

Summary
Ca2+ mobilization and cytoskeletal reorganization are key hallmarks of T-cell activation, and their interdependence has long been recognized. Recent advances in the field have elucidated the molecular pathways that underlie these events and have revealed several points of intersection. Ca2+ signaling can be divided into two phases: initial events leading to release of Ca2+ from endoplasmic reticulum stores, and a second phase involving STIM 1 (stromal interaction molecule 1) clustering and CRAC (calcium-release activation calcium) channel activation. Cytoskeletal dynamics promote both phases. During the first phase, the actin cytoskeleton promotes T-cell receptor mechanotransduction and serves as a dynamic scaffold for microcluster assembly. Proteins that drive actin polymerization such as WASp (Wiskott-Aldrich syndrome protein) and HS1 (hematopoietic lineage cell-specific protein 1) promote signaling through PLCγ1 (phospholipase Cγ1) and release of Ca2+ from endoplasmic reticulum stores. During the second phase, the WAVE (WASP-family verprolin homologous protein) complex and the microtubule cytoskeleton promote STIM 1 clustering at sites of plasma membrane apposition, opening Orai channels. In addition, gross cell shape changes and organelle movements buffer local Ca2+ levels, leading to sustained Ca2+ mobilization. Conversely, elevated intracellular Ca2+ activates cytoskeletal remodeling. This can occur indirectly, via calpain activity, and directly, via Ca2+-dependent cytoskeletal regulatory proteins such as myosin II and L-plastin. While it is true that the cytoskeleton regulates Ca2+ responses and vice versa, interdependence between Ca2+ and the cytoskeleton also encompasses signaling events that occur in parallel, downstream of shared intermediates. Inositol cleavage by PLCγ1 simultaneously triggers both endoplasmic reticulum store release and diacylglycerol-dependent microtubule organizing center reorientation, while depleting the pool of phosphatidylinositol-4,5-bisphosphate, an activator of multiple actin regulatory proteins. The close interdependence of Ca2+ signaling and cytoskeletal dynamics in T cells provides positive feedback mechanisms for T-cell activation and allows for finely tuned responses to extracellular cues.
Keywords: Actin, myosin, microtubules, T cells, immunological synapse, calcium
Introduction
Calcium (Ca2+) signaling and cytoskeletal responses in T cells are functionally intertwined in complex ways, creating feedback loops that promote T-cell activation and direct effector functions. Correlative evidence for a linkage between cytoskeletal remodeling and Ca2+ mobilization dates back to the earliest single-cell studies of T-cell activation. In the 1970s and 1980s, it was noted that target cell lysis required extracellular Ca2+ and entailed a programmed series of cell shape changes (reviewed in 1). Soon thereafter, T-cell receptor (TCR) engagement was shown to induce an increase in intracellular Ca2+ levels (2). Moreover, two distinct sets of cytoskeletal rearrangements were observed: first, antigen-presenting cell (APC) binding-induced the polymerization of F-actin and recruitment of actin binding proteins such as talin to the cell-cell contact site, and second, the T-cell microtubule organizing center (MTOC), associated Golgi complex, and lytic granules reoriented to face the APC. Causal linkage between Ca2+ mobilization and cytoskeletal remodeling was established by subsequent pharmacological studies. Using pharmacological agents to disrupt actin dynamics, several groups showed that TCR-induced polymerization of F-actin is needed for Ca2+ mobilization (3–5). Reciprocal experiments showed that Ca2+ responses are needed for cytoskeletal remodeling as well. Although initial studies showed that chelation of extracellular Ca2+ using EGTA does not inhibit TCR-induced polymerization of actin or recruitment of talin to the immunological synapse (IS) (1, 6), treatment of T cells with a combination of EGTA and BAPTA-AM to deplete both extracellular and intracellular Ca2+ showed that Ca2+ elevation is required for actin-dependent spreading (7). In addition, elevated intracellular Ca2+ was also found to be required for MTOC reorientation toward the APC (8).
Recent years have seen considerable progress in our understanding of the mutual regulation of cytoskeletal remodeling and Ca2+ signaling during T-cell activation. Many of the key proteins that control both actin dynamics and Ca2+ mobilization have been identified, making it possible to define specific points where the two pathways intersect. Moreover, as we begin to grasp how the cytoskeletal network functions as a unit, we are gaining new insights into the role of cell shape changes, mechanotransduction and other higher order signaling events. In this review, we address the mechanisms underlying the complex interaction between cytoskeletal reorganization and Ca2+ signaling in T cells and highlight important areas for future investigation.
General features of calcium signaling
To understand the interplay between cytoskeletal dynamics and Ca2+ signaling, it is important to review the mechanisms by which TCR engagement leads to Ca2+ mobilization. Broadly speaking, the T cell Ca2+ signals reflect two distinct but interrelated processes: triggering Ca2+ release from endoplasmic reticulum (ER) stores, and activation of calcium release-activated calcium (CRAC) channels (Orai1) in the plasma membrane (PM) (Fig. 1). Release of Ca2+ from the ER by TCR engagement results from the formation of sub-micron scale signaling microclusters (MCs), enriched in TCRs as well as kinases and adapter proteins (9). The early tyrosine phosphorylation events that take place within MCs have been reviewed extensively elsewhere (10, 11). Briefly, the Src kinase Lck phosphorylates inducible tyrosine activation motifs (ITAMs) on the ζ chains of the receptor complex, which serve as docking sites for the Syk kinase ζ-chain-associated protein of 70 kDa (Zap70). Zap70 then phosphorylates linker for activation of T cells (LAT) and SH2 domain-containing leukocyte protein of 76 kDa (SLP-76). Cooperative assembly of these and other MC components culminates in the recruitment and subsequent activation of phospholipase C γ1 (PLCγ1). Upon activation, PLCγ1 cleaves phosphotidylinositide 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG activates the Ras pathway, while IP3 triggers its receptors in the ER membrane, leading to Ca2+ release from ER stores. This early phase of Ca2+ mobilization ensues within seconds of TCR engagement and requires engagement of only a few TCRs (12). ER stores are emptied within seconds, and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps in the ER membrane immediately begin to return cytoplasmic Ca2+ to the ER. Thus, on its own, this phase represents a transient and relatively small rise in intracellular Ca2+, lasting on the order of minutes.
Fig. 1. F-actin remodeling and Ca2+ mobilization downstream of TCR signaling.

TCR stimulation by peptide-loaded MHC triggers Lck-mediated phosphorylation of ITAMs in the intracellular regions of the TCR complex. Once phosphorylated, these sites recruit Zap70, which phosphorylates LAT and subsequently SLP-76. These adapter proteins cooperatively serve as a docking site for PLCγ1. SLP-76 also recruits two other key regulators of downstream signaling: Vav1 and Itk. Vav1 promotes F-actin polymerization by activating Rac and Cdc42, which in turn activate WAVE and WASp. Itk phosphorylates and activates PLCγ1, which then cleaves PIP2, generating DAG and IP3. IP3 binds to IP3 receptors on the ER membrane, inducing the release of Ca2+ from ER stores. This process is opposed by SERCA pumps, which refill ER stores on an ongoing basis. ER store depletion triggers oligomerization of STIM 1 in the ER membrane, which facilitates STIM 1 delivery to the plasma membrane at specialized sites of ER-PM apposition. There, STIM 1 interacts with and activates the CRAC channel Orai1 to allow the influx of extracellular Ca2+ into the cytosol. Sustained Ca2+ mobilization activates the phosphatase calcineurin, which then activates NFAT and allows its shuttling into the nucleus to initiate T-cell reprogramming at the level of gene expression.
Release of Ca2+ from the ER initiates events that lead to a more prolonged increase in cytoplasmic Ca2+ levels. IP3-induced depletion of Ca2+ from ER stores and the resulting decrease in ER Ca2+ concentration leads to dissociation of Ca2+ from N-terminal EF hand domains of stromal interaction molecule 1 (STIM 1), a Ca2+ sensor that spans the ER membrane (13). A subsequent conformational change in STIM 1 leads to its oligomerization and subsequent delivery to the sites of ER-PM juxtaposition, probably through its interaction with microtubule +TIP tracking proteins (14, 15). There, STIM 1 associates with trans-membrane Orai1 (CRAC) channels and activates them. This allows the entry of Ca2+ from the extracellular space via a process termed store-operated calcium entry (SOCE). The resulting sustained elevation of cytoplasmic Ca2+ is responsible for the activation of T-cell transcriptional machinery. A key target of elevated intracellular Ca2+ is calcineurin, which dephosphorylates the transcription factor NFAT, allowing its translocation to the nucleus (16). Once in the nucleus, NFAT promotes transcription of interleukin-2 (IL-2) and other proteins that lead to T-cell activation.
The two phases of T-cell Ca2+ mobilization are not temporally segregated. Thus, during the initial rise in Ca2+ due to release from the ER, extracellular Ca2+ enters via Orai1 and ‘floods’ the cytoplasm. Without ongoing TCR signaling leading to continued IP3 receptor activation, SERCA pumps in the ER membrane quickly replenish ER stores, STIM 1 disengages Orai1 and relocalizes away from the PM, and Orai1 channels close (17, 18). In support of the requirement for ongoing signaling leading to ER store release, disruption of PLCγ1 activation during the sustained phase of signaling correlates with a concomitant drop in intracellular Ca2+ levels (19). The finely tuned nature of this 2-phase system becomes apparent when one considers the magnitude of changes in the intracellular Ca2+ concentration upon TCR triggering. In resting T cells, the cytosolic Ca2+ concentration is 50–100 nM, while Ca2+ concentrations in the ER and the extracellular space are 800–1000 μM and 2 mM, respectively. Upon TCR engagement, cytoplasmic Ca2+ levels rise 10-fold, to ~1 μM. Efficient Ca2+ influx is facilitated by the steep concentration gradient across the PM (20). As discussed below, cytoskeletal dynamics play a key role in positioning signaling molecules and organelles to modulate this process.
