

Abstract
Background
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by chronic nonmalignant lymphoproliferation, accumulation of double-negative T cells, hypergammaglobulinemia G and A, and autoimmune cytopenia.
Objectives
Although mostly associated with FAS mutations, different genetic defects leading to impaired apoptosis have been described in patients with ALPS, including the FAS ligand gene (FASLG) in rare cases. Here we report on the first case of complete FAS ligand deficiency caused by a homozygous null mutant.
Methods
Double-negative T-cell counts and plasma IL-10 and FAS ligand concentrations were determined as ALPS markers. The FASLG gene was sequenced, and its expression was analyzed by means of Western blotting. FAS ligand function was assessed based on reactivation-induced cell death.
Results
We describe a patient born to consanguineous parents who presented with a severe form of ALPS caused by FASLG deficiency. Although the clinical presentation was compatible with a homozygous FAS mutation, FAS-induced apoptosis was normal, and plasma FAS ligand levels were not detectable. This patient carries a homozygous, germline, single-base-pair deletion in FASLG exon 1, leading to a premature stop codon (F87fs x95) and a complete defect in FASLG expression. The healthy parents were each heterozygous for the mutation, confirming its recessive trait.
Conclusion
FAS ligand deficiency should be screened in patients presenting with ALPS features but lacking the usual markers, including plasma soluble FAS ligand and an in vitro apoptotic defect. An activation-induced cell death test could help in discrimination.
Keywords: Autoimmune lymphoproliferative syndrome, Fas ligand, diagnosis
Patients with autoimmune lymphoproliferative syndrome (ALPS)1-3 present with early-onset, chronic, nonmalignant polyclonal lymphoproliferation that is usually associated with autoimmune features and increased susceptibility to malignancies.4 The biological features of ALPS include hypergammaglobulinemia, increased soluble plasma FAS ligand5 and IL-106 levels, and the specific accumulation of double-negative (DN) T cells (T-cell receptor αβ–positive CD4−CD8− T cells).7 This disorder of lymphocytic homeostasis was first identified in lpr and gld mice presenting with fas and faslg mutations, respectively.8 FAS (also known as CD95, Apo1, and TNFRSF6) is a member of the TNF receptor superfamily. It is essential for apoptosis of chronically activated lymphocytes and prevention of the accumulation of self-reactive T and B lymphocytes.9 Oligomerization of FAS is induced by its interaction with the FAS ligand (CD178) and leads to formation of a death-inducing signaling complex. The death-inducing signaling complex then triggers a caspase activation cascade in permissive cells.10
In human subjects approximately two thirds of cases of ALPS are associated with germline (ALPS-FAS)3,11-15 or somatic (ALPS-sFAS)16 heterozygous FAS mutations or combined germline and somatic mutations.17 However, no mutations are found in the remaining third of patients.13 Homozygous and heterozygous FAS mutations are associated with severe and milder phenotypes, respectively. A FASLG mutation has been identified in just 3 cases of ALPS (ALPS-FASLG). In 2 American patients heterozygous FASLG mutations (1 missense mutation and 1 in-frame deletion) caused systemic lupus erythematosus or mild ALPS through a dominant negative effect.18,19 In a third case a Spanish patient carried a homozygous, autosomal recessive, FASLG missense mutation. This mutation impaired FAS ligand function, despite normal FAS ligand expression.20 Here we report on the first case of a complete FAS ligand deficiency (detected in a patient hailing from the Indian subcontinent).
METHODS
The patient
Blood samples were obtained from the patient and his relatives after the provision of informed written consent and in accordance with the principles of the Declaration of Helsinki.
Determination of the proportion of DN T cells and the plasma IL-10 and Fas ligand concentrations
These 3 ALPS markers were determined as described elsewhere.5
Apoptosis assays
Apoptosis and activation-induced cell death (AICD) assays were performed at days 9 and 12 on T cells that had been activated for 2 days with toxic shock syndrome toxin 1 (TSST-1; 1 μg/mL) and then cultured with IL-2 (100 IU/mL), as previously described.21 In the apoptosis assay T cells were cultivated with 10 or 100 ng/mL of the agonistic anti-Fas antibody Apo1.3. Apo1.3 was then cross-linked with a rabbit anti-mouse antibody (10 μg/mL). For the AICD assay, cells were cultured with 1 μg/mL TSST-1. Cell death was detected by means of hypodiploid nuclei quantification by using fluorescence-activated cell-sorting analysis after propidium iodide staining.
