

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurological disorder characterized by the degeneration of motor neurons and typically results in death within 3–5 years from onset. Familial ALS (FALS) comprises 5%–10% of ALS cases, and the identification of genes associated with FALS is indispensable to elucidating the molecular pathogenesis. We identified a Japanese family affected by late-onset, autosomal-dominant ALS in which mutations in genes known to be associated with FALS were excluded. A whole- genome sequencing and parametric linkage analysis under the assumption of an autosomal-dominant mode of inheritance with incomplete penetrance revealed the mutation c.2780G>A (p. Arg927Gln) in ERBB4. An extensive mutational analysis revealed the same mutation in a Canadian individual with familial ALS and a de novo mutation, c.3823C>T (p. Arg1275Trp), in a Japanese simplex case. These amino acid substitutions involve amino acids highly conserved among species, are predicted as probably damaging, and are located within a tyrosine kinase domain (p. Arg927Gln) or a C-terminal domain (p. Arg1275Trp), both of which mediate essential functions of ErbB4 as a receptor tyrosine kinase. Functional analysis revealed that these mutations led to a reduced autophosphorylation of ErbB4 upon neuregulin-1 (NRG-1) stimulation. Clinical presentations of the individuals with mutations were characterized by the involvement of both upper and lower motor neurons, a lack of obvious cognitive dysfunction, and relatively slow progression. This study indicates that disruption of the neuregulin-ErbB4 pathway is involved in the pathogenesis of ALS and potentially paves the way for the development of innovative therapeutic strategies such using NRGs or their agonists to upregulate ErbB4 functions.
Main Text
Amyotrophic lateral sclerosis (ALS) is a devastating neurological disorder in which the degeneration of motor neurons leads to progressive weakness and wasting of limb, bulbar, and respiratory muscles. Familial ALS (FALS) comprises 5%–10% of ALS cases, and the remaining cases are simplex cases of ALS (SALS). To date, more than 20 genes have been shown to be associated with ALS,1 and these account for 75% of FALS and 14% of SALS cases.2 Mutations that are found in FALS-associated genes but that are also identified in individuals with SALS are considered mutations with reduced penetrance or de novo mutations. Further discovery of genes associated with FALS is indispensable to elucidating the molecular backgrounds of both FALS and SALS.
Identification of genes associated with familial diseases has been accomplished through identification of the disease loci on the chromosomes by linkage analysis of large pedigrees and subsequent positional cloning of the genes. The majority of the FALS pedigrees, however, are not large and do not have multiple affected members as a result of the poor prognosis of the disease and the late age of onset, which makes it difficult to sufficiently narrow the candidate regions by linkage analyses and means that it takes a tremendous effort to identify the genes associated with FALS. The recent development of massively parallel sequencing technologies has allowed us to overcome the difficulty by means of whole-genome sequencing (WGS) or exome analysis.
We identified a Japanese family with three affected siblings presenting with late-onset ALS (Figure 1A and Table 1). The familial history indicated that the mode of inheritance is probably an autosomal-dominant one. Mutational analysis of the proband (II-4) employing direct nucleotide sequence analysis, a microarray-based resequencing, or a repeat-primed PCR analysis excluded SOD1[MIM 147450], ALS2[MIM 606352], DCTN1[MIM 601143], CHMP2B[MIM 609512], ANG[MIM 105850], TARDBP[MIM 605078], FUS[MIM 137070] and C9ORF72[MIM 1614260] as the genes associated with FALS.3,4 To identify a gene associated with FALS, we applied WGS in combination with a linkage analysis to the pedigree. Written informed consent was obtained from all the participants. This study was approved by the institutional review board at the University of Tokyo.
Figure 1.