Actin function in TCR signaling
Actin dynamics are intimately involved in basic mechanical interactions and spatio-temporal control of signaling events leading to Ca2+ mobilization. Actin promotes early steps of TCR signaling at two levels: via effects on the TCR itself, and via the assembly of MCs that transduce and amplify TCR signals. In both cases, the actin cytoskeleton is not just a static scaffold or a conventional link in a chain of signaling events. Instead, actin exerts forces and orchestrates molecular movements needed for Ca2+ signaling.
Initial TCR triggering
In mature T cells, TCR engagement leads to association of phosphorylated ITAMs in the TCR complex with the actin cytoskeleton (21, 22). Moreover, the CD3 complex reportedly binds to Nck, a component of a protein complex that drives actin polymerization (23). Linkage of the TCR complex to actin filaments is almost certainly indirect, and the exact molecular mechanisms remain controversial. Nonetheless, there is broad consensus that centripetal TCR movement at the IS is driven by actin dynamics (24–26). The dynamic association of the TCR complex with the actin cytoskeleton has two important implications for signaling events leading to Ca2+ mobilization. First, the actin-dependent movement of the TCR impacts the kinetics of TCR-pMHC interactions. Recent studies have shown that the outcome of TCR engagement is controlled by receptor-ligand kinetics, rather than by t1/2 or KD of antigen/TCR binding alone (27, 28), and ligands with fast on-rates can bind and rebind the same TCR several times. Thus, depending on the particular TCR-pMHC interaction, continuous movement of the TCR at the IS may either facilitate serial receptor encounters with rare agonist pMHCs or minimize opportunities for pMHC rebinding. Huppa et al. (29), demonstrated that the synaptic TCR-pMHC dissociation rate is decreased significantly upon treatment of T cells with actin depolymerizing agents, consistent with idea that actin-driven TCR movement promotes its dissociation from pMHC complexes. Since ligand mobility is an important variable in this model, it is important to point out that these experiments were done using stimulatory planar lipid bilayers where pMHC mobility is essentially unrestricted. Mobility of pMHC complexes and costimulatory ligands on the APC membrane is modulated by the APC cytoskeleton (our unpublished data). Thus, it will be important to determine to what extent this affects TCR-pMHC binding kinetics.
Another mechanism through which the actin cytoskeleton may directly affect TCR signaling involves mechanotransduction. Recent studies indicate that the TCR is a mechano-receptor that depends on physical force to propagate signals across the membrane (30, 31). Thus, interaction of the TCR complex with the actin cytoskeleton could promote TCR signaling through mechanical tension, produced by active cytoskeletal flow on the one side and ligand binding on the other (32–34). To account for the role of actin network flow in TCR signaling, Ma and Finkel (35, 36) have proposed the receptor deformation model. Building upon the earlier work showing that TCR stimulation is greatly increased by the immobilization of agonist pMHCs, they showed that effective TCR triggering depends on T-cell adhesion to the stimulatory surfaces and an intact T-cell cytoskeleton; lack of either of those factors precludes efficient Ca2+ mobilization. Based on this evidence, the authors postulated that actin flow at the T cell IS provides a force that is counteracted by molecular interactions at the T-cell-APC interface. The resulting tension on the TCR elicits structural changes within the complex to facilitate downstream signaling. The specific mechanism of TCR triggering is not fully understood (37, 38), and the contribution of force-induced TCR deformation is controversial. Arguably, ITAMs in the TCR complex are fully exposed without applied force, such that any role for mechanotransduction must lie downstream of the TCR itself. Nonetheless, the involvement of mechanical tension at some stage in TCR signaling is supported by evidence that depletion of F-actin abrogates signaling (39, 40). Moreover, work from our laboratory has shown that a static actin scaffold is insufficient to sustain TCR-induced Ca2+ signaling, pointing to the necessity for ongoing actin polymerization and/or centripetal flow of the branched actin network at the IS (19). Additional support for mechanical tension in T-cell signaling comes from studies of T cells interacting with TCR stimulatory beads, where Ca2+ mobilization is enhanced by moving the attached bead away from the IS (41).
One important and understudied question in this arena is the role played by the stimulatory APC. Ligand mobility and surface stiffness have both been implicated in modulating TCR signaling (32, 34, 42, 43). Consequently, determinants of these variables on APCs could significantly impact both receptor-ligand binding kinetics and mechanotransduction (27). Since there is evidence that actin is recruited to the dendritic cell side of the IS (44), it will be important to understand how the APC cytoskeleton impacts these aspects of T-cell activation.
TCR microcluster assembly and maintenance
In addition to its role in TCR triggering, the actin cytoskeleton regulates the assembly of TCR-proximal signaling complexes at the T-cell-APC interface. These complexes form MCs containing receptors, kinases, and adapter molecules, many of which contain actin-binding and actin-regulatory domains. Studies of the IS using surrogate planar stimulatory surfaces have greatly advanced our understanding of cytoskeletal function in the assembly and maintenance of signaling MCs. TCR MCs arise at initial sites of T-cell contact with stimulatory surfaces, concomitant with the initiation of intracellular Ca2+ signaling (45–47). Multiple actin-regulatory molecules are also recruited to these earliest sites of TCR signaling (24, 48). This process, in turn, induces T-cell spreading and formation of a well-defined lamellipodial region rich in branched actin filaments, and an inner lamellar region that contains prominent acto-myosin II bundles (19, 49, 50). The ongoing polymerization of actin at the cell periphery, coupled with the organizing forces generated by myosin II contractility, results in persistent actin centripetal flow at the IS. After maximal spreading has been achieved, nucleation of TCR MCs persists in the lamellar region and they are swept inward in parallel with the cytoskeletal flow (39, 45, 47, 51).
The actin cytoskeleton is essential for stabilizing newly formed MCs. Key evidence from the Dustin laboratory (39) showed that the integration of signaling molecules into the cytoskeletal scaffold greatly increases the lifetime of nascent MCs and promotes T-cell stimulation. When fully spread T cells were treated with the actin destabilizing agent Latrunculin A, the newly-formed peripheral MCs dissolved, while the mature central MCs persisted for more than 10 min after drug treatment, presumably because they were stabilized by higher order interactions among MC components. More recent work from the lab indicates that TCR engagement triggers the formation of TCR MC-associated actin patches, which are distinct from the lamellipodial actin pool (Kumari S, and Dustin ML, manuscript submitted). Presumably, this MC-associated F-actin pool is responsible for stabilizing newly-formed MCs, but how this works is yet unknown. Two complementary explanations seem likely. First, forces exerted by the actin cytoskeleton may be needed to induce conformational change in MC proteins, exposing sites for tyrosine phosphorylation and/or protein-protein interactions. Second, since many molecular adapters in TCR MCs bind to actin filaments, actin may provide a scaffold that promotes initial macromolecular interactions, stabilizing the complex until it reaches some critical mass. In support of the latter idea, Gomez et al. (52) showed that Vav1 recruitment to the IS occurs via a positive feedback loop, whereby Vav1 promotes actin polymerization, which in turn stabilizes Vav1 recruitment.
The Varma study (39) showed that the MCs that persist upon the depletion of F-actin are not sufficient to sustain signaling, since Ca2+ was reduced to baseline levels within minutes of Latrunculin A treatment. Consistent with this observation, we reported that Rho kinase inhibition (with Y-27632) to inhibit myosin II activity and actin stabilization (with Jasplakinolide) in T cells resulted in complete F-actin immobilization with a concomitant drop in intracellular Ca2+ levels (19). In addition to immobilizing the F-actin network, this pharmacological manipulation inhibited new MC formation and immobilized existing MCs. Thus, two interpretations are possible: either ongoing actin-dependent assembly of new MCs or application of actin-dependent mechanical force on existing MCs is needed to sustain Ca2+ signaling. While these two possibilities would be technically challenging to tease apart, it is clear that the existence of a static actin scaffold is insufficient to support Ca2+ signaling. Rather, a dynamic actin network is needed, reflecting a requirement either for continued actin polymerization or for centripetal flow of the acto-myosin II network. Interestingly, we found that immobilization of the F-actin network did not affect tyrosine phosphorylation of Zap70 or SLP-76, suggesting that this phenomenon does not stem from effects on the TCR per se. However, we did find that PLCγ1 phosphorylation was diminished. This finding is in agreement with our studies on HS1-deficient T cells, where defects in lamellipodial actin and MC dynamics correlated with diminished association of PLCγ1 with the insoluble cytoskeletal fraction and unstable recruitment of pPLCγ1 to the IS (53, 54). Similarly, inhibition of WASp function leads to dissolution of actin patches and destabilization of PLCγ1 association with TCR MCs (Kumari and Dustin, manuscript submitted). Given that the active, phosphorylated pool of PLCγ1 preferentially associates with the cytoskeleton (53, 54), it is interesting to speculate that PLCγ1 is a relatively direct point of intersection between the T-cell cytoskeleton and the signaling cascade leading to ER store release.