DNA isolation and sequencing
DNA was isolated from whole blood by using standard methods (proteinase K digestion and phenol-chloroform extraction). The FASLG gene’s 4 exons were amplified and sequenced according to standard methods and by using the pairs of primers listed in Table I.
TABLE I.
Primers used for DNA and cDNA amplification and sequencing
| Forward | Reverse | |
|---|---|---|
| Genomic | ||
| FASLG | ||
| Exon 1 | GTCAGCAACAGGGTCCCGTCC | TTGAAAAGCACTTT GCAAGCC |
| Exon 2 | CAGGGCTTGGTTTATTTGACG | AAGCAGCTCAAGGA ATCATAG |
| Exon 3 | TTAGACTGTTGCCATTTACGG | TCTACAAGTGCCAG GATTGAG |
| Exon 4 | CTTACTGCAATGGATTACGGG | AGAGTTCTATGTTCT TCCGTC |
| cDNA | ||
| FASLG | CTTGGTAGGATTGGGCCTGG | TGCAAGATTGACCC CGGAAG |
| HPRT | GGGAGGCCATCACATTGTAGC | CTGGCGATGTCAAT AGGACTC |
HPRT, Hypoxanthine phosphoribosyltransferase.
RNA isolation and cDNA synthesis
Total RNA was extracted by using standard methods. cDNA synthesis was performed as described elsewhere.17 The cDNA was then amplified by means of PCR with the primers listed in Table I.
Western blot analysis
Proteins were extracted from 3 million activated T cells in standard radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors. Lysates were resolved with 12% SDS-PAGE and transferred onto a polyvinylidine difluoride membrane (Immobilon P; Millipore SAS, Molsheim, France). FAS ligand protein was detected by using overnight incubation with a murine anti-human FAS ligand antibody (clone G247-4). The immunoblot was developed by using the ECL Plus Western Blotting Detection System (GE Healthcare, Velizy, France). After stripping, the membrane was probed with a rabbit anti-human glyceraldehyde-3-phosphate dehydrogenase antibody (clone 14C10).
RESULTS
Clinical and immunologic presentation
At 2 months of age, the patient (born to consanguineous parents) presented with abdominal distension (since birth), repeated episodes of fever, and upper respiratory tract infections. At 8 months, he presented with massive hepatosplenomegaly (with the liver and spleen extending 7-8 cm and 12-14 cm below the costal margin, respectively; Fig 1, A), multiple small cervical lymph nodes (1-3 cm), and bilateral lung infiltrates with right upper lobe consolidation. His white blood cell (WBC) count was 157,000/mm3, 90% of which were lymphocytes. Investigations aimed at ruling out acute leukemia were inconclusive. Height and weight were both less than the third percentile. The child was readmitted a month later (ie, at the age of 9 months) for right upper lobe pneumonia. He was treated with intravenous antibiotics for 7 days and then discharged. Mycophenolate mofetil (MMF) was introduced at this time in view of an increased total WBC count of 80,700/mm3. Two months later, the patient (aged 11 months) had fever and abdominal distension. A chest radiograph found enlarged mediastinal lymph nodes (Fig 1, B). A computed tomographic scan revealed multiple enlarged retroperitoneal lymph nodes. We suspected MMF toxicity and withdrew the drug. After 3 months of follow-up, the child (aged 14 months) was found to have a liver and spleen extending 10 and 12 cm below the costal margin, respectively. The total WBC count was increased (51,500/mm3). Hence MMF (300 mg/m2/d) was reintroduced.
FIG 1.

Massive tumor syndrome in the patient with ALPS-FASLG. A, At 8 months of age, the patient showed massive hepatosplenomegaly. B, A chest radiograph obtained at the age of 11 months highlighted mediastinal enlargement caused by persistent lymph nodes.
At the time of writing, the patient has been on MMF for almost 11 months. He continues to have growth restriction. The child’s liver and spleen extend 5 and 6 cm below the costal margin, respectively, and the WBC count is 24,000/mm3. No clinical manifestation of autoimmunity has been observed. Although all relatives were asymptomatic, the family’s medical history indicated that 3 other siblings had died very early (in utero and 3 days and 7 months after birth, respectively) from unknown causes (Fig 2).
FIG 2.