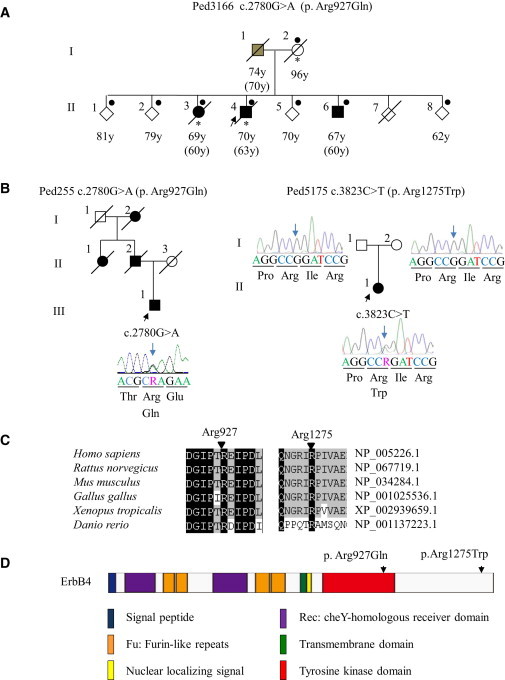
Pedigrees of ALS and Characterization of Mutations
(A) Pedigree charts of the index family.
Filled symbols indicate affected individuals. The arrow indicates the proband. For confidentiality purposes, all unaffected siblings are indicated by diamonds. Dots or asterisks indicate individuals included in the linkage study or WGS, respectively. Age at present or age at death is shown under each individual, and ages at onset are shown in parentheses. The box with gray shading indicates that the individual’s clinical information obtained from the family members strongly supports the diagnosis of ALS, although detailed neurological evaluations have not been conducted for this individual.
(B) Additional Canadian (Ped255) and Japanese (Ped5175) pedigrees with ERBB4 mutations. The electropherograms of mutational data are shown beside each member. Nucleotide colors correspond to the colors in the electropherograms. The amino acids are designated below the nucleotide sequences. The blue arrows indicate the nucleotide positions of the mutations. In the electropherograms (Ped5175), nucleotide sequences of the reverse complementary strand are shown.
(C) Amino acid conservation. The amino acids Arg927 and Arg1275 are highly conserved among species.
(D) The protein structure along with the locations of amino acid substitutions are shown; amino acid substitutions are indicated by arrows. The amino acid substitution p. Arg927Gln resides in the tyrosine kinase domain, which mediates the key functions of ErbB4. The amino acid substitution p. Arg1275Trp resides in the C-terminal domain in the vicinity of multiple phosphorylation sites, which mediate downstream signaling pathways.
Table 1.
Clinical Characteristics of Affected Individuals
| Pedigree Number | Pedigree 3166 | Pedigree 255 | Pedigree 5175 | |||
|---|---|---|---|---|---|---|
| Ethnicity | Japanese | Canadian | Japanese | |||
| Inheritance | familial (autosomal dominant) | familial (autosomal dominant) | simplex | |||
| Mutation | c.2780G>A | c.2780G>A | c.3823C>T | |||
| Amino acid substitution | p. Arg927Gln | p. Arg927Gln | p. Arg1275Trp | |||
| Members | I-1 | II-3 | II-4 (proband) | II-6 | III-3 | II-1 |
| Age at onset | 70 | 60 | 63 | 60 | 67 | 45 |
| Initial symptoms | bulbar | N.D. | upper limbs | respiration | upper limbs | upper limbs |
| Diagnostic criteriaa | N.D. | N.D. | definite | definite | probable | probable |
| Progression | unable to walk after 3 years | ventilator -dependent after 5 years, locked-in state after 8 years | locked-in state after 5 years | ventilator- dependent after 1 year, locked-in state after 5 years | slow progression that significantly decelerated and finally stopped after 8 years | wheelchair- bound, MRS 1-2/5 in upper extremities after 5 years |
| Cognitive function | N.D. | N.D. | normal | normal | N.D. | normal |
| Age at death | 74 | 69 | 70 | 66 | N/A | N/A |
Abbreviations are as follows: N.D., not described; MRS, manual muscle testing rating scale; and N/A, not applicable.
El Escorial and Airlie House revised criteria.