Calcium-regulatory roles of individual actin-regulatory proteins
In view of the multiple levels at which cytoskeletal remodeling affects Ca2+ signaling in T cells, one would expect that loss of important actin-regulatory molecules would affect Ca2+ mobilization, and this is indeed the case. As in other cell types, the formation of a branched F-actin network in T cells is driven by the seven subunit Arp2/3 complex, which directs the formation of new actin filaments on the sides of pre-existing filaments. The Arp2/3 complex is activated by one or more nucleation-promoting factors (NPFs), including WASp, WAVE2, and HS1 (55). WASp and WAVE2 function downstream of the Rho GTPases Cdc42 and Rac1, which are, in turn, activated by guanine exchange factors such as Vav1. Superimposed on this branched actin network is a higher level of organization; myosin II induces bundling and sliding of filaments within lamellar regions of the IS. Analysis of signaling defects in cells lacking individual actin-regulatory proteins is instructive in defining the mechanisms though which cytoskeletal dynamics influence Ca2+ mobilization.
Nucleation-promoting factors
The first actin-regulatory molecule to be carefully studied in T cells was WASp, the protein defective in the immunodeficiency disorder Wiskott-Aldrich syndrome. T cells lacking WASp exhibit defects in actin dynamics, although the magnitude of such defects is variable (56–58), possibly due to overlapping function of the closely related protein N-WASp (59), or even more distantly related proteins such as WAVE2. However, WASp-deficient T cells show a significant reduction in Ca2+ influx, which is associated with defective nuclear translocation of NFAT and diminished T cell activation (57, 60, 61). Conversely, mutations that perturb ubiquitin-dependent degradation of WASp lead to increased Ca2+ influx (62). In a recent study, Calvez et al. (63) explored the relationship between WASp function and T-cell Ca2+ responses. They showed that T cells from WAS patients formed conjugates with APCs at normal frequency but exhibited disorganized actin responses and asymmetric polarization of the MTOC, culminating in reduced proliferation in response to superantigen-charged APCs. In keeping with the idea that WASp manages signaling dynamics at the IS, the authors show that in comparison with control T cells, WAS T cells show diminished focusing of phosphotyrosine at the IS. Interestingly, Ca2+ mobilization during the sustained phase of signaling was erratic and sometimes pulsatory, a phenotype that the authors attribute to the unstable nature of the T cell-APC contact. While this study does not directly test whether the Ca2+ defects in these cells occur at the level of ER store release or CRAC channel function, the observed alterations in tyrosine phosphorylation patterns suggest that early signaling steps leading to IP3 generation and ER Ca2+ release are perturbed. These findings showing that WASp is important for synapse organization are consistent with a model proposed by Dustin and coworkers (64) in which WASp controls synapse symmetry by opposing the activity of PKCθ. According to this view, synapse stabilization and symmetry may be required for efficient integration of TCR (and, possibly, also costimulatory) signals.
Signaling defects similar to those observed in WASp-deficient T cells are seen in T cells lacking HS1, the hematopoietic homologue of cortactin. Like WASp, HS1 can activate Arp2/3 complex-dependent formation of branched actin filaments, and additionally, it can stabilize F-actin by binding to it. We showed that T cells lacking HS1 exhibit defects in TCR engagement-induced actin dynamics as well as Ca2+ mobilization and NFAT dependent transcriptional activation (52). In cell spreading assays, HS1-deficient T cells exhibit unstable lamellipodial protrusions, and in conjugates, they show loss of F-actin accumulation at the IS within a few minutes of cell-cell contact. Although we did not observe destabilization of adhesion in HS1-deficient T cells, defects in integrin-dependent adhesion and signaling were noted in conjugates formed with HS1-deficient NK cells (65). Single cell analysis of T-cell Ca2+ responses showed that release from ER stores is inhibited. Moreover, defects in Ca2+ signaling are rescued by treatment with the SERCA pump inhibitor thapsigargin, indicating that CRAC channel activity is intact (53). Further analysis of TCR signaling pathways revealed that PLCγ1 phosphorylation and recruitment to the IS is intact, but that dynamics of PLCγ1 MCs and cytoskeletal association of phospho-PLCγ1 was perturbed (53). Thus, in the case of HS1, it seems clear that defects in stabilization of branched actin filaments lead to unstable lamellipodial protrusions and aberrant dynamics of TCR-induced signaling MCs, resulting in defective induction of ER store release. Numerous technical differences in the analysis of T cells deficient for HS1 and WASp make it hard to make direct comparisons. Nonetheless, there are many phenotypic similarities, consistent with the observation that these two proteins frequently work together to generate and stabilize branched actin networks.
Like WASp and HS1, WAVE2 functions together with WAVE complex components to activate Arp2/3 complex-dependent formation of branched actin filaments in response to TCR engagement (58). In comparison with T cells lacking WASp or HS1, T cells lacking WAVE2 show much more profound defects in TCR-induced actin polymerization and lamellipodial protrusion. WAVE2 deficiency abrogates spreading, and T-cell-APC conjugates show virtually no actin polymerization at the IS. Similar defects are observed in T cells lacking Abi proteins, components of the WAVE complex that are needed for translocation of WAVE to the IS (66). As with loss of other Arp2/3 complex activators, loss of WAVE2 leads to significant blunting of Ca2+ mobilization. Surprisingly, however, WAVE2-deficient cells differ from cells lacking HS1 in that the initial release of Ca2+ from ER stores is intact. Moreover, defects are not bypassed by thapsigargin treatment. This phenotype points clearly to a requirement for WAVE2 in signaling for extracellular Ca2+ influx. Whether WAVE2 is needed for Orai1 function per se or for facilitating STIM 1/Orai1 interactions remains to be determined. In other cell types, WAVE2 has been shown to be targeted to sites of lamellipodial protrusion via interactions with proteins that control microtubule dynamics such as EB1 and stathmin (67), raising the possibility that WAVE2 promotes microtubule-dependent dynamics of STIM 1.
Upstream regulators of actin nucleation
As one might expect, molecules that regulate activation of WASp, WAVE2, and HS1 downstream of the TCR are required for both actin responses and Ca2+ mobilization. For example, Rac GTPases, which activate WAVE2, are needed for both processes (68, 69). Often, however, the relevant signaling proteins are large, modular molecules with several interdependent functions. Thus, it can be difficult to distinguish the extent to which they affect Ca2+ signaling by regulating actin dynamics. Several recent reviews address T-cell signaling pathways leading to actin polymerization in detail (70–72). Here, we discuss key molecules that illustrate the complexities associated with upstream regulation of actin and Ca2+ responses.
A good example of this type of functional complexity is Vav1. Upon TCR engagement, Vav1 is recruited to the IS and behaves as a guanine exchange factor (GEF) for the Rho family GTPases Rac1 and Cdc42 (73), which activate WAVE2 and WASp, respectively. Vav1 function is essential for actin polymerization at the IS (74–76). Other Vav family members are ubiquitously expressed and participate redundantly in the activation of small GTPases (77, 78). T cells lacking Vav1 show profound defects in Ca2+ mobilization. Interestingly, however, Vav1’s role in regulating actin and Ca2+ responses may be distinct. In particular, the GEF activity of Vav1 seems to be dispensable for Ca2+ mobilization, since T cells from knockin mice bearing a Vav1 mutant lacking GEF activity display normal Ca2+ and proliferative responses. Furthermore, Vav2 and Vav3 cannot mediate Ca2+ elevation in stimulated T cells, indicating that the isoforms are not completely redundant. In a recent study, Li et al. (79) isolated a 20 amino acid region in the N-terminus of Vav1 that is indispensable for Ca2+ mobilization independently of GEF activity. The authors argue that this sequence within the calponin homology domain is essential for calmodulin binding and recruitment to the sites of active signaling (79, 80), highlighting the scaffolding function of Vav1 in TCR-induced Ca2+ mobilization.
A second example of multidomain complexity is seen in the Tec family kinase Itk. Like Vav1, Itk is a component of the TCR signalosome. Itk phosphorylates PLCγ1, and so plays a critical role in signaling Ca2+ release from ER stores. Through its SH2 domain, Itk interacts with HS1 and recruits it to the IS (53), and T cells deficient for Itk show defective actin responses similar to those of HS1-deficient T cells (81–83). Via HS1, Itk also promotes Vav1 recruitment to the IS, so that Itk-deficient T cells also have defects in TCR engagement-induced activation of Cdc42 and WASp (82). Interestingly, it appears that the domains of Itk responsible for regulating actin and Ca2+ responses are largely distinct. PLCγ1 activation requires kinase activity, while actin regulation requires the SH2 domain and is unaffected by mutations that abrogate kinase activity (83). Even so, this separation may not be complete. Binding of SLP-76 to the SH2 domain of Itk has been shown to activate Itk kinase activity (84), and the same may be true for interactions with HS1. Thus, there could be a direct linkage between Ca2+ signaling and actin-regulatory pathways, even though actual actin remodeling is not involved.
In addition to the complexities introduced by multidomain signaling molecules, higher order molecular organization intertwines actin polymerization and Ca2+ signaling. Signalosome components are held together by multiple low affinity molecular interactions, such that loss of any one component disrupts interactions among the others and perturbs T-cell signaling (85–87). As part of this process, actin scaffolds generated by these signaling molecules stabilize newly formed signaling complexes (46), thereby generating a positive feedback loop to facilitate T-cell activation. As discussed further below, there is evidence that this process requires ongoing actin polymerization, rather than a static actin scaffold (19). Interestingly, PLCγ1 activation appears to be an important control point for this higher order cytoskeletal control of Ca2+ signaling (19, 53).