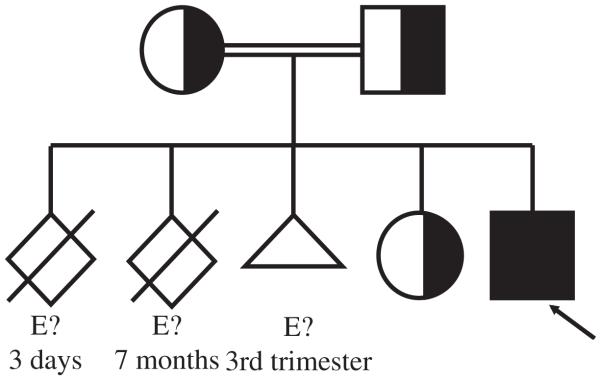
Family pedigree of the patient with ALPS-FASLG. E?, DNA material unavailable.
Severe ALPS phenotype associated with a homozygous FASLG mutation
The patient exhibited a massively increased WBC count (mostly composed of DN T cells) and markedly increased serum IgG levels (Table II). However, soluble FAS ligand could not be detected in the patient’s plasma (in contrast to the situation in patients with ALPS-FAS), suggesting that the protein was either not secreted or not recognized by the antibody used in the ELISA. We thus screened for FASLG mutations using direct gene sequencing (Fig 3, A). The patient exhibited a homozygous single-base-pair deletion in exon 1 (259 del T). The frameshift resulted in a premature stop codon (F87fsx95), leading to a short protein devoid of a transmembrane region and resulting in a null mutant. This mutation was not found in databases (National Center for Biotechnology Information, 1000 genomes). The patient’s parents (Fig 3, A) and a sister (data not shown) were found to be heterozygous for this mutation and are all healthy. Accordingly, the increased plasma IL-105,6 and vitamin B1222 concentrations usually associated with ALPS were found in the patient but not in his healthy relatives (Table II). Moreover, soluble CD25 concentrations was also markedly increased in the patient (Table II), as previously described by Del-Rey et al20 in a homozygous patient.
TABLE II.
Biological characteristics of the patient and his relatives
| Mother | Father | Adult normal values | Sister | Patient | Normal value | |
|---|---|---|---|---|---|---|
| Clinical phenotype | ||||||
| Age (mo) | NA | NA | NA | 14 | ||
| Age at onset (mo) | – | – | – | Birth | ||
| Splenomegaly | – | – | – | +++* | ||
| Hepatomegaly | – | – | – | +++* | ||
| Lymphadenopathy | – | – | – | +* | ||
| Autoimmunity | ||||||
| Clinical features | None | None | None | None | ||
| Autoantibodies (anti-) | ND | ND | ND | None | ||
| Immunologic phenotype | ||||||
| WBC count (/mm3) | 5,200 | 4,700 | 4,000-10,000 | 10,300 | 51,500 | 8,000-12,000 |
| Lymphocyte count (/mm3) | 3,500 | 3,300 | 1,000-4,000 | 6,100 | 40,700 | 3,500-5,000 |
| Monocyte count (/mm3) | 400 | 500 | 200-700 | 700 | 5,800 | 200-1,000 |
| Granulocyte count (/mm3) | 1,300 | 900 | 1,700-7,500 | 3,500 | 5,000 | 3,500-6,000 |
| Hemoglobin concentration (g/dL) | 11.6 | 11.2 | 12-18 | 10.6 | 6.2 † | 11-15 |
| IgG (g/L): | 16.7 | 17.5 | 6.7-12.8 | 16 | 38.1 | 4.5-8 |
| IgA (g/L): | 2.48 | 1.74 | 0.7-3.5 | 1.76 | 2.47 | 0.4-1.22 |
| IgM (g/L): | 1.09 | 1.08 | 0.5-1.5 | 0.96 | 1.72 | 0.5-1.5 |
| DN T cells (% of TCRαβ+ cells) | 1.5 | 4 | <3 | 2.5 | 87 | <3 |
| Biological phenotype | ||||||
| Plasma Fas ligand (ng/mL) | <0.07 | <0.07 | <0.2 | <0.07 | <0.07 | <0.2 |
| Plasma IL-10 (pg/mL) | <4 | <4 | <20 | <4 | 330 | <20 |
| Plasma vitamin B12 (pg/mL) | 220 | 248 | 299 | 1,890 | 220-1,100 | |
| Plasma CD25 (pg/mL) | ND | ND | ND | 17,000 | 880 ± 110 | |
Values in boldface indicate the major variations.
NA, Not available; ND, not determined; TCR, T-cell receptor.
Described in the Results section.
Direct Coombs negative.
FIG 3.

