. Cilia from mutant fibroblasts were not shortened but unexpectedly appeared longer than those of the controls (Figure 3A). Similarly, we found that cilia from the remaining kidney tubules of individual NPH2218 also appeared longer (Figure S3). We next assessed the ciliary composition in the three human fibroblast cell lines (A2052-21, NPH2161, and NPH2218) and observed a partial loss of the IFT-A protein IFT140 along the cilium, accompanied by an accumulation of IFT140 at the basal body (Figure 3B). Interestingly, we also observed a significant decrease in adenylyl cyclase III (ACIII) staining along the axonemes of all three human fibroblast cell lines (Figure 3C). Reduced ciliary ACIII can result in defective cyclic AMP (cAMP) signaling and consequent reduction of the activity of protein kinase A (PKA), a negative regulator of mammalian Sonic hedgehog signaling. Indeed, we observed an abnormal accumulation of the catalytic subunits of PKA (PKAc) at the base of the cilium in affected individuals’ fibroblasts that were treated with the adenylate cyclase activator forskolin (Figure S4), suggesting altered cAMP-PKA signaling. Together, these results indicate that fibroblasts from affected individuals exhibit an altered ciliary composition and potentially lead to defects in ciliary trafficking and cilia-mediated signaling.
Figure 3.
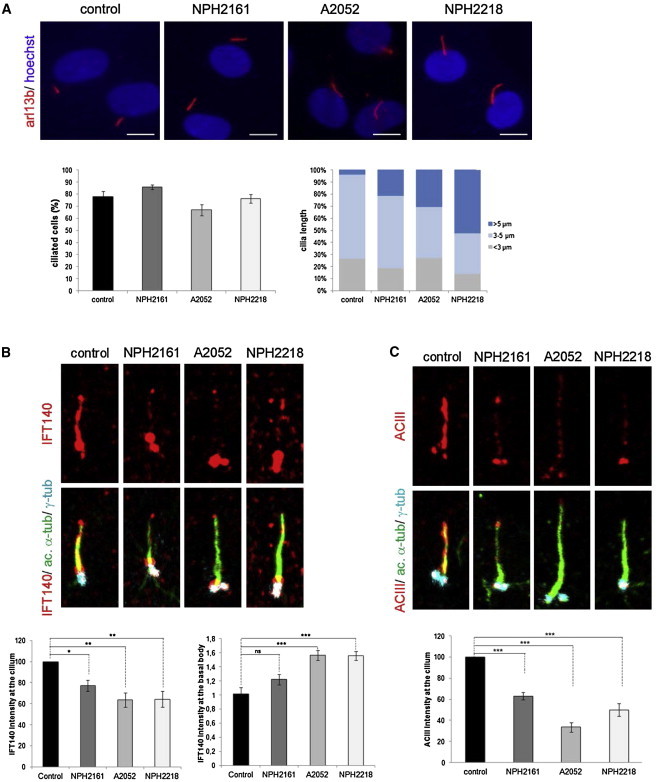
Alteration of Ciliogenesis and Ciliary Composition in Human Mutant Fibroblasts
Control and mutant fibroblasts from individuals NPH2161, A2052-21, and NPH2218 were starved for 48 hr for inducing ciliogenesis and were fixed with MetOH.
(A) Staining of ARL13B (polyclonal rabbit antibody, Proteintech; 1:400), quantification of ciliated cells, and measurement of cilia length with the use of Lucia G on Nikon DXM 1200 Software. Compared to controls, mutant fibroblasts displayed elongated cilia. The scale bar represents 10 μm.
(B) Staining of acetylated-tubulin (mouse monoclonal antibody, Sigma Aldrich; 1:10,000), γ-tubulin (goat polyclonal antibody, Santa Cruz; 1:200), and IFT140 (polyclonal rabbit antibody, Proteintech; 1:100) showed a decrease in ciliary and an increase in basal body IFT140 staining intensity in mutant fibroblasts compared to controls.
(C) Staining of adenylyl cyclase III (ACIII, rabbit polyclonal antibody, Santa Cruz; 1:100) showed a decrease in ciliary ACIII-staining intensity in mutant fibroblasts compared to controls.
Images in (B) and (C) were recorded with a Leica SP8 confocal microscope and analyzed with ImageJ. All graphs show the mean ± SEM of at least three independent experiments. “ns” stands for not significant. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 were calculated via Dunn’s Multiple Comparison Test after the analysis of variance ANOVA test.