Myosin IIA function in the T-cell calcium response
Another actin interacting protein that has gained recent interest in the field is myosin IIA. This motor protein is the sole representative of non-muscle myosin II family in mouse primary T cells (88) and the predominant isoform expressed in human T cells (19). Results of recent studies are conflicting as to the exact role that myosin IIA plays at the IS both in terms of actin dynamics and Ca2+ signaling (50). Initial work by Ilani et al. (89) concluded that myosin IIA is indispensable for centripetal movement of TCR microclusters at the IS (presumably driven by actin dynamics), as well as for both initial and sustained Ca2+ signaling. However, subsequent detailed studies yielded disparate results. In studies of murine TCR transgenic T cells responding to stimulatory planar bilayers, Yu et al. (90) found that inhibition of myosin light chain kinase with ML-7 resulted in a profound decrease in Ca2+ mobilization, while Kumari et al. (91) observed only a modest dampening of the Ca2+ response upon RNAi-mediated suppression of myosin IIA. Our laboratory tested the effects of treating Jurkat T cells spreading on stimulatory glass surfaces with the with ROCK inhibitor Y-27632 (19). While this treatment effectively inhibited myosin II-dependent contractility at the IS, it had no effect on Ca2+ signaling. However, we also observed that loss of myosin II activity destabilized the architecture of the IS, so it remains possible that myosin IIA contributes to Ca2+ signaling at late stages of T-cell-APC interaction. We have proposed that the contribution of myosin IIA to TCR signaling may vary depending on the nature of the activating stimulus, including stimulatory substrate rigidity and/or ligand mobility (19, 50). Additional work will be needed to test this idea and to define role of myosin IIA under physiological conditions.
Regulation of calcium entry by cytoskeletal control of organelle positioning
In addition to promoting the immediate signaling events that take place downstream of TCR, the cytoskeleton directs the higher order organization of cellular organelles. This, too, contributes to sustained Ca2+ signaling. Organelles known to modulate Ca2+ signaling in a cytoskeleton-dependent fashion include the ER, mitochondria, and the plasma membrane.
Remodeling of the endoplasmic reticulum
The ER is distributed throughout the cytoplasm via its interactions with cytoskeleton. While comparatively little is known about ER remodeling in T cells, analysis in other cell types has shown that ER organization is determined by the balance between movement toward the cell periphery driven by microtubule motors and attachment to growing microtubule tips, and movement toward the cell body driven by acto-myosin (92, 93). The net result of these processes is the formation of specialized sites where the ER membrane comes in close proximity to the PM, and it is at these sites where the ER protein STIM 1 engages Orai1 in the PM, promoting Ca2+ influx (94). STIM 1 associates with the MT tip tracking protein EB3, which directs the accumulation of Stim1 to sites where the ER interacts with microtubule plus ends. Indeed, Grigoriev et al. (14) investigated the role of Stim1 in ER remodeling in HeLa cells and MRC5 fibroblasts and found that the interaction between Stim1 and EB3 (and to lesser extent EB1) promotes ER tubule extension. Upon depletion of ER Ca2+ stores, Stim1 oligomerizes and moves within the plane of the ER, triggering the formation of Stim1-ORAI1 clusters at ER-PM junctions. Consistent with the notion that the microtubule cytoskeleton positions the ER to promote Stim1-Orai1 interactions, depolymerization of microtubules with nocodazole inhibits SOCE (95).
Recruitment of mitochondria to the immunological synapse
Mitochondria are delivered to the IS along microtubules by kinesin-1 (96). Besides their canonical role as ‘cellular powerhouses’, mitochondria are well-adapted for Ca2+ buffering in their immediate vicinity. This turns out to be important for the function of the Orai1 channel, which becomes auto-inhibited if the local concentration of Ca2+ reaches high levels (97). Mitochondria at the IS buffer Ca2+, preventing its accumulation at the channel mouth, thereby ensuring that the CRAC channels remain active. Mitochondria then release Ca2+ away from the IS so that it can propagate the signaling cascade. In this way, mitochondria set up a narrow gradient of Ca2+ ions near the sites of TCR signaling.
Polarization of mitochondria to the IS is microtubule dependent. Contento et al. (98) reported that MTOC and mitochondrial polarization to the IS occurs via outside-in LFA-1 signaling, providing a mechanism for costimulatory signaling. However, microtubules alone are not sufficient. Mitochondria are large organelles that are fused into intricate networks, impeding their navigation through intracellular space. Thus, regulated remodeling of mitochondria by fusion and fission facilitates their delivery to the IS and promotes their interaction with CRAC channels, as well as delivery of ATP to the TCR signaling machinery. TCR stimulation triggers activation of dynamin-related protein 1 (Drp1), a GTPase essential for mitochondrial fission (99). Depletion of Drp1 in T cells interacting with APCs leads to defects in mitochondrial polarization and TCR dynamics (100). Once at the IS, mitochondrial fragments are fused into large structures by mitofusin and become enriched at the pSMAC region (Fig. 2). This localization is most likely regulated by the interplay between microtubule- and actin-dependent motors; cytoplasmic dynein binding to mitochondria would drive centripetal movement, as would pushing by actin retrograde flow. Centripetal movement of mitochondria at the IS may be opposed by kinesin-1 and myosin V, which have been shown to bind to mitochondria and direct their migration toward the cell periphery in other cell types (101, 102). Interestingly, kinesin-1 binding to the mitochondrial surface is regulated by Ca2+ (103). This provides a potential feedback mechanism that could couple mitochondrial to CRAC activity at the IS.
Fig. 2. Morphological changes and organelle remodeling associated with T-cell polarization in response to TCR triggering.
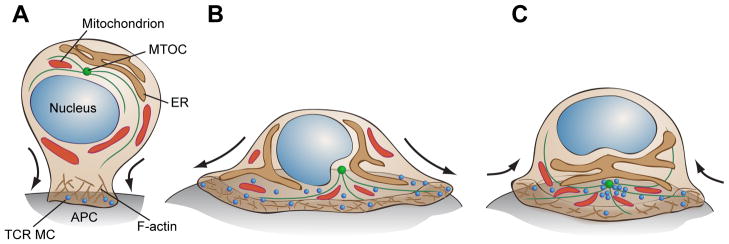
(A) Upon encountering an APC bearing cognate pMHC, a T cell undergoes polarization towards the site of antigen presentation. Actin polymerization at sites of TCR engagement stabilizes newly formed signaling MCs, and lamellipodial F-actin polymerization induces T-cell spreading on the APC surface. (B) TCR MCs form in the actin-rich periphery of the IS and are continuously delivered to the central region in parallel with actin retrograde flow, a process that involves actin polymerization at the cell periphery coupled with contraction of the acto-myosin network. For at least some MC components, microtubule-dependent motor activity also contributes to centripetal movement. Concomitant with T-cell spreading, the MTOC is recruited to the cell-cell contact zone, which establishes tracks for retrograde traffic of signaling molecules to the cSMAC region, and anterograde movement of the ER and mitochondria to the IS. Mitochondria undergo fission and fusion to enhance their trafficking to the sites of active TCR signaling. (C) Once the machinery is set in place, T cells undergo partial acto-myosin II-dependent contraction to focus receptors and develop a mature IS.
Plasma membrane flattening
Because the Ca2+ buffering efficiency of mitochondria is limited to short distances, it is essential that these organelles be juxtaposed against the IS within 200 nm of the PM (104). Therefore, flattening of the T cell PM at the IS is important to facilitate signaling. The Hoth laboratory (104) investigated the importance of cell morphology during T-cell activation. They discovered that Ca2+ signaling was augmented in cells that underwent actin-dependent PM flattening upon TCR signaling and that these changes promoted mitochondrial delivery to the peripheral contact zone (Fig. 2). Furthermore, they demonstrated that pre-incubation of T cells with non-stimulatory adhesive beads (a process that deforms the PM) greatly increased their ability to respond to soluble TCR ligands.
Regulation of cytoskeletal dynamics by Ca2+ signaling
Although most of the available literature focuses on cytoskeletal control of Ca2+ mobilization, it is clear that Ca2+ signaling also impacts remodeling of both actin filaments and microtubules. Perhaps more importantly, cytoskeletal dynamics and Ca2+ signaling are inextricably linked at the level of inositol metabolism (Fig. 3).
Fig. 3. Parallel control of cytoskeletal remodeling and calcium signaling via lipid metabolism.
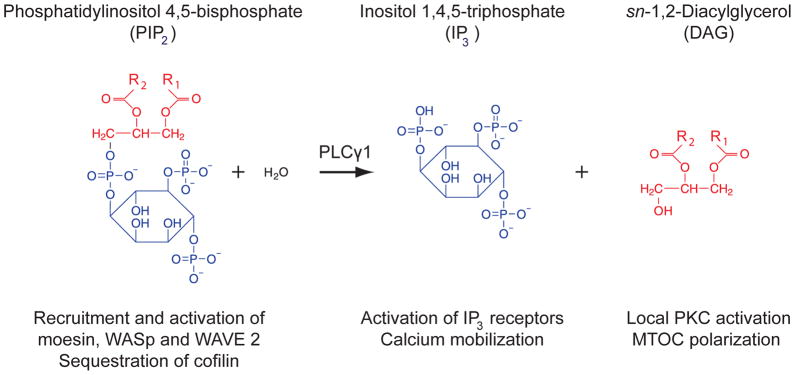
Phosphatidylinositol 4,5-bisphosphate (PIP2) is required for recruitment and regulation of key actin-regulatory proteins, such as moesin, WASp, WAVE 2, and cofilin. PLCγ1 activity cleaves PIP2 at the IS to generate inositol 1,4,5-triphosphate (IP3) and sn-1,2-Diacylglycerol (DAG). The release of IP3 triggers receptors on the ER membrane, resulting in release of stored Ca2+. The release of DAG stimulates local protein kinase C activation, defining the site for MTOC polarization, as well as enhancing Ras signaling and other processes. PLCγ1 also affects F-actin remodeling at the IS by transiently consuming PIP2. This leads to release and inactivation of moesin and downregulates activity of WASp and WAVE2, as well as other signaling proteins including Vav1 and Itk. The actin-severing protein cofilin is sequestered by binding to PIP2 and is released in an active form by PLCγ1 activity.