WGS was performed on three individuals (I-2, II-3 and II-4, as shown in Figure 1A) in the index pedigree. Paired-end DNA libraries were generated and subjected to massively parallel sequencing with a GAII Illumina Genome Analyzer in accordance with the manufacturer’s instructions. The short read sequences obtained were aligned to the reference genome (NCBI37/hg19 assembly) via the Burrows-Wheeler Aligner.5 Downstream analyses in which potential PCR duplicates were removed were processed with SAMtools.6 Aligned reads were viewed on an Integrative Genomics Viewer.7 Genomic sequence variations were identified with the SAMtools pileup command and annotated with Refseq, dbSNP135, 1000 Genomes, personal genome databases, the NHLBI GO Exome Sequencing Project (NHLBI-ESP) database, and an in-house variant database containing 41 whole genomes and 1,408 exomes in the Japanese population. The numbers of nonsynonymous variants that were identified in individuals I-2, II-3, and II-4 but that were not present in any of the databases (hereafter, variants not found in the databases are referred to as “novel”) were 411, 404, and 382, respectively (Table S1). No novel nonsynonymous variants in genes known to be associated with FALS were included. Among the identified variants, 57 were identified both in the proband and in the affected sibling, but not in the mother, and were subjected to further analysis.
The individuals indicated by dots in Figure 1A were genotyped with Genome-Wide Human SNP Array 6.0 (Affymetrix). Linkage analysis and haplotype reconstruction were conducted with the pipeline software SNP-HiTLink8 and Allegro version 29 under the assumption of an autosomal-dominant mode of inheritance and a disease-allele frequency of 0.000001. Parametric multipoint linkage analysis under the assumption of complete penetrance revealed three loci spanning 23.6 Mb on chromosomes 1, 6, and 13, having a maximum LOD score of 1.8 (Figure S1; penetrance = 1.0), and containing 88 annotated genes. However, no novel nonsynonymous variants were identified in the candidate regions. We then considered the possibility of reduced penetrance. When penetrance was reduced to 0.8 (Figure S1), seven additional loci had LOD scores > 0.7 and were thus shown to support linkage; these loci contained 809 annotated genes. Three heterozygous novel nonsynonymous variants were identified in these regions; among these variants, only c.2780G>A (p. Arg927Gln; dbSNP SubSNP ID ss831884245) substituting glutamine for arginine at codon 927 (p. Arg927Gln) in v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) (ERBB4 [MIM 600543; RefSeq accession number NM_005235.2]) was not present in 477 controls (Table S2). When we allowed further reduced penetrance, we identified 19 additional loci with LOD > 0; these loci contained 1,265 annotated genes. In these regions, we identified seven heterozygous novel nonsynonymous variants, among which three variants in OR2D3 (RefSeq NM_001004684.1), FTCD (MIM 606806; RefSeq NM_206965.1), and TJP2 (MIM 607709; RefSeq NM_001170414.2) were not present in 477 controls (Table S2). OR2D3 is an olfactory receptor gene; the substituted amino acid in OR2D3 is not conserved, and the substitution is predicted as benign by PolyPhen-2 analysis. FTCD and TJP2 are associated with autosomal-recessive glutamate formiminotransferase deficiency (MIM 229100) and familial hypercholanemia (MIM 607748), respectively, and heterozygous carriers have not been described as exhibiting ALS. Taken together, the results pointed to c.2780G>A in ERBB4 as the most likely pathogenic mutation.