To recapitulate the human phenotype and investigate defective IFT172 in vivo, we performed zebrafish knockdown with two morpholino oligonucleotides (MOs) targeting the exon 1-intron 1 and intron 1-exon 2 splice sites. Both MOs led to a similar phenotype comparable to that of the previously described ift172 zebrafish mutants,31,32 displaying ventral body-axis curvature, formation of kidney cysts, otolith defects, and hydrocephalus (Figure 4B and Figures S5C and S5E). In addition, morphants exhibited cartilage defects, demonstrated by Alcian-blue staining of the craniofacial skeleton (Figure 4B). Furthermore, upon knockdown of ift172 in a rhodopsin-GFP transgenic zebrafish line, we were able to visualize the impact on retinal rhodopsin expression. We thereby demonstrated that the level of rhodopsin-GFP was lower in ift172 morphants than in controls, suggesting retinal degeneration (Figure S5A). Lastly, by scanning electron microscopy of the olfactory placode, we observed ciliogenesis defects, including shortened and truncated cilia, in ift172 morphants compared to controls (Figure S5B). Ciliogenesis defects with fewer and shorter cilia have been previously described in ift172 mutant zebrafish embryos,33 as well as in the ift172 mouse mutant wim.17 In contrast to these observations, the fibroblasts from the three individuals with IFT172 mutations were normally ciliated. This discrepancy might be due to the nature of the identified human mutations, each carrying at least one hypomorphic allele that might partially conserve IFT172 function and thus result in an overall milder phenotype.
Figure 4.
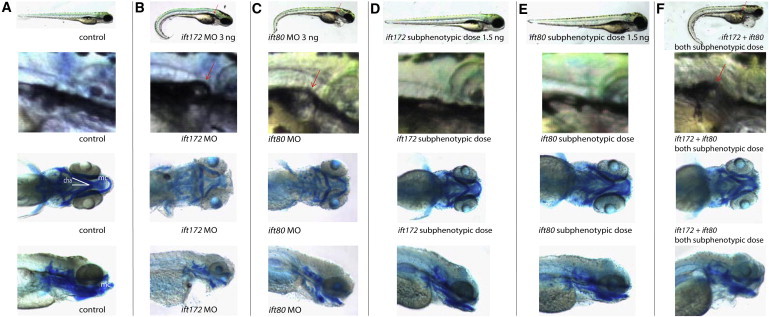
Knockdown of ift172 and ift80 and Genetic Epistasis between ift172 and ift80 in Zebrafish
(A–C) When compared to the control (A), both ift172 (B) and ift80 (C) morphants displayed similar ciliopathy phenotypes, including ventral body-axis curvature (first row), formation of renal cysts (red arrows, second row), and cartilage defects with hypoplasia of the Meckel’s cartilage (mc) and widening of ceratohyal angle (cha), as shown by Alcian-blue staining (third and fourth row).
(D–E) Zebrafish injected with subphenotypic doses of either ift172 (D) or ift80 (E) MO appeared no different than the control (A).
(F) Similar to a full dose of each MO alone, combined injection of subphenotypic doses of ift172 MO and subphenotypic doses of ift80 MO resulted in body-axis curvature, formation of renal cysts, and cartilage defects.
IFT80, a 777 amino acid protein, shows striking similarities to IFT172. Both proteins are part of the peripheral, non-core, IFT-B complex, feature multiple N-terminal WD-40 domains, and are implicated in individuals with ATD when defective.16 Therefore, we investigated whether IFT172 and IFT80 interact genetically with each other. When comparing the phenotypes of both zebrafish morphants, we observed striking similarities with regard to body-axis curvature, formation of kidney cysts, and pattern of cartilage defects (Figures 4B–4F). Moreover, the combined injection of subphenotypic doses of both ift80 (1.5 ng) and ift172 (1.5 ng) MOs resulted in the same phenotype as did injection of a full dose (3 ng) of each MO alone, indicating genetic interaction (Figures 4B–4F and Figures S5C and S5G).