Ca2+ control of the actin cytoskeleton
With respect to the actin cytoskeleton, Bunnell et al. (7) showed that chelation of extracellular and intracellular Ca2+ blocked actin-dependent T-cell spreading on TCR-stimulatory surfaces. The defect was profound and mirrored that of blocking Src kinase activity. This observation revealed the necessity for elevated intracellular Ca2+ early in the sequence of events leading to actin polymerization and cell spreading. Although the mechanisms involved have not been elucidated in detail, many actin-regulatory proteins are Ca2+-sensitive. These include proteins such as myosin II, L-plastin and gelsolin that are regulated directly by Ca2+, as well as proteins such as talin, ezrin, and WASp that are sensitive to cleavage by the Ca2+-dependent protease calpain.
One proposed mechanism by which Ca2+ signaling may modulate T-cell cytoskeletal function involves cleavage of key actin regulatory proteins by the Ca2+-activated proteases calpain 1 and 2. Such a mechanism has been proposed to explain seemingly unique functions of ezrin, which is calpain sensitive, versus the closely related protein moesin, which lacks a calpain cleavage site (105, 106). Similarly, calpain has been implicated in degradation of WASp under conditions where it is not assembled with WIP (107). Finally, calpain has been proposed to regulate LFA-1 activation and consequent adhesion and migration via cleavage of talin or other proteins such as α-actinin and filamin A (108–110). However, while these proteins are calpain substrates, recent work has cast doubt about the physiological significance of these cleavage events for T-cell function. In the case of ezrin, our laboratory recently generated mice with conditional deletion of ezrin in mature T cells, in hopes of uncovering ezrin-specific aspects of T cell function. We found minimal ezrin-specific defects in these cells; instead, our results pointed to overlapping, dose-dependent function of ezrin and moesin in T-cell activation, adhesion, and migration (111, 112). With respect to WASp, Ca2+ influx downstream of TCR stimulation leads to calpain-dependent cleavage of WASp followed by proteasomal degradation, and treatment with the calpain inhibitor calpeptin reportedly prolonged high F-actin content post TCR stimulation (107, 113). However only a minor fraction of WASp is cleaved in WT cells, and significant cleavage was shown only in activated T cells from patients with destabilizing WASp mutations (107, 113). Lastly, the Huttenlocher laboratory looked closely at calpain-dependent activation of LFA-1 in T cells. Unlike previous studies that relied on pharmacological inhibition of calpain, this group generated mice with deletion of calpain 4 in mature T cells (114). Though T cells from these mice expressed very low levels of both calpain 1 and 2 (both of which rely on assembly with calpain 4 for stabilization) and exhibited diminished talin proteolysis, the T cells showed normal LFA-1 dependent adhesion and migration. Binding to APCs, recruitment of F-actin to the IS, and proliferation were also unperturbed. This study provides strong evidence that calpain-dependent proteolysis is not a major mechanism by which Ca2+ levels affect actin remodeling in T cells.
While the role of calpain is in question, other cytoskeletal regulatory proteins are clearly Ca2+-dependent. One of the best examples is the actin bundling protein L-plastin (reviewed in detail elsewhere in this volume, 115). Although multiple plastin isoforms are expressed throughout the body, L-plastin is the sole isoform expressed in T cells (116) and seems to be the only one that is Ca2+-dependent (117). All plastin isoforms contain two N-terminal Ca2+-binding sites and two C-terminal actin-binding sites, which enable plastins to bundle actin filaments. Actin-bundling activity is thought to be negatively regulated by Ca2+ (117). Thus, in low Ca2+ concentrations (~ 100 nM), L-plastin is able to bundle actin, while Ca2+ concentrations in the micromolar range perturb its bundling activity. Given the Ca2+ concentrations associated with T-cell activation, it seems that L-plastin would be active prior to TCR signaling, when intracellular Ca2+ levels are low, and would be inactivated upon Ca2+ elevation during early TCR signaling. This hypothesis is in line with evidence that early TCR-dependent phosphorylation events and Ca2+ influx are intact in T cells lacking L-plastin (118). However, L-plastin-deficient T cells have defects in IS maturation and polarization, indicating that L-plastin promotes later stages of T-cell activation, perhaps by regaining activity as cytoplasmic Ca2+ levels decay. Finally, as discussed below, Ca2+ signaling regulates the balance between T-cell migration and stopping, and L-plastin is poised to play an important role in that context. In keeping with that idea, L-plastin-deficient T cells exhibit migration defects (119).
Ca2+ control of microtubule dynamics
Ca2+ signaling also plays a role in MTOC reorientation to the IS interface. Early work by the Kupfer laboratory (8) demonstrated that the MTOC polarization is Ca2+-dependent. This finding was confirmed by Weiss laboratory (120, 121), who showed that MTOC reorientation is dependent on signaling through PLCγ1, and on the presence of extracellular Ca2+. The relevant Ca2+-dependent molecules were not identified, although involvement of calcineurin and CaMK was ruled out. More recently, however, the Huse laboratory found that MTOC reorientation is Ca2+-independent, and depends instead on local production of DAG (thereby explaining the dependence on PLCγ1) (122). In that study, T cells pretreated with Ca2+ blockade solution (EGTA plus BAPTA-AM) polarized their MTOCs to the IS just as efficiently as control cells and maintained a high degree of polarization for the first 10 min of stimulation. One explanation that could reconcile these conflicting findings is the temporal differences in the experimental conditions. While the earlier work concentrated on prolonged T-cell-APC contact (15–60 min), the Huse laboratory assessed MTOC reorientation by live-cell microscopy immediately post TCR stimulation. Thus, it may be that Ca2+ is not needed for initial MTOC recruitment to the IS, but is required for retention at later times. Finally, as detailed elsewhere in this volume (123), MTOC reorientation requires both pulling forces provided by cytoplasmic dynein as well as pushing forces produced by myosin II (124, 125). Thus, the requirement for myosin II function could explain the Ca2+ dependence of MTOC reorientation in some experimental settings.
Parallel control via inositol metabolism
While one tends to think about serial signaling pathways in which cytoskeletal dynamics regulate Ca2+ mobilization or vice versa, it is important to point out that these two processes can be signaled in parallel, via a common mediator, PIP2 (Fig. 3). Thus, the observed coordinate control is at least partially attributable to mutual dependence on inositol metabolism, since cytoskeletal regulatory pathways are highly sensitive to inositol lipids, especially PIP2 and its metabolite, diacylglycerol. Many actin-regulatory proteins including WASp, WAVE2, moesin, cofilin, and Vav1 interact with and are activated by PIP2 in the PM (126–130). PLCγ1-dependent cleavage of PIP2 simultaneously stimulates ER store release by generating IP3 and consumes a key upstream regulator of actin dynamics. For example, the actin-tethering protein moesin binds to PIP2 (131), which activates its ability to link PM proteins to the actin cytoskeleton. Cleavage of PIP2 leads to moesin inactivation, resulting in diminished cortical stiffness, and allowing redistribution of moesin-linked PM proteins (128, 132). In contrast, cleavage of PIP2 by PLCγ1 releases and activates cofilin, leading to enhanced severing activity and providing free monomer and uncapped barbed ends for new filament growth (133). In the case of WASp and WAVE2, which are activated by both Rho GTPases and PIP2 binding, consumption of PIP2 may serve to attenuate or terminate the response that was initially activated by TCR-dependent activation of Rho GTPases. The interplay between inositol metabolism, Ca2+ signaling and cytoskeletal reorganization is even clearer in the case of microtubule reorganization. Here, local accumulation of DAG produced by PIP2 cleavage activates protein kinase C-dependent events leading to MTOC reorientation. Details of that pathway are reviewed elsewhere in this volume (123).
Higher level complexity: the transition from migration to activation
The complex interplay between cytoskeletal dynamics and Ca2+ signaling sets the stage for finely tuned changes in response to environmental cues. For example, since Ca2+ signaling also controls migratory responses downstream of chemokine receptors, it is poised to coordinate the ‘stop’ signal that occurs when T cells migrating within lymphoid organs encounter APCs bearing rare agonist pMHCs. In migrating T cells, extrinsic factors such as chemokines and integrin ligands induce F-actin polymerization in the leading edge of the cell and myosin II contractility to form a trailing uropod (134). The MTOC localizes behind the nucleus, stabilizing the uropod and establishing directional persistence (135, 136). Upon encounter with an APC, intracellular Ca2+ levels rise, and the T cell stops migrating, rounds up, and polarizes actin filaments and the MTOC toward the APC. This series of events occurs in mature T cells encountering antigen in peripheral lymphoid organs (137) and also during thymic development. Using two-photon microscopy, Bhakta et al. (138) showed that naive thymocytes are highly mobile when intracellular Ca2+ concentration are low. However, upon an increase in intracellular Ca2+ levels, thymocytes become immobile and eventually undergo positive selection. By artificially manipulating Ca2+ levels, the group could show that elevation of intracellular Ca2+ is sufficient to inhibit cell migration, prolonging interaction with antigen-bearing stromal cells and promoting genetic reprogramming and positive selection. This study highlights the importance of crosstalk between Ca2+ signaling and cytoskeletal dynamics for T-cell development.
