

Abstract
We have investigated the membrane destabilizing properties of synthetic amphiphilic cationic peptides, MAX1 and MAX35, which have the propensity to form β-hairpin structures under certain conditions, and a control non-β-hairpin-forming peptide MAX8V16E. All three peptides bind to liposomes containing a mixture of zwitterionic POPC and negatively charged POPS lipids as determined by Zeta potential measurements. Circular dichroism measurements indicated folding of MAX1 and MAX35 in the presence of the POPC/POPS liposomes, whereas no such folding was observed with MAX8V16E. There was no binding or folding of these peptides to liposomes containing only POPC. MAX1 and MAX35 induced release of contents from negatively charged liposomes, whereas MAX8V16E failed to promote solute release under identical conditions. Thus, MAX1 and MAX35 bind to, and fold at the surface of negatively charged liposomes adopting a lytic conformation. We ruled out leaky fusion as a mechanism of release by including 2 mol % PEG-PE in the liposomes, which inhibits aggregation/fusion but not folding of MAX or MAX-induced leakage. Using a concentration-dependent quenching probe (calcein), we determined that MAX-induced leakage of liposome contents was an all-or-none process. At MAX1 concentrations, which cause release of ∼50% of the liposomes that contain small (Rh <1.5 nm) markers, only ∼15% of those liposomes release a fluorescent dextran of 40 kDa. A multimeric model of the pore is presented based on these results. Atomistic molecular dynamics simulations show that barrels consisting of 10 β-hairpin MAX1 and MAX35 peptides are relatively more stable than MAX8V16E barrels in the bilayer, suggesting that barrels of this size are responsible for the peptides lytic action.
Introduction
Antimicrobial peptides (AMPs) are a unique and diverse group of molecules, which target and disrupt the function of cell membranes (1). These peptides are capable of killing bacteria (2), fungi (3), and cancer cells (4–6). We designed a class of hydrogel materials from self-assembling peptides whose surfaces display antibacterial activity (7). Interestingly, the primary sequence and amphiphilic secondary structure of these self-assembling peptides resemble classical AMPs. However, distinct from AMPs is their ability to self-assemble into macroscopic gels. Our peptides, initially unordered when dissolved in aqueous solution, can be triggered to fold into an amphiphilic β-hairpin conformation that rapidly self-assembles to form a network of fibrils that constitute the formation of a hydrogel. For example, the peptide MAX1 is composed of N- and C-terminal β-strands containing alternating hydrophobic (valine) and hydrophilic (lysine) residues (Table 1). A central four-residue sequence (-VDPPT-) connects the two strands. At neutral pH and solution of low ionic strength, electrostatic repulsion between protonated lysine side chains keeps the peptide unfolded. Increasing the ionic strength of a neutral solution with NaCl to 150 mM (8) or simply increasing the pH to 9 reduces the positive charge (9,10), allowing the peptide to fold into a facially amphiphilic β-hairpin that rapidly self-assembles. We have reported earlier that the surface of the resulting macroscopic gel confers antibacterial activity. As will be shown, we have found that negatively charged surfaces, such as that provided by model liposomes can also induce the folding of MAX1. Here, electrostatic interactions between the positively charged lysine side chains and negatively charged lipid headgroups drive the folding of the peptide into an amphiphilic hairpin. When the peptides fold at the surface of liposome membrane it is unable to undergo gelation. Rather, the folded conformer acts to destabilize the membrane resulting in liposome leakage. Herein, we use a combination of biophysical techniques and peptide design to probe the mechanism of action. The influence of peptide hydrophobic content is assessed via the study of MAX35, a derivative of MAX1 that contains four isoleucine residues in place of four original valine side chains. Finally, a control peptide, MAX8V16E, was prepared to study the importance of membrane-induced folding with respect to the peptide’s membrane-disrupting action. MAX8V16E contains two sequential glutamate residues that render it incapable of folding at the surface of the membrane.
Table 1.