In summary, we have identified mutations in IFT172 as an additional cause of ATD and MZSDS in humans. In contrast to “classical” ATD, which is due to mutated IFT80 and DYNC2H1 and which typically manifests without any extraskeletal symptoms,16,34 ATD in these subjects with mutations in IFT172 is characterized by a complex phenotype and is frequently associated with extraskeletal involvement, notably NPHP, liver fibrosis, retinal degeneration, obesity, and rarely, cerebellar vermis hypoplasia (in A2052-21, A2052-22, and NPH2218). None of the previously identified genes associated with ATD or MZSDS have been implicated in CNS dysplasia, underlying the special role of IFT172 in mammalian brain development as demonstrated by another ift172-null mouse model (slb).35 Because of the significant overlap between the phenotypic features and other forms of NPHP-RC, we introduce the alias “NPHP17” for IFT172. The most similar phenotype, however, results from recessive mutations in IFT140 (MIM 266920), encoding one of six IFT-A subunits. Defective IFT140 is a frequent cause of MZSDS and ATD with multiple extraskeletal involvements, including NPHP, retinal degeneration, and liver anomalies.11,36 Consistently, IFT172 is the only IFT-B protein shown to interact with IFT140 in a series of pull-down experiments in mice.37 In that context, we have demonstrated here that mutations in IFT172 lead to partial delocalization of IFT140 in fibroblasts of affected individuals, suggesting the necessity of functional IFT172 for sufficient IFT140 to enter the cilium. Taken together with the Chlamydomonas and Tetrahymena data on the importance of IFT172 for the transition from anterograde to retrograde transport and for the turnaround at the flagellar tip,28,30 our data strengthen the hypothesis that IFT172 is indeed involved in an interaction between the two subcomplexes, IFT-A and IFT-B. By characterization of IFT172 as an additional gene associated with ATD and as the second gene identified to be associated with MZSDS in humans,11,36 we link a subset of peripheral IFT-B proteins, consisting of IFT172 and IFT80, to a phenotype that was also described in individuals with mutations in genes encoding IFT-A proteins. We hereby provide a further piece to the puzzle of correlating protein complexes to certain clinical phenotypes.
The BBSome was the first protein complex whose members were defined as defective in a distinct ciliopathy phenotype, BBS.38 Similarly, the majority of NPHP- and JBTS-related proteins were mapped to four distinct protein modules located around the ciliary transition zone.39 IFT-A has only recently been linked to a variety of human ciliopathies that specifically involve skeletal dysplasia. We have now shown that defects in the second IFT-B component, IFT172, also result in a well-defined group of ciliopathies with skeletal involvement. We therefore hypothesize that complete or partial loss of function of other IFT-B members might equally either lead to a bone-related disorder or turn out to be embryonically lethal.
Acknowledgments
We are grateful to all individuals with nephronophthisis-related ciliopathies, asphyxiating thoracic dystrophy, and Mainzer-Saldino syndrome and their family members for their participation. We further thank the investigators of the UK10K Consortium (www.uk10k.org) and the FORGE Canada Consortium, as well as the following funding agencies that supported this work: the Howard Hughes Medical Institute, the National Institutes of Health, the Agence Nationale de la Recherche, the Fondation pour la Recherche Médicale, the Institute National de la Santé et de la Recherche Médicale, the Imagine Institute, the Wellcome Trust, the Dutch Kidney Foundation, the European Community, the Royal Children’s Hospital Brisbane Foundation, the Newlife Foundation for Disabled Children UK, and the Action Medical Research UK. Detailed Supplemental Acknowledgments can be found in the Supplemental Data.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Sophie Saunier, Email: sophie.saunier@inserm.fr.
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
BEAGLE 3.3.2, http://faculty.washington.edu/browning/beagle/beagle_3.3.2_31Oct11.pdf
CASAVA, http://www.illumina.com/software/genome_analyzer_software.ilmn
CiliaProteome, http://www.ciliaproteome.org
Complete Genomics, http://www.completegenomics.com/public-data/
Condel, http://bg.upf.edu/condel/home
Family-based Exome Variants Analysis (FEVA), www.exome.info
hapFabia (Bioconductor), http://www.bioconductor.org/packages/2.11/bioc/html/hapFabia.html
Human Gene Mutation Database (HGMD) Professional, http://www.biobase-international.com/product/hgmd
The Human Protein Atlas, http://www.proteinatlas.org/
HomozygosityMapper, http://www.homozygositymapper.org/HomozygosityMapper/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Novoalign, http://www.novocraft.com/main/page.php?s=novoalign
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
SIFT, http://sift.jcvi.org/
UCSC Human Genome Browser, http://genome.ucsc.edu/
UK10K project, http://www.uk10k.org
UniProt, http://www.uniprot.org/uniprot/
References