Subsequent work from the Krummel laboratory (139) extended the investigation of the relationship between Ca2+ and cell migration using stimulatory planar lipid bilayers. Their findings showed that the amplitude of the Ca2+ response is dependent on the density of the presented antigen. Consistent with previous in vivo observations (140, 141), the laboratory found that TCR stimulation slowed the T cells but did not strictly halt their migration. T-cell migration was inversely proportional to Ca2+ spikes. Thus, while T cells with high intracellular Ca2+ concentrations stopped migration, cells with intermediate Ca2+ signaling showed a graded response in motility. Furthermore, the average speed of migrating T cells underwent step changes between high, intermediate, and low motile cells. In a related study, Marangoni et al. (142) compared the Ca2+ responses required for diminished T-cell motility with those required for translocation of NFAT to the nucleus and found that NFAT translocation requires high intracellular Ca2+ levels associated with migratory arrest. In tumor-infiltrating T cells, nuclear NFAT was maintained for several minutes in cells with diminished intracellular Ca2+ and unstable APC contacts, a condition that was associated with induction of T-cell tolerance. Taken together, these studies demonstrate the integration of Ca2+ mobilization and T-cell migration during TCR signaling, and emphasize the importance of these events for T-cell development and effector function.
Concluding remarks
The interdependence of Ca2+ signaling and cytoskeletal remodeling has been evident since the earliest single studies of T-cell activation. Over the past several years, we have identified many of the molecules that control these two processes and placed them into major regulatory pathways. This has revealed key points of intersection within the signaling network. Cytoskeletal influence on Ca2+ signaling is simultaneously exerted at various scales, ranging from single molecule conformational changes to changes in cell morphology. The tight association between Ca2+ signaling and the cytoskeleton provides mechanisms by which environmental cues can tune the T-cell response, such as when the presence of cognate antigen induces stopping of T cells trafficking through lymphoid organs. In addition, the complex interplay between Ca2+ and the cytoskeleton provides a basis for positive and negative feedback loops. For example, minute bursts of actin polymerization may promote early TCR signaling. The resulting rise in cytoplasmic Ca2+ may then sustain actin remodeling during T-cell spreading. Finally, cell shape changes or forces generated by T-cell spreading may, in turn, promote sustained Ca2+ elevation.
While key points of intersection have been identified, many mechanistic questions remain. Does mechanotransduction occur at the level of the TCR? If so, how is force transmitted to the TCR, and what, if any, is the contribution of the APC cytoskeleton? Why is activation of PLCγ1 sensitive to perturbation of actin dynamics – is there mechanotransduction at the level of signaling MCs? How, exactly, do WAVE2 and microtubules affect CRAC coupling? And, how are the effects of cell shape changes, documented using in vitro assays, manifested in T cells interacting with APCs within lymphoid tissues? In addition to these molecular questions, there are many unanswered questions about higher order interactions and feedback events. Going forward, the challenge for the field is to understand this integrated complexity, a task that will require novel approaches ranging from protein conformational biosensors to multi-photon analysis of signaling dynamics during an in vivo immune response.
Acknowledgments
The authors thank Dr. Bruce Freedman for teaching us that calcium (like actin) is at the center of everything. We thank Dr. Freedman and members of the Burkhardt laboratory for critical reading of the manuscript and many helpful discussions. This work was supported by National Institutes of Health grants T32AR7442-25 (to A.B.), and R01AI065644 and P01CA093615 (to J.K.B.). The authors have no conflicts of interest to declare.
References
- 1.Kupfer A, Singer SJ. Cell biology of cytotoxic and helper T cell functions: immunofluorescence microscopic studies of single cells and cell couples. Annu Rev Immunol. 1989;7:309–337. doi: 10.1146/annurev.iy.07.040189.001521. [DOI] [PubMed] [Google Scholar]
- 2.Ostergaard H, Clark WR. The role of Ca2+ in activation of mature cytotoxic T lymphocytes for lysis. J Immunol. 1987;139:3573–3579. [PubMed] [Google Scholar]
- 3.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delon J, Bercovici N, Liblau R, Trautmann A. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur J Immunol. 1998;28:716–729. doi: 10.1002/(SICI)1521-4141(199802)28:02<716::AID-IMMU716>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.Liu SJ, Hahn WC, Bierer BE, Golan DE. Intracellular mediators regulate CD2 lateral diffusion and cytoplasmic Ca2+ mobilization upon CD2-mediated T cell activation. Biophys J. 1995;68:459–470. doi: 10.1016/S0006-3495(95)80207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phatak PD, Packman CH. Engagement of the T-cell antigen receptor by anti-CD3 monoclonal antibody causes a rapid increase in lymphocyte F-actin. J Cell Physiol. 1994;159:365–370. doi: 10.1002/jcp.1041590220. [DOI] [PubMed] [Google Scholar]
- 7.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 8.Kupfer A, Dennert G, Singer SJ. The reorientation of the Golgi apparatus and the microtubule-organizing center in the cytotoxic effector cell is a prerequisite in the lysis of bound target cells. J Mol Cell Immunol. 1985;2:37–49. [PubMed] [Google Scholar]
- 9.Yokosuka T, Saito T. The immunological synapse, TCR microclusters, and T cell activation. Current topics in microbiology and immunology. 2010;340:81–107. doi: 10.1007/978-3-642-03858-7_5. [DOI] [PubMed] [Google Scholar]
- 10.Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. doi: 10.1146/annurev-biophys-042910-155238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller CL, Braciale VL, Samelson LE. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol Rev. 2003;191:220–236. doi: 10.1034/j.1600-065x.2003.00004.x. [DOI] [PubMed] [Google Scholar]
- 12.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 13.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grigoriev I, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barr VA, et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19:2802–2817. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rao A. Signaling to gene expression: calcium, calcineurin and NFAT. Nat Immunol. 2009;10:3–5. doi: 10.1038/ni0109-3. [DOI] [PubMed] [Google Scholar]
- 17.Alonso MT, Manjarres IM, Garcia-Sancho J. Privileged coupling between Ca(2+) entry through plasma membrane store-operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Mol Cell Endocrinol. 2012;353:37–44. doi: 10.1016/j.mce.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Smyth JT, Dehaven WI, Bird GS, Putney JW., Jr Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121:762–772. doi: 10.1242/jcs.023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babich A, Li S, O’Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol. 2012;197:775–787. doi: 10.1083/jcb.201201018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robert V, Triffaux E, Savignac M, Pelletier L. Calcium signaling in T lymphocytes. Med Sci (Paris) 2012;28:773–779. doi: 10.1051/medsci/2012288020. [DOI] [PubMed] [Google Scholar]
- 21.Rozdzial MM, Pleiman CM, Cambier JC, Finkel TH. pp56Lck mediates TCR zeta-chain binding to the microfilament cytoskeleton. J Immunol. 1998;161:5491–5499. [PubMed] [Google Scholar]
- 22.Rozdzial MM, Malissen B, Finkel TH. Tyrosine-phosphorylated T cell receptor zeta chain associates with the actin cytoskeleton upon activation of mature T lymphocytes. Immunity. 1995;3:623–633. doi: 10.1016/1074-7613(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 23.Kesti T, et al. Reciprocal regulation of SH3 and SH2 domain binding via tyrosine phosphorylation of a common site in CD3epsilon. J Immunol. 2007;179:878–885. doi: 10.4049/jimmunol.179.2.878. [DOI] [PubMed] [Google Scholar]
- 24.Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol. 2005;6:80–89. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- 25.Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A. 2009;106:12729–12734. doi: 10.1073/pnas.0902621106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys J. 2008;94:3286–3292. doi: 10.1529/biophysj.107.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8724–8729. doi: 10.1073/pnas.1000966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang J, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim ST, et al. The alphabeta T cell receptor is an anisotropic mechanosensor. The Journal of biological chemistry. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim ST, et al. TCR Mechanobiology: Torques and Tunable Structures Linked to Early T Cell Signaling. Front Immunol. 2012;3:76. doi: 10.3389/fimmu.2012.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng SY, Liu M, Dustin ML. CD80 cytoplasmic domain controls localization of CD28, CTLA-4, and protein kinase Ctheta in the immunological synapse. J Immunol. 2005;175:7829–7836. doi: 10.4049/jimmunol.175.12.7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feigelson SW, et al. Occupancy of lymphocyte LFA-1 by surface-immobilized ICAM-1 is critical for TCR- but not for chemokine-triggered LFA-1 conversion to an open headpiece high-affinity state. Journal of immunology. 2010;185:7394–7404. doi: 10.4049/jimmunol.1002246. [DOI] [PubMed] [Google Scholar]