Physical properties of liposomes
| Liposome formulation | Physical characteristic | No peptide | MAX35 | MAX1 | MAX8V16E |
|---|---|---|---|---|---|
| POPC | Diameter (nm) | 97 ± 19 | 112 ± 3 | 104 ± 10 | 110 ± 7 |
| Zeta potential (mV) | −7.5 ± 2.0 | −3.0 ± 2.0 | −3.7 ± 1.5 | −5.7 ± 1.2 | |
| POPC/POPS (1:1) | Diameter (nm) | 79 ± 1 | 106 ± 14 | 109.0 ± 24 | 96.0 ± 16 |
| Zeta potential (mV) | −35.4 ± 2.2 | −19.7a ± 1.6 | −23.9a ± 2.3 | −30.4 ± 2.4 | |
| Sequence | MAX1: VKVKVKVKVDPPTKVKVKVKV-NH2 | ||||
| MAX35: IKVKIKVKVDPPTKIKVKIKV-NH2 | |||||
| MAX8V16E: VKVKVKVKVDPPTKVEEKVKV-NH2 | |||||
Diameter and Zeta potential of liposome with or without MAX peptides. The sequences of the MAX peptides are shown in the bottom row.
Values were significantly different from no peptide control according to an analysis of variance test, p ≤ 0.05, n = 2.
We used unilamellar vesicles (liposomes) with an average diameter of 100–300 nm consisting of a neutral phospholipid, 1-palmitoyl-2-oleoyl-sn-glycero-phosphocholine (POPC)#, and a negatively charged phospholipid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphor-L-serine (POPS), at various mole ratios. Circular dichroism (CD) spectroscopy was employed to assess the β-hairpin forming propensity of these peptides as a function of negative charge on the liposomes. Release of liposome contents as a result of interaction with MAX peptides was monitored using different molecular weight markers encapsulated into the liposomes. Our data show that addition of MAX1 and MAX35 to liposomes containing a sufficient negative charge (>10% PS) results in a rapid release of small molecules in a dose and negative surface charge-dependent manner. At MAX1 concentrations, which cause release of ∼50% of the liposomes that contain small (Rh <1.5 nm) markers, only ∼15% of those liposomes release a fluorescent dextran of 40 kDa. We surmise that the 15% value represents the formation of very large (diameter >10 nm) pores and the remaining liposomes release their contents via pores that can discriminate between molecules of different sizes. Atomistic molecular dynamics (MD) simulations are consistent with the notion that these peptides form β-barrels in the lipid bilayer, which are relatively stable and allow permeation of molecules with defined size limitations.
Materials and Methods
Phospholipids POPC, POPS, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (PEG-PE) were purchased from Avanti Polar Lipids (Alabaster, AL). Tb (III) Chloride Hexahydrate (TbCl3·6H2O), 2,6-Pyridinedicarboxylic acid (Dipicolinic acid), octyl β-D-glucopyranoside (OG), and other reagents were purchased from Sigma-Aldrich (St. Louis, MO). SephadexG-50 and Sepharose CL-6B were purchased from GE Healthcare Biosciences AB (Uppsala, Sweden). Texas red dextran 40,000 (TRD-40 k) was purchased from Invitrogen. Appropriately side-chain protected Fmoc-amino acids were purchased from Novabiochem (Billerica, MA). 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyl-aminium hexafluorophosphate (HCTU) was purchased from Peptides International (Louisville, KY). 3H-inulin was purchased from Perkin Elmer (Boston, MA). Buffer 1, 10 mM TES, 100 mM NaCl, 1 mM EDTA (pH 7.4); Buffer 2, 10 mM TES, 100 mM NaCl (pH 7.4); Stock solutions: Tb/DPA, 7.5 mM TbCl3, 75 mM Na citrate, 75 mM Na dipicolinate, 10 mM TES (pH 7.4). Inulin solution was prepared in buffer 1 (pH 6.6) at a concentration of 1% weight by volume by incubation for 2 h at 55°C followed by cooling at room temperature. Any insoluble material was removed by centrifugation and the clear supernatant was used further. Calcein solution was prepared at a final concentration of 100 mM (pH 7.4).