- 1.Baker K., Beales P.L. Making sense of cilia in disease: the human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 4.Huber C., Cormier-Daire V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:165–174. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 5.Czarnecki P.G., Shah J.V. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arts H.H., Bongers E.M.H.F., Mans D.A., van Beersum S.E.C., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A.M., Schuurs-Hoeijmakers J.H.M., de Leeuw N. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 7.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S.A., von der Haar S., Zenker M., Zahnleiter D., Stöss H., Beinder E. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz-Perez V.L., Ide S.E., Strom T.M., Lorenz B., Wilson D., Woods K., King L., Francomano C., Freisinger P., Spranger S. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000;24:283–286. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Perez V.L., Tompson S.W., Blair H.J., Espinoza-Valdez C., Lapunzina P., Silva E.O., Hamel B., Gibbs J.L., Young I.D., Wright M.J., Goodship J.A. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am. J. Hum. Genet. 2003;72:728–732. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 17.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 18.Murcia N.S., Richards W.G., Yoder B.K., Mucenski M.L., Dunlap J.R., Woychik R.P. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- 19.Chaki M., Hoefele J., Allen S.J., Ramaswami G., Janssen S., Bergmann C., Heckenlively J.R., Otto E.A., Hildebrandt F. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 2011;80:1239–1245. doi: 10.1038/ki.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 21.Lehman A.M., Eydoux P., Doherty D., Glass I.A., Chitayat D., Chung B.Y.H., Langlois S., Yong S.L., Lowry R.B., Hildebrandt F., Trnka P. Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy. Am. J. Med. Genet. A. 2010;152A:1411–1419. doi: 10.1002/ajmg.a.33416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halbritter J., Porath J.D., Diaz K.A., Braun D.A., Kohl S., Chaki M., Allen S.J., Soliman N.A., Hildebrandt F., Otto E.A., GPN Study Group Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casteels I., Demandt E., Legius E. Visual loss as the presenting sign of Jeune syndrome. Eur. J. Paediatr. Neurol. 2000;4:243–247. doi: 10.1053/ejpn.2000.0313. [DOI] [PubMed] [Google Scholar]
- 24.Tüysüz B., Bariş S., Aksoy F., Madazlı R., Ungür S., Sever L. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. Am. J. Med. Genet. A. 2009;149A:1727–1733. doi: 10.1002/ajmg.a.32962. [DOI] [PubMed] [Google Scholar]
- 25.McInerney-Leo A.M., Schmidts M., Cortés C.R., Leo P.J., Gener B., Courtney A.D., Gardiner B., Harris J.A., Lu Y., Marshall M., UK10K Consortium Short-Rib Polydactyly and Jeune Syndromes Are Caused by Mutations in WDR60. Am. J. Hum. Genet. 2013;93:515–523. doi: 10.1016/j.ajhg.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Troelstra C., van Gool A., de Wit J., Vermeulen W., Bootsma D., Hoeijmakers J.H.J. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 28.Pedersen L.B., Miller M.S., Geimer S., Leitch J.M., Rosenbaum J.L., Cole D.G. Chlamydomonas IFT172 is encoded by FLA11, interacts with CrEB1, and regulates IFT at the flagellar tip. Curr. Biol. 2005;15:262–266. doi: 10.1016/j.cub.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 29.Friedland-Little J.M., Hoffmann A.D., Ocbina P.J.R., Peterson M.A., Bosman J.D., Chen Y., Cheng S.Y., Anderson K.V., Moskowitz I.P. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet. 2011;20:3725–3737. doi: 10.1093/hmg/ddr241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsao C.-C., Gorovsky M.A. Different effects of Tetrahymena IFT172 domains on anterograde and retrograde intraflagellar transport. Mol. Biol. Cell. 2008;19:1450–1461. doi: 10.1091/mbc.E07-05-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 32.Sukumaran S., Perkins B.D. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009;49:479–489. doi: 10.1016/j.visres.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lunt S.C., Haynes T., Perkins B.D. Zebrafish ift57, ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect hedgehog signaling. Dev. Dyn. 2009;238:1744–1759. doi: 10.1002/dvdy.21999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidts M., Arts H.H., Bongers E.M.H.F., Yap Z., Oud M.M., Antony D., Duijkers L., Emes R.D., Stalker J., Yntema J.-B.L., UK10K Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 2013;50:309–323. doi: 10.1136/jmedgenet-2012-101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorivodsky M., Mukhopadhyay M., Wilsch-Braeuninger M., Phillips M., Teufel A., Kim C., Malik N., Huttner W., Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev. Biol. 2009;325:24–32. doi: 10.1016/j.ydbio.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidts M., Frank V., Eisenberger T., Al Turki S., Bizet A.A., Antony D., Rix S., Decker C., Bachmann N., Bald M. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum. Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Follit J.A., Xu F., Keady B.T., Pazour G.J. Characterization of mouse IFT complex B. Cell Motil. Cytoskeleton. 2009;66:457–468. doi: 10.1002/cm.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peränen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C., Jackson P.K. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 39.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.