The FASLG mutation and its effect on the expression and function of FAS ligand protein. A, Sequencing profiles of genomic FASLG exon 1 obtained from blood cells. B and C, RT-PCR products of FASLG and hypoxanthine phosphoribosyltransferase (HPRT) amplified from cDNA (Fig 3, B) or immunoblots of FAS ligand and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from protein lysates of activated T cells reactivated (+) or not (−; Fig 3, C). D, A functional apoptosis assay was performed on activated T cells from a healthy control subject, the patient, the patient’s relatives, and a patient with ALPS carrying a homozygous null FAS mutation (ALPS-FAS-HMZ). The histogram is representative of 3 independent experiments.
FASLG expression defect
To assess the patient’s FASLG gene expression at the transcription level, we first performed RT-PCR analysis on RNA extracted from activated T cells. We found that the level of FASLG RNA synthesis (measured against the ubiquitously expressed hypoxanthine phosphoribosyltransferase [HPRT] gene) was slightly lower than that observed in a control subject (Fig 3, B). We then determined the extent of FAS ligand protein expression by performing Western blotting on proteins extracted from activated T cells. In control cells FAS ligand expression was upregulated after 6 hours of reactivation with TSST-1 (Fig 3, C), as expected. In contrast, we observed a complete absence of FAS ligand protein expression by the patient’s T cells (both before and after TSST-1 reactivation), confirming that the genetic defect was a null mutation. Under the same experimental conditions, we observed slightly below-normal levels of FAS ligand protein in cells from relatives carrying the heterozygous FASLG mutation.
Functional apoptosis induced by agonistic anti-Fas antibody was normal, whereas reactivation-induced cell death was impaired
To determine the FASLG mutation’s effect on the FAS-FASLG signaling pathway, we performed an apoptosis assay on activated T cells (by using an agonistic anti-Fas antibody called Apo1.3) and a reactivation-induced cell death assay. Activated T cells from the patient and his parents exhibited control levels of apoptosis on FAS stimulation (Fig 3, D), thus confirming that the FAS pathway was intact. In contrast, AICD was profoundly lower than control values for the patient’s T cells and slightly lower for the parent’s T cells (Fig 3, D), indicating impaired FAS ligand–induced apoptosis. However, the AICD defect was partial. Approximately 30% of the patient’s cells died, as seen for T cells from a patient with ALPS-FAS carrying a null homozygous FAS mutation (Fig 3, D). These results indicated that a FAS-FASLG–independent pathway accounts for the partial cell death of FAS ligand-deficient T cells, as has been previously described for FAS-deficient T cells.21
DISCUSSION
Although more than 200 cases of ALPS-FAS have been described to date, only 3 patients with ALPS-FASLG have previously been reported.18-20 Two patients carried heterozygous dominant FASLG mutations. The first FASLG mutation was associated with systemic lupus erythematosus and lymphadenopathy in an adult patient.19 Although the second patient presented with an ALPS phenotype, the mutation’s functional effect was determined in a cytotoxicity test, but the AICD functional test was not examined.18 More recently, a homozygous FASLG missense mutation has been reported in a girl presenting with typical ALPS symptoms.20 The mutation herein described is the sole example of a complete FAS ligand expression defect (ie, a null mutation). The lack of clinical features in heterozygous relatives suggests autosomal recessive inheritance; this is consistent with a lower frequency of ALPS-FASLG than ALPS-FAS. Comparisons of these cases are depicted in Fig 4.
FIG 4.

Schema of the location of the 4 FASLG mutations described to date in human subjects and comparison of biological characteristics of the 4 patients. Heterozygous and homozygous mutations are depicted in blue or red, respectively. The table summarizes the main biological characteristics of the 4 patients. SLE, Systemic lupus erythematosus; TM, transmembrane domain.
The 2 homozygous patients presented with early-onset, severe clinical manifestations of ALPS, as seen in patients carrying the homozygous FAS mutation.2 In contrast, the reported heterozygous cases of FASLG mutations were associated with variable phenotypes, suggesting that modifying factors might be involved (again, as in cases of ALPS-FAS, cases of heterozygous mutations and partial clinical penetrance). The 4 described FASLG mutations were found in variable domains (Fig 4) and did not suggest phenotype-genotype correlation. Given the few examples of FASLG mutations, it is reasonable to assume that the latter are either recessive or dominant with very low penetrance.
The patients with homozygous ALPS-FASLG lacked increases in soluble FAS ligand levels (Table II and data not shown). However, they both exhibited plasma IL-10, vitamin B12, and soluble CD25 concentration increase (Table II). Our present findings confirmed that FASLG genetic defects should be screened for in typical ALPS presentations lacking FAS mutations associated with some ALPS markers (increased DN T-cell counts, IL-10, vitamin B12, and soluble CD25 concentrations) but eventually without increased plasma soluble FAS ligand.