We used a direct nucleotide sequence analysis method to conduct mutational analysis of ERBB4 in 364 FALS and 818 SALS individuals by using an ABI 3100 sequencer and BigDye Terminator ver3.1 (Applied Biosystems). We used the ExonPrimer website to design oligonucleotide primers (Table S3). The mutation c.2780G>A was also identified in one Canadian FALS individual (Figure 1B). Unfortunately, DNA from other family members was not available to confirm segregation. To investigate a possibility that the c.2780G>A mutation identified in the Japanese and Canadian families is a common founder mutation, we compared the haplotypes with the c.2780G>A mutation in ERBB4 of the Japanese and Canadian families (Figure S2). Different SNPs were observed 14 kbp and 5 kbp centromeric and telomeric to the mutation, respectively, indicating that disease haplotypes of the Japanese and Canadian families are different and that mutation occurred independently. We identified a de novo mutation of c.3823C>T (dbSNP SubSNP ID ss831884246), substituting tryptophan for arginine at codon 1275 (p. Arg1275Trp), in a Japanese SALS individual (Figure 1B) in whom a biological parent-descendant relationship was confirmed (Table S4) by the PLINK10 algorithm. These mutations were neither present in the 477 Japanese controls nor registered in the in-house database containing 41 whole genomes and 1408 exomes, the 1000 Genomes database, or the NHLBI-ESP database, containing 6503 exomes. Furthermore, c.2780G>A was not present in 190 Canadian controls. The identification of c.2780G>A in two independent families of different ethnic backgrounds strongly supported c.2780G>A as the causative mutation for ALS. Given that de novo mutation rates have been estimated to be 1.20 × 10−8 per nucleotide per generation11 and less than one nonsynonymous single-nucleotide variant (SNV)/generation,12 the observation of the de novo mutation further supports the idea that c.3823C>T is likely to be the causative mutation for ALS in this individual. The mutation’s substituted arginine residues, Arg927 and Arg1275, are highly conserved among species (Figure 1C), and the substitutions are predicted to be probably damaging by PolyPhen-2 analysis. The amino acid residue Arg927 resides in a tyrosine kinase domain, which is essential for the receptor tyrosine kinase activity, and Arg1275 is located in a C-terminal domain in the vicinity of multiple phosphorylation sites, which mediate downstream signaling pathways (Figure 1D). The clinical presentations of these ALS individuals with the ERBB4 mutations are summarized in Table 1. The common clinical characteristics of the individuals included both upper and lower motor-neuron involvement diagnosed as definite or probable ALS according to El Escorial and Airlie House revised criteria, relatively slow disease progression, and no obvious cognitive impairment. The individuals with the c.2780G>A mutation were characterized by relatively late onset (the ages at onset ranged from 60–70 years) and a slightly reduced penetrance. In contrast, the individual with the c.3823C>T mutation was characterized by early onset (45 years of age).
ErbB4 is a member of the epidermal growth factor (EGF) subfamily of receptor tyrosine kinases (RTKs). It forms a homodimer or a heterodimer with ErbB2 or ErbB3 and is activated upon binding of neuregulins (NRGs) to the extracellular ligand-binding domain of ErbB4.13 Activation of ErbB4 is mediated by increased tyrosine kinase activity upon NRG binding, resulting in autophosphorylation of the C-terminal tail.14 To determine how the two mutations identified in the ALS individuals affect ErbB4 functions, we investigated the autophosphorylation of ErbB4 in cells expressing either wild-type or mutant (c.2780G>A or c.3823C>T) ERBB4 in the presence of NRG-1. The ERBB4 mutations were introduced into the pBABE-puroERBB4JM-aCYT-2HA plasmid encoding HA-tagged ErbB4 JM-a CYT-215 by site-directed mutagenesis according to the protocol described in the Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific). After mutagenesis, all the constructs were verified by sequencing. The plasmids were transiently transfected into COS-7 cells via FuGENE 6 transfection reagent (Roche) in accordance with the manufacturer’s instructions. Transfected cells were starved of serum overnight and stimulated with 0 or 50 ng/ml NRG-1 (R&D Systems) for 10 min at 37°C. After stimulation, the cells were lysed, and samples equivalent to 50 μg of total protein were separated through 8% SDS-PAGE gels. For detectiin of ErbB4 phosphorylation and total ErbB4 protein levels, immunoblotting was performed antibodies against phospho-ErbB4 (Tyr1284) (Cell Signaling) and HA-tag (Abcam), respectively. The two amino acid substitutions, p. Arg927Gln and p. Arg1275Trp, showed a clearly reduced autophosphorylation of ErbB4 (Figure 2). On the basis of these genetic and functional data, we concluded that the two mutations are causative mutations for ALS (ALS19).