Peptides were prepared on PL-Rink Resin in 0.25 mM quantities via automated Fmoc peptide synthesis employing an ABI 433A peptide synthesizer (Applied Biosystems, Snoqualmie, WA) and HCTU activation. The resulting dry resin-bound peptides were cleaved and side-chain-deprotected using a TFA: thioanisole: ethanedithiol: anisole (90:5:3:2) cocktail. Crude peptides were purified by RP-HPLC (Agilent technologies, Santa Clara, CA) using preparative Grade C18 peptide/protein column (Vydac, Hesperia, CA). Peptides were purified using a linear gradient from solvent A (0.1% TFA in water) to solvent B (90% Acetonitrile, 9.9% water, 0.1% TFA). The following gradients were used for each peptide: MAX1 15% to 100% B over 159 min; MAX35 18% to 100% B over 170 min; MAX8V16E 16% to 100% B over 175 min. MAX1 electrospray ionization-mass spectrometry (ESI-MS, Shimadzu, Columbia, MD): 2231.4 [(M+H)+calculated 2229.96]; MAX35 ESI-MS: 2287.4 [(M+H)+ calculated 2286.1]; MAX8V16E ESI-MS: 2262.2 [(M+H)+ calculated 2260.88]. More detailed methods including liposome preparation, size and Zeta potential measurements, CD studies, solute release assays, and atomistic MD simulations are available in the Supporting Material, Document S1.
Results
Structural changes for MAX peptides upon binding to negatively charged liposomes
For our studies on MAX peptide-liposome interactions we used POPC (zwitterionic) and/or POPS (negatively charged) as the prototype lipids. We characterized these liposomes with respect to their size and surface charge (Table 1). We performed a one way analysis of variance of the liposome size and Zeta potentials listed in Table 1 (see Document S1 in the Supporting Material). The measured parameters with peptide were tested against the liposomes without the peptide (controls). This analysis shows no significant changes in size of liposomes (POPC alone or POPC/POPS) in the presence or absence of MAX peptides. Therefore, we conclude that the binding of MAX peptides to POPC and POPS/POPC liposomes does not affect their size distribution. These data rule out MAX-induced aggregation or fusion of these liposomes. There were no significant changes in Zeta potentials of POPC liposomes in the presence of the MAX peptides and of POPS/POPC liposomes in the presence of MAX8VE16. By contrast, Zeta potentials of POPS/POPC liposomes were significantly different in the presence of MAX1 and MAX35 indicating binding of these peptides to these liposomes. Because binding and folding of MAX1 and MAX35 peptides are electrostatically driven, there are much higher probabilities for these peptides to bind anionic lipid liposome compared to zwitter-ionic POPC vesicles.
CD spectroscopy shows that MAX1 and MAX35 fold at the surface of negatively charged liposomes. Fig. 1 A shows that when MAX1 is dissolved in buffer alone, it remains unfolded, but folds when negatively charged POPS-containing liposomes are present as evident by the increase in negative mean residue ellipticity at ∼218 nm. In contrast, when neutral POPC liposomes are present, the peptide remains unfolded. The data suggest that MAX1 binds to and folds at the surface of negatively charged liposomes and that the driving force for folding is electrostatically driven. Presumably, the lysine side chains of the peptide can form salt bridge pairs with the POPS headgroups to drive the folding event, which leads to the formation of an amphiphilic hairpin. When the peptide initially folds at the surface of the liposome, the lysine-rich face of the hairpin is engaged with the lipid headgroup region and its hydrophobic face is exposed to bulk water. Solvating this hydrophobic surface area with water is an energetically unfavorable proposition. In response, the peptide partitions into the membrane to release ordered water and solvate its hydrophobic face with lipid’s hydrophobic core. Fig. 1 B shows that MAX35, the more hydrophobic peptide behaves in a similar fashion. However, Fig. 1 C shows that the control peptide, MAX8V16E is unable to fold at the surface of negatively charged liposomes. Here, the anionic glutamates, through charge repulsion with the negatively charged liposome, disfavors binding (see Table 1) and folding. The CD data taken together indicate that both MAX1 and MAX35 fold at the surface of negatively charged liposomes, adopting amphiphilic hairpin conformations. The exact mechanism by which the peptide partitions into the membrane after the initial binding and folding events is not known. But as will be shown, once partitioned into the membrane, the folded amphiphiles can induce liposomal leakage.
Figure 1.