- 34.Hsu CJ, et al. Ligand mobility modulates immunological synapse formation and T cell activation. PLoS One. 2012;7:e32398. doi: 10.1371/journal.pone.0032398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Z, Finkel TH. T cell receptor triggering by force. Trends Immunol. 2010;31:1–6. doi: 10.1016/j.it.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Z, Janmey PA, Finkel TH. The receptor deformation model of TCR triggering. FASEB J. 2008;22:1002–1008. doi: 10.1096/fj.07-9331hyp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dushek O. Elementary steps in T cell receptor triggering. Front Immunol. 2011;2:91. doi: 10.3389/fimmu.2011.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 39.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci U S A. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li YC, et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. Journal of immunology. 2010;184:5959–5963. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- 42.Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T lymphocyte activation. Biophysical journal. 2012;102:L5–7. doi: 10.1016/j.bpj.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Connor RS, et al. Substrate rigidity regulates human T cell activation and proliferation. Journal of immunology. 2012;189:1330–1339. doi: 10.4049/jimmunol.1102757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Alwan MM, et al. Cutting edge: dendritic cell actin cytoskeletal polarization during immunological synapse formation is highly antigen-dependent. J Immunol. 2003;171:4479–4483. doi: 10.4049/jimmunol.171.9.4479. [DOI] [PubMed] [Google Scholar]
- 45.Bunnell SC, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokosuka T, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 48.Singleton KL, et al. Itk controls the spatiotemporal organization of T cell activation. Sci Signal. 2011;4:ra66. doi: 10.1126/scisignal.2001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yi J, Wu XS, Crites T, Hammer JA., 3rd Actin Retrograde Flow and Acto-Myosin II Arc Contraction Drive Receptor Cluster Dynamics at the Immunological Synapse in Jurkat T-Cells. Molecular biology of the cell. 2012 doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammer JA, 3rd, Burkhardt JK. Controversy and consensus regarding myosin II function at the immunological synapse. Curr Opin Immunol. 2013 doi: 10.1016/j.coi.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen K, Sylvain NR, Bunnell SC. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity. 2008;28:810–821. doi: 10.1016/j.immuni.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 52.Gomez TS, et al. HS1 Functions as an Essential Actin-Regulatory Adaptor Protein at the Immune Synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrizosa E, et al. Hematopoietic lineage cell-specific protein 1 is recruited to the immunological synapse by IL-2-inducible T cell kinase and regulates phospholipase Cgamma1 Microcluster dynamics during T cell spreading. J Immunol. 2009;183:7352–7361. doi: 10.4049/jimmunol.0900973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patsoukis N, et al. RIAM regulates the cytoskeletal distribution and activation of PLC-gamma1 in T cells. Sci Signal. 2009;2:ra79. doi: 10.1126/scisignal.2000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Higgs HN, Pollard TD. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J Biol Chem. 1999;274:32531–32534. doi: 10.1074/jbc.274.46.32531. [DOI] [PubMed] [Google Scholar]
- 56.Badour K, et al. The Wiskott-Aldrich Syndrome Protein Acts Downstream of CD2 and the CD2AP and PSTPIP1 Adaptors to Promote Formation of the Immunological Synapse. Immunity. 2003;18:141–154. doi: 10.1016/s1074-7613(02)00516-2. [DOI] [PubMed] [Google Scholar]
- 57.Cannon JL, Burkhardt JK. Differential roles for Wiskott-Aldrich syndrome protein in immune synapse formation and IL-2 production. J Immunol. 2004;173:1658–1662. doi: 10.4049/jimmunol.173.3.1658. [DOI] [PubMed] [Google Scholar]
- 58.Nolz JC, et al. The WAVE2 Complex Regulates Actin Cytoskeletal Reorganization and CRAC-Mediated Calcium Entry during T Cell Activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cotta-de-Almeida V, et al. Wiskott Aldrich syndrome protein (WASP) and N-WASP are critical for T cell development. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15424–15429. doi: 10.1073/pnas.0706881104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dupre L, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity. 2002;17:157–166. doi: 10.1016/s1074-7613(02)00360-6. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med. 1999;190:1329–1342. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reicher B, Joseph N, David A, Pauker MH, Perl O, Barda-Saad M. Ubiquitylation-dependent negative regulation of WASp is essential for actin cytoskeleton dynamics. Mol Cell Biol. 2012;32:3153–3163. doi: 10.1128/MCB.00161-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calvez R, Lafouresse F, De Meester J, Galy A, Valitutti S, Dupre L. The Wiskott-Aldrich syndrome protein permits assembly of a focused immunological synapse enabling sustained T-cell receptor signaling. Haematologica. 2011;96:1415–1423. doi: 10.3324/haematol.2011.040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sims TN, et al. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell. 2007;129:773–785. doi: 10.1016/j.cell.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 65.Butler B, Kastendieck DH, Cooper JA. Differently phosphorylated forms of the cortactin homolog HS1 mediate distinct functions in natural killer cells. Nat Immunol. 2008;9:887–897. doi: 10.1038/ni.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zipfel PA, et al. Role for the abi/wave protein complex in T cell receptor-mediated proliferation and cytoskeletal remodeling. Curr Biol. 2006;16:35–46. doi: 10.1016/j.cub.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 67.Takahashi K. WAVE2 Protein Complex Coupled to Membrane and Microtubules. Journal of oncology. 2012;2012:590531. doi: 10.1155/2012/590531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu H, Leitenberg D, Li B, Flavell RA. Deficiency of small GTPase Rac2 affects T cell activation. J Exp Med. 2001;194:915–926. doi: 10.1084/jem.194.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arrieumerlou C, Randriamampita C, Bismuth G, Trautmann A. Rac is involved in early TCR signaling. J Immunol. 2000;165:3182–3189. doi: 10.4049/jimmunol.165.6.3182. [DOI] [PubMed] [Google Scholar]
- 70.Gomez TS, Billadeau DD. T cell activation and the cytoskeleton: you can’t have one without the other. Adv Immunol. 2008;97:1–64. doi: 10.1016/S0065-2776(08)00001-1. [DOI] [PubMed] [Google Scholar]
- 71.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 72.Fooksman DR, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dumont C, et al. Rac GTPases play critical roles in early T-cell development. Blood. 2009;113:3990–3998. doi: 10.1182/blood-2008-09-181180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bustelo XR. Vav proteins, adaptors and cell signaling. Oncogene. 2001;20:6372–6381. doi: 10.1038/sj.onc.1204780. [DOI] [PubMed] [Google Scholar]
- 75.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 76.Tybulewicz VL, Ardouin L, Prisco A, Reynolds LF. Vav1: a key signal transducer downstream of the TCR. Immunol Rev. 2003;192:42–52. doi: 10.1034/j.1600-065x.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 77.Cao Y, Janssen EM, Duncan AW, Altman A, Billadeau DD, Abraham RT. Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. Embo J. 2002;21:4809–4819. doi: 10.1093/emboj/cdf499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zakaria S, et al. Differential regulation of TCR-mediated gene transcription by Vav family members. J Exp Med. 2004;199:429–434. doi: 10.1084/jem.20031228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li SY, et al. The N-terminal 20-amino acid region of guanine nucleotide exchange factor Vav1 plays a distinguished role in T cell receptor-mediated calcium signaling. J Biol Chem. 2013;288:3777–3785. doi: 10.1074/jbc.M112.426221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou Z, Yin J, Dou Z, Tang J, Zhang C, Cao Y. The calponin homology domain of Vav1 associates with calmodulin and is prerequisite to T cell antigen receptor-induced calcium release in Jurkat T lymphocytes. J Biol Chem. 2007;282:23737–23744. doi: 10.1074/jbc.M702975200. [DOI] [PubMed] [Google Scholar]
- 81.Grasis JA, Browne CD, Tsoukas CD. Inducible T cell tyrosine kinase regulates actin-dependent cytoskeletal events induced by the T cell antigen receptor. J Immunol. 2003;170:3971–3976. doi: 10.4049/jimmunol.170.8.3971. [DOI] [PubMed] [Google Scholar]
- 82.Labno CM, et al. Itk functions to control actin polymerization at the immune synapse through localized activation of Cdc42 and WASP. Current Biology. 2003;13:1619–1624. doi: 10.1016/j.cub.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dombroski D, et al. Kinase-independent functions for Itk in TCR-induced regulation of Vav and the actin cytoskeleton. J Immunol. 2005;174:1385–1392. doi: 10.4049/jimmunol.174.3.1385. [DOI] [PubMed] [Google Scholar]
- 84.Bogin Y, Ainey C, Beach D, Yablonski D. SLP-76 mediates and maintains activation of the Tec family kinase ITK via the T cell antigen receptor-induced association between SLP-76 and ITK. Proc Natl Acad Sci U S A. 2007;104:6638–6643. doi: 10.1073/pnas.0609771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hartgroves LC, Lin J, Langen H, Zech T, Weiss A, Harder T. Synergistic assembly of linker for activation of T cells signaling protein complexes in T cell plasma membrane domains. J Biol Chem. 2003;278:20389–20394. doi: 10.1074/jbc.M301212200. [DOI] [PubMed] [Google Scholar]
- 86.Barda-Saad M, et al. Cooperative interactions at the SLP-76 complex are critical for actin polymerization. EMBO J. 2010 doi: 10.1038/emboj.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Houtman JC, et al. Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nature structural & molecular biology. 2006;13:798–805. doi: 10.1038/nsmb1133. [DOI] [PubMed] [Google Scholar]
- 88.Jacobelli J, Chmura SA, Buxton DB, Davis MM, Krummel MF. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat Immunol. 2004;5:531–538. doi: 10.1038/ni1065. [DOI] [PubMed] [Google Scholar]