Of note, AICD was impaired in patients with ALPS-FASLG (present work).18-20 These data highlight the usefulness of this functional assay combined with a Fas-induced apoptosis assay to discriminate cases of ALPS-FAS and ALPS-FASLG.
Clinical implications: We report on a severe ALPS phenotype associated with a homozygous FASLG null mutation. ALPS markers as a plasma soluble FAS ligand and apoptosis defect are lacking, but the AICD assay helped for diagnosis.
Acknowledgments
Supported by grants from INSERM, Agence Nationale de la Recherche (ANR; grant no. 08-GENO-015-01), the E-Rare 2007 programme (EPINOSTICS), the Ligue National Contre le Cancer, the European STREP (AUTOROME), and an advanced grant from the European Research Council (ERC). A.M.-C. was funded by the European STREP, the Centre de Référence des Déficits Immunitaires Héréditaires (CEREDIH) and the ERC. N.L. was awarded a doctoral fellowship by INSERM.
We thank Manuel Del-Rey and Luis Allende for providing the plasma of the first described patient with homozygous FASLG mutation and Stéphanie Vicca for assessment of serum immunoglobulin levels.
Abbreviations used
- AICD
Activation-induced cell death
- ALPS
Autoimmune lymphoproliferative syndrome
- DN
Double-negative
- FASLG
FAS ligand gene
- MMF
Mycophenolate mofetil
- TSST-1
Toxic shock syndrome toxin 1
- WBC
White blood cell
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Canale VC, Smith CH. Chronic lymphoadenopathy simulating malignant lymphoma. J Pediatr. 1967;70:891–9. doi: 10.1016/s0022-3476(67)80262-2. [DOI] [PubMed] [Google Scholar]
- 2.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and auto-immunity. Science. 1995;268:1347–9. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 3.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 4.Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 5.Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, et al. FAS-L, IL-10, and double-negative CD4- CD8- TCR alpha/beta1 T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–30. doi: 10.1182/blood-2008-09-179630. [DOI] [PubMed] [Google Scholar]
- 6.Lopatin U, Yao X, Williams RK, Bleesing JJ, Dale JK, Wong D, et al. Increases in circulating and lymphoid tissue interleukin-10 in autoimmune lymphoproliferative syndrome are associated with disease expression. Blood. 2001;97:3161–70. doi: 10.1182/blood.v97.10.3161. [DOI] [PubMed] [Google Scholar]
- 7.Bleesing JJ, Brown MR, Dale JK, Straus SE, Lenardo MJ, Puck JM, et al. TcR-alpha/beta(1) CD4(−)CD8(−) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol. 2001;100:314–24. doi: 10.1006/clim.2001.5069. [DOI] [PubMed] [Google Scholar]
- 8.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 9.Hao Z, Duncan GS, Seagal J, Su YW, Hong C, Haight J, et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29:615–27. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–92. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Worth A, Thrasher AJ, Gaspar HB. Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol. 2006;133:124–40. doi: 10.1111/j.1365-2141.2006.05993.x. [DOI] [PubMed] [Google Scholar]
- 12.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, et al. Clinical, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–8. [PubMed] [Google Scholar]
- 13.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–33. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 14.Kasahara Y, Wada T, Niida Y, Yachie A, Seki H, Ishida Y, et al. Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol. 1998;10:195–202. doi: 10.1093/intimm/10.2.195. [DOI] [PubMed] [Google Scholar]
- 15.Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–14. doi: 10.1086/302333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–18. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 17.Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 2011;121:106–12. doi: 10.1172/JCI43752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi LL, Pan G, Atkinson TP, Zheng L, Dale JK, Makris C, et al. Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) Type Ib. BMC Med Genet. 2007;8:41. doi: 10.1186/1471-2350-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;98:1107–13. doi: 10.1172/JCI118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del-Rey M, Ruiz-Contreras J, Bosque A, Calleja S, Gomez-Rial J, Roldan E, et al. A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood. 2006;108:1306–12. doi: 10.1182/blood-2006-04-015776. [DOI] [PubMed] [Google Scholar]
- 21.Mateo V, Menager M, de Saint-Basile G, Stolzenberg MC, Roquelaure B, Andre N, et al. Perforin-dependent apoptosis functionally compensates Fas deficiency in activation-induced cell death of human T lymphocytes. Blood. 2007;110:4285–92. doi: 10.1182/blood-2007-05-088286. [DOI] [PubMed] [Google Scholar]
- 22.Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125:946–9. e6. doi: 10.1016/j.jaci.2009.12.983. [DOI] [PMC free article] [PubMed] [Google Scholar]