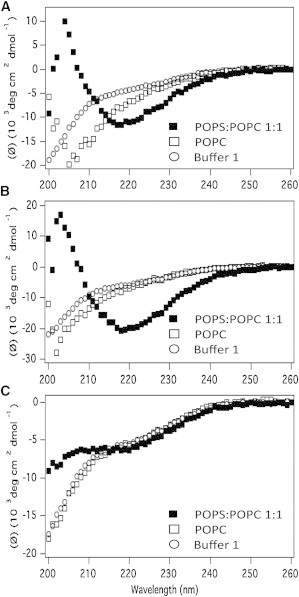
CD spectra of MAX peptides in buffer 1 (○), POPC liposomes (□), and POPC/POPS (1:1) liposomes (▪) for (A) MAX1 (B) MAX35, and (C) MAX8V16E. All samples contain 50 μM peptide and a lipid/peptide ratio of 50:1.
β-hairpin peptides induced membrane permeabilization
To monitor the permeabilization of liposomes upon interaction with MAX peptides, we have employed a fluorescence-based Tb3+/DPA assay (11). Tb3+/DPA chelates, which are ∼10,000 times more fluorescent than free Tb3+, were encapsulated into liposomes. Upon the release of liposome contents into a medium containing EDTA, fluorescence decreases due to the dissociation of the Tb3+/DPA complexes. Maximum release was calculated by adding OG to the medium. Fig. 2 shows the kinetics of leakage of POPC/POPS liposomes upon addition of MAX1 and MAX35. Strikingly, the addition of MAX1 (Fig. 2 A) and MAX35 (Fig. 2 B) resulted in a rapid release of liposome contents reaching a steady state within ∼3 s.
Figure 2.
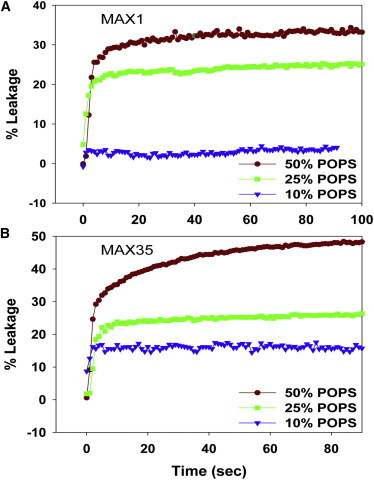
Kinetics of leakage of Tb/DPA from POPC/POPS liposomes containing varying % of POPS viz; 10% (▾), 25% (▪), 50% (●) upon addition of (A) MAX1 and (B) MAX35 peptides at a lipid/peptide ratio of 24:1. To see this figure in color, go online.
The CD and leakage data taken together indicate that formation of β-structure is required for the permeabilization of the liposome membrane and that the interaction of positively charged peptide with negatively charged lipids is necessary but not sufficient. Various mechanisms have been invoked to account for the mode of action of lytic peptides on membrane permeabilization. One such mechanism posits that the peptide induces aggregation/fusion of liposomes that is accompanied by leakage of liposome contents (leaky fusion) (12). However, aggregation/fusion of liposomes is prevented by inclusion of pegylated lipid into liposomes (12). CD spectra indicate that folding of the MAX peptides was identical in our liposome formulations prepared with or without inclusion of 2 mol % PEG-PE (data not shown). Fig. 3 shows similar leakage for pegylated (right panels) versus non-pegylated (left panels) liposomes ruling out leaky fusion as a mechanism for MAX peptide-induced membrane permeabilization.
Figure 3.
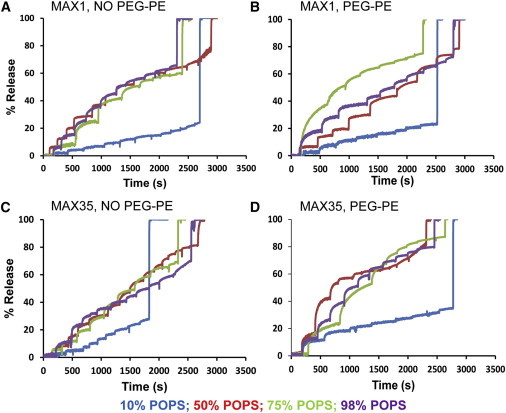
Sequential addition of MAX peptides to the same liposomal formulation (nonpegylated and pegylated 2 mol % PEG-PE) of POPC/POPS cause additional Tb/DPA leakage. Each liposome formulation contains a different % of POPS. MAX1 and MAX35 were added at a concentration of 0.25 μM successively to each liposomal formulation after a certain interval of time resulting in Tb/DPA leakage at each addition. Each successive increase in the profile shows Tb/DPA leakage corresponding to the peptide addition. The last addition corresponds to the addition of detergent OG to obtain maximal leakage.