Figure 2.
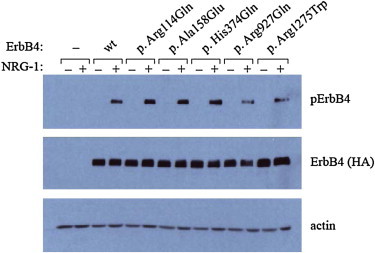
Functional Analysis of Wild-Type and Mutant ErbB4 upon Neuregulin-1 Stimulation
COS-7 cells transfected with an empty-vector control or plasmids encoding either wild-type (wt) or mutant HA-tagged ErbB4 (p. Arg114Gln, p. Ala158Glu, p. His374Gln, p. Arg927Gln, or p. Arg1275Trp) were stimulated with or without NRG-1, and the autophosphorylation activity of ErbB4 was analyzed by immunoblot analysis with antibodies against phospho-ErbB4 (Tyr1284) (Cell Signaling) and HA tag (Abcam), respectively. For loading controls, immunoblotting was performed with an anti-actin antibody (Santa Cruz Biotechnology). Three amino acid substitutions, including p.Arg114Gln, p.Ala158Glu, and p.His374Gln (rs760369), identified through mutational analysis of FALS and SALS individuals, were included in autophosphorylation assay. The substitutions p.Arg114Gln and p.Ala158Glu were not considered to be relevant to ALS because neither recurrence nor cosegregation was confirmed.
This study revealed that a reduced autophosphorylation of ErbB4 upon NRG-1 stimulation is involved in the pathogenesis of ALS. Erbb4 is specifically expressed in the soma of large motor neurons of the rat spinal cord.16 The lack of Erbb4 is embryonically lethal in mice, which displayed the derangement of motor-neuron axon guidance and pathfinding during embryogenesis.17 Heterozygous-null mice showed a reduced body weight and delayed motor development, and brain-specific conditional knock-out mice demonstrated reduced spontaneous motor activity and grip strength of the hindlimbs.18 Mice lacking cysteine-rich domain (CRD) isoforms of Nrg-1 (CRD-NRG-1−/−) die perinatally as a result of respiratory failure, lack detectable limb movement, and exhibit a loss of ∼60% of spinal motor neurons.19 Similarly, motor and sensory neuron-specific conditional Nrg-1 knockout mice die at birth and showed marked retraction of motor-neuron axons.20 Furthermore, a decrease in the amount of CRD-NRG-1 has been detected in the spinal motor neurons in FALS and SALS individuals and Sod1 mutant mice at disease onset,21 raising the possibility that disruption of the NRG-ErbB pathway is commonly involved in the motor-neuron degeneration underlying ALS. This study provides insight into ALS pathogenesis and is expected to pave the way for the development of innovative therapeutic strategies such as using NRGs or their agonists to upregulate ErbB4 functions.
Consortia
Consortium members of JaCALS include Ryoichi Nakamura, Hazuki Watanabe, Yuishin Izumi, Ryuji Kaji, Mitsuya Morita, Kotaro Ogaki, Akira Taniguchi, Ikuko Aiba, Koichi Mizoguchi, Koichi Okamoto, Kazuko Hasegawa, Masashi Aoki, Akihiro Kawata, Imaharu Nakano, Koji Abe, Masaya Oda, Masaaki Konagaya, Takashi Imai, Masanori Nakagawa, Takuji Fujita, Hidenao Sasaki, and Masatoyo Nishizawa.