- 89.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu Y, Fay NC, Smoligovets AA, Wu HJ, Groves JT. Myosin IIA modulates T cell receptor transport and CasL phosphorylation during early immunological synapse formation. PLoS One. 2012;7:e30704. doi: 10.1371/journal.pone.0030704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumari S, et al. T Lymphocyte Myosin IIA is Required for Maturation of the Immunological Synapse. Front Immunol. 2012;3:230. doi: 10.3389/fimmu.2012.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. Journal of Cell Biology. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Waterman-Storer CM, Desai A, Bulinski JC, Salmon ED. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr Biol. 1998;8:1227–1230. doi: 10.1016/s0960-9822(07)00515-5. [DOI] [PubMed] [Google Scholar]
- 94.Shen WW, Frieden M, Demaurex N. Remodelling of the endoplasmic reticulum during store-operated calcium entry. Biology of the cell/under the auspices of the European Cell Biology Organization. 2011;103:365–380. doi: 10.1042/BC20100152. [DOI] [PubMed] [Google Scholar]
- 95.Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J Cell Sci. 2007;120:3762–3771. doi: 10.1242/jcs.015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Contento RL, Campello S, Trovato AE, Magrini E, Anselmi F, Viola A. Adhesion shapes T cells for prompt and sustained T-cell receptor signalling. EMBO J. 2010;29:4035–4047. doi: 10.1038/emboj.2010.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baixauli F, et al. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 2011;30:1238–1250. doi: 10.1038/emboj.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J Neurosci. 2010;30:8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quintana A, Kummerow C, Junker C, Becherer U, Hoth M. Morphological changes of T cells following formation of the immunological synapse modulate intracellular calcium signals. Cell Calcium. 2009;45:109–122. doi: 10.1016/j.ceca.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 105.Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol. 2007;179:733–746. doi: 10.1083/jcb.200707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shcherbina A, Bretscher A, Kenney DM, Remold-O’Donnell E. Moesin, the major ERM protein of lymphocytes and platelets, differs from ezrin in its insensitivity to calpain. FEBS Lett. 1999;443:31–36. doi: 10.1016/s0014-5793(98)01674-3. [DOI] [PubMed] [Google Scholar]
- 107.de la Fuente MA, et al. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP) Proc Natl Acad Sci U S A. 2007;104:926–931. doi: 10.1073/pnas.0610275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stewart MP, McDowall A, Hogg N. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J Cell Biol. 1998;140:699–707. doi: 10.1083/jcb.140.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bleijs DA, van Duijnhoven GC, van Vliet SJ, Thijssen JP, Figdor CG, van Kooyk Y. A single amino acid in the cytoplasmic domain of the beta 2 integrin lymphocyte function-associated antigen-1 regulates avidity-dependent inside-out signaling. J Biol Chem. 2001;276:10338–10346. doi: 10.1074/jbc.M008967200. [DOI] [PubMed] [Google Scholar]
- 110.Cairo CW, Mirchev R, Golan DE. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 2006;25:297–308. doi: 10.1016/j.immuni.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 111.Chen EJ, Shaffer MH, Williamson EK, Huang Y, Burkhardt JK. Ezrin and moesin are required for efficient T cell adhesion and homing to lymphoid organs. PLoS One. 2013;8:e52368. doi: 10.1371/journal.pone.0052368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shaffer MH, et al. Ezrin and moesin function together to promote T cell activation. J Immunol. 2009;182:1021–1032. doi: 10.4049/jimmunol.182.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watanabe Y, et al. T-cell receptor ligation causes Wiskott-Aldrich syndrome protein degradation and F-actin assembly downregulation. J Allergy Clin Immunol. 2013 doi: 10.1016/j.jaci.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 114.Wernimont SA, Simonson WT, Greer PA, Seroogy CM, Huttenlocher A. Calpain 4 is not necessary for LFA-1-mediated function in CD4+ T cells. PLoS One. 2010;5:e10513. doi: 10.1371/journal.pone.0010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morley C. The actin-bundling protein L-plastin supports T cell motility and activation. Immunological Reviews. 2013 doi: 10.1111/imr.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heng TS, Painter MW Immunological Genome Project C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 117.Namba Y, Ito M, Zu Y, Shigesada K, Maruyama K. Human T cell L-plastin bundles actin filaments in a calcium-dependent manner. J Biochem. 1992;112:503–507. doi: 10.1093/oxfordjournals.jbchem.a123929. [DOI] [PubMed] [Google Scholar]
- 118.Wabnitz GH, et al. Sustained LFA-1 cluster formation in the immune synapse requires the combined activities of L-plastin and calmodulin. Eur J Immunol. 2010;40:2437–2449. doi: 10.1002/eji.201040345. [DOI] [PubMed] [Google Scholar]
- 119.Freeley M, et al. L-plastin regulates polarization and migration in chemokine-stimulated human T lymphocytes. J Immunol. 2012;188:6357–6370. doi: 10.4049/jimmunol.1103242. [DOI] [PubMed] [Google Scholar]
- 120.Lowin-Kropf B, Shapiro VS, Weiss A. Cytoskeletal polarization of T cells is regulated by an immunoreceptor tyrosine-based activation motif-dependent mechanism. J Cell Biol. 1998;140:861–871. doi: 10.1083/jcb.140.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kuhne MR, et al. Linker for activation of T cells, zeta-associated protein-70, and Src homology 2 domain-containing leukocyte protein-76 are required for TCR-induced microtubule-organizing center polarization. J Immunol. 2003;171:860–866. doi: 10.4049/jimmunol.171.2.860. [DOI] [PubMed] [Google Scholar]
- 122.Quann EJ, Merino E, Furuta T, Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nature immunology. 2009;10:627–635. doi: 10.1038/ni.1734. [DOI] [PubMed] [Google Scholar]
- 123.Huse M, Le Floc’h A, Liu X. From lipid second-messengers to molecular motors: microtubule-organizing center reorientation in T cells. Immunological Reviews. 2013 doi: 10.1111/imr.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Combs J, et al. Recruitment of dynein to the Jurkat immunological synapse. Proc Natl Acad Sci U S A. 2006;103:14883–14888. doi: 10.1073/pnas.0600914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu X, Kapoor TM, Chen JK, Huse M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1306180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun Y, Dandekar RD, Mao YS, Yin HL, Wulfing C. Phosphatidylinositol (4,5) bisphosphate controls T cell activation by regulating T cell rigidity and organization. PLoS One. 2011;6:e27227. doi: 10.1371/journal.pone.0027227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Higgs HN, Pollard TD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol. 2000;150:1311–1320. doi: 10.1083/jcb.150.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ben-Aissa K, et al. Activation of moesin, a protein that links actin cytoskeleton to the plasma membrane, occurs by phosphatidylinositol 4,5-bisphosphate (PIP2) binding sequentially to two sites and releasing an autoinhibitory linker. J Biol Chem. 2012;287:16311–16323. doi: 10.1074/jbc.M111.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Han J, et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. doi: 10.1126/science.279.5350.558. [DOI] [PubMed] [Google Scholar]
- 130.Yonezawa N, Nishida E, Iida K, Yahara I, Sakai H. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem. 1990;265:8382–8386. [PubMed] [Google Scholar]
- 131.Hirao M, et al. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Delon J, Kaibuchi K, Germain RN. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity. 2001;15:691–701. doi: 10.1016/s1074-7613(01)00231-x. [DOI] [PubMed] [Google Scholar]
- 133.van Rheenen J, et al. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol. 2007;179:1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barreiro O, Vicente-Manzanares M, Urzainqui A, Yanez-Mo M, Sanchez-Madrid F. Interactive protrusive structures during leukocyte adhesion and transendothelial migration. Front Biosci. 2004;9:1849–1863. doi: 10.2741/1285. [DOI] [PubMed] [Google Scholar]
- 135.Ratner S, Sherrod WS, Lichlyter D. Microtubule retraction into the uropod and its role in T cell polarization and motility. J Immunol. 1997;159:1063–1067. [PubMed] [Google Scholar]
- 136.Takesono A, Heasman SJ, Wojciak-Stothard B, Garg R, Ridley AJ. Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS One. 2010;5:e8774. doi: 10.1371/journal.pone.0008774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wei SH, Safrina O, Yu Y, Garrod KR, Cahalan MD, Parker I. Ca2+ signals in CD4+ T cells during early contacts with antigen-bearing dendritic cells in lymph node. J Immunol. 2007;179:1586–1594. doi: 10.4049/jimmunol.179.3.1586. [DOI] [PubMed] [Google Scholar]
- 138.Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol. 2005;6:143–151. doi: 10.1038/ni1161. [DOI] [PubMed] [Google Scholar]
- 139.Beemiller P, Jacobelli J, Krummel MF. Integration of the movement of signaling microclusters with cellular motility in immunological synapses. Nature immunology. 2012 doi: 10.1038/ni.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Friedman RS, Beemiller P, Sorensen CM, Jacobelli J, Krummel MF. Real-time analysis of T cell receptors in naive cells in vitro and in vivo reveals flexibility in synapse and signaling dynamics. J Exp Med. 2010;207:2733–2749. doi: 10.1084/jem.20091201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Skokos D, et al. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat Immunol. 2007;8:835–844. doi: 10.1038/ni1490. [DOI] [PubMed] [Google Scholar]
- 142.Marangoni F, et al. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity. 2013;38:237–249. doi: 10.1016/j.immuni.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]