Even though sufficient peptide is available at L/P = 24 to form at least one pore (see below) in all the liposomes, the extent of leakage only reached ∼50% (Fig. 2, A and B). We therefore examined whether adding more peptide would cause additional leakage. Indeed, at POPS/POPC ratios ≥50%, more extensive leakage is realized (Fig. 3). However, at 10 mol % POPS the extent of leakage did not go beyond ∼30% even at low L/P ratios. These data indicate that a threshold of POPS in the liposome is required for membrane permeabilization.
Fig. 4 shows dose-response curves for the extent of leakage induced by MAX1 (Fig. 4 A) and MAX35 (Fig. 4 B) peptides, respectively. Overall, the activities of the two peptides do not appear to be very different except for higher activity of MAX35 at 10 mol % POPS (see above) in the liposomes. MAX activities in liposomes containing 50 and 75 mol % POPS seem to be higher than in 98% POPS liposomes (Fig. 3), whereas in liposomes containing 10 mol % POPS the maximal release even at high peptide concentrations remains in the range of 15–40%. Fitting the curves to a Langmuir isotherm (see legend) yields maximal extents of release and the peptide concentrations for half-maximal activity. In Fig. 4 C we show the maximal extents of leakage for MAX1, MAX35, and MAX8V16E as a function of mol % POPS in the liposomes. Whereas the leakage activities of MAX1 and MAX35 are similar at POPS >25 mol %, it appears that at 10 mol % POPS in the liposomes, MAX35 induces higher leakage activity. This may be the result of greater hydrophobicity of MAX35 resulting in a higher probability of pore formation. Interestingly, a decrease is seen in the extent of leakage from liposomes containing very high POPS levels suggesting that initial peptide binding is not sufficient for membrane disruption. A similar observation has been reported by Ramamoorthy and co-workers (13) who showed that the antimicrobial peptide pexiganan cannot disrupt purely anionic lipid bilayers like POPG or POPS as they bind strongly to the bilayer surface. MAX8V16E had no significant binding activity (Table 1) and did not induce leakage from liposomes at all POPS concentrations tested.
Figure 4.
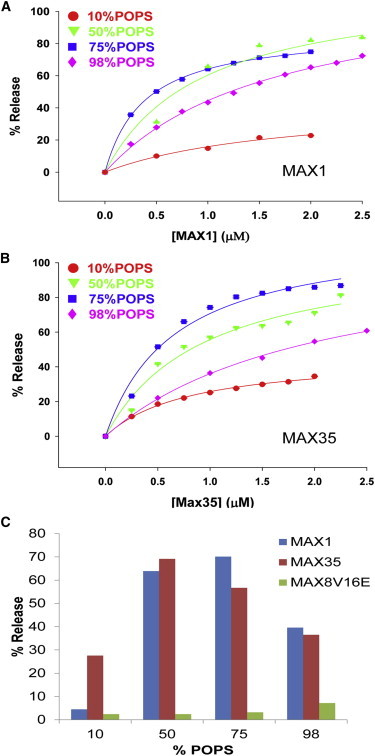
(A) Dose response curve for the extent of Tb/DPA leakage induced by the addition of MAX1 peptide to liposomes of POPC/POPS/PEG-PE. Each liposome formulation contains a different % of POPS. (B) Dose response curve for the extent of Tb/DPA leakage induced by the addition of MAX35 peptide to liposomes of POPC/POPS/PEG-PE. Each liposome formulation contains a different % of POPS. The data were fitted to a Langmuir isotherm f = a∗x/(b+x), where f is extent of release, a represents maximal release, and b is the concentration of peptide at half-maximal release. (C) The maximal extents of release are plotted against %POPS in the liposome. Blue bars: MAX1; brown bars: MAX35; green bars: MAX8V16E.