Acknowledgments
We thank all the family members for participating in this study. This study was supported in part by KAKENHI (Grants-in-Aid for Scientific Research on Innovative Areas [22129001 and 22129002]) to S.T.; the Global COE Program from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and a grant-in-aid (H23-Jitsuyoka [Nanbyo]-Ippan-004) from the Ministry of Health, Labour, and Welfare, Japan to S.T. We acknowledge support to R.H.B. from ALS Therapy Alliance, Project ALS, P2ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, the ALS Family Charitable Foundation, and grant 1R01NS050557 from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health and support to G.A.N. from the MND Research Institute of Australia. P.G.-P. was supported by the Alfonso Martin Escudero Foundation (Madrid).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project Database, http://www.1000genomes.org/
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
NCBI37/hg19 assembly, http://genome.ucsc.edu/
NHLBI GO Exome Sequencing Project (NHLBI-ESP), https://esp.gs.washington.edu/drupal
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Personal genome databases, http://www.sequenceontology.org/resources/10Gen.html
PLINK algorithm, http://pngu.mgh.harvard.edu/purcell/plink/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
UCSC Human Genome Browser, http://genome.ucsc.edu/
Accession Numbers
The dbSNP accession numbers for the c. 2780G>A and c. 3823C>T mutations reported for ERBB4 in this paper are ss831884245 and ss831884246, respectively.
References
- 1.Al-Chalabi A., Jones A., Troakes C., King A., Al-Sarraj S., van den Berg L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124:339–352. doi: 10.1007/s00401-012-1022-4. [DOI] [PubMed] [Google Scholar]
- 2.Andersen P.M., Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi Y., Seki N., Ishiura H., Mitsui J., Matsukawa T., Kishino A., Onodera O., Aoki M., Shimozawa N., Murayama S. Development of a high-throughput microarray-based resequencing system for neurological disorders and its application to molecular genetics of amyotrophic lateral sclerosis. Arch. Neurol. 2008;65:1326–1332. doi: 10.1001/archneur.65.10.1326. [DOI] [PubMed] [Google Scholar]
- 4.Ishiura H., Takahashi Y., Mitsui J., Yoshida S., Kihira T., Kokubo Y., Kuzuhara S., Ranum L.P., Tamaoki T., Ichikawa Y. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch. Neurol. 2012;69:1154–1158. doi: 10.1001/archneurol.2012.1219. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda Y., Nakahara Y., Date H., Takahashi Y., Goto J., Miyashita A., Kuwano R., Adachi H., Nakamura E., Tsuji S. SNP HiTLink: a high-throughput linkage analysis system employing dense SNP data. BMC Bioinformatics. 2009;10:121. doi: 10.1186/1471-2105-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 10.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plowman G.D., Green J.M., Culouscou J.M., Carlton G.W., Rothwell V.M., Buckley S. Heregulin induces tyrosine phosphorylation of HER4/p180erbB4. Nature. 1993;366:473–475. doi: 10.1038/366473a0. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter G. ErbB-4: mechanism of action and biology. Exp. Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 15.Sundvall M., Korhonen A., Vaparanta K., Anckar J., Halkilahti K., Salah Z., Aqeilan R.I., Palvimo J.J., Sistonen L., Elenius K. Protein inhibitor of activated STAT3 (PIAS3) protein promotes SUMOylation and nuclear sequestration of the intracellular domain of ErbB4 protein. J. Biol. Chem. 2012;287:23216–23226. doi: 10.1074/jbc.M111.335927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearson R.J., Jr., Carroll S.L. ErbB transmembrane tyrosine kinase receptors are expressed by sensory and motor neurons projecting into sciatic nerve. J. Histochem. Cytochem. 2004;52:1299–1311. doi: 10.1177/002215540405201006. [DOI] [PubMed] [Google Scholar]
- 17.Gassmann M., Casagranda F., Orioli D., Simon H., Lai C., Klein R., Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 18.Golub M.S., Germann S.L., Lloyd K.C.K. Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav. Brain Res. 2004;153:159–170. doi: 10.1016/j.bbr.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Wolpowitz D., Mason T.B., Dietrich P., Mendelsohn M., Talmage D.A., Role L.W. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- 20.Yang X., Arber S., William C., Li L., Tanabe Y., Jessell T.M., Birchmeier C., Burden S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 21.Song F., Chiang P., Wang J., Ravits J., Loeb J.A. Aberrant neuregulin 1 signaling in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2012;71:104–115. doi: 10.1097/NEN.0b013e3182423c43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.