Release mechanisms
Liposomes containing differentially sized markers were incubated with MAX1 and fractionated through a sizing column to separate liposome-containing macromolecules from the free macromolecules. Fig. 5 A shows that MAX1 induced ∼33% leakage of inulin (mw. ∼5000; Rh ∼1.39 nm) from POPC/POPS liposomes. To test whether MAX peptide-induced leakage may result from formation of pores that can discriminate between molecules of different sizes, we encapsulated both calcein (mw. ∼622.55; Rh ∼0.74 nm) and Texas red dextran 40,000 (TRD-40 k, mw. ∼40,000; Rh ∼5 nm) into the same liposomes and then examined the differential release of both molecules induced by MAX1. Under conditions that cause release of ∼50% of the liposomes that contain calcein, only ∼15% of those liposomes released the TRD-40 k (Fig. 5 B). The data indicate that under these experimental conditions 15% of the liposomes release their contents as a result of very large (diameter >10 nm) pore formation, whereas the remaining liposomes contain pores that allow permeation of molecules with defined size limitations.
Figure 5.
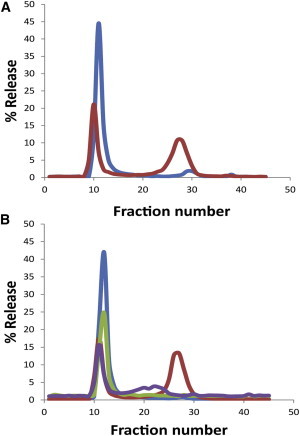
Release of macromolecules of different hydrodynamic radii (Rh). Release from POPC/POPS/PEG-PE (1:1:0.02, mole ratio) liposomes was determined upon addition of MAX1 peptide at L/P = 24. (A) % Release of 3H-inulin (mw. ∼5000; Rh ∼1.39 nm), blue line denotes untreated liposomes and red line denotes liposomes treated with MAX1. (B) Coencapsulated TRD-40 k (mw. ∼40,000; Rh ∼5 nm) and calcein (mw. ∼622.55; Rh ∼0.74 nm). The fluorescence intensity was measured at λex = 490 nm and λem = 517 nm (calcein) for untreated (blue line) and MAX1 treated (red line), respectively, and at λex = 590 nm and λem = 615 nm (TRD-40 k) for untreated (green line) and MAX1 treated (purple line), respectively. The peaks at higher fraction numbers represent released molecules. The % release was calculated from the formula; 100 × [total counts (3H or fluorescence) in the released fractions]/[the total counts in all fractions]. The average release calculated from two separate experiments was 51 ± 2, 33.3 ± 8.3, and 15.5 ± 1.5% for calcein, inulin, and TRD-40 k, respectively.
The question is whether leakage via the (remaining) pores is graded or all or none. In other words when we measure 50% leakage do all vesicles leak 50% of their contents or do 50% of the vesicles leak all of their contents and the remaining vesicles remain intact. In the former case the pores will open for a very short amount of time, insufficient to allow complete depletion of vesicle contents. We followed the procedure developed by Weinstein et al. (14) to make this determination. Because these molecules exhibit a concentration-dependent quenching mechanism, quenching ratios (Q) following MAX-induced leakage and repassing the vesicles through a column should indicate the appropriate mechanism. In the case of all or none the recovered vesicles should exhibit the same quenching ratios as the original vesicles, whereas in the graded case the quenching ratios should change. Fig. 6 clearly shows that the recovered vesicles closely follow the original curve indicating an all-or-none mechanism. The data suggest the formation of a very stable pore allowing enough time for the peptide-challenged vesicles to deplete their contents.
Figure 6.
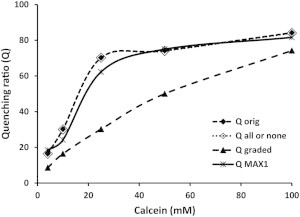
MAX1-induced relief of self-quenching of calcein. The quenching ratio (Q) in each POPC/POPS/PEG-PE (1:1:0.02, mole ratio) liposome preparation that encapsulated calcein at different concentrations was determined by measuring fluorescence at λex = 490 nm and λem = 517 nm before and after adding 1% OG. Black diamonds: Q of the original liposomes; White diamonds: calculated Q for an all or none release mechanism; Dark triangles: calculated Q for a graded mechanism; Crosses: experimentally determined Q after treatment of liposomes with MAX1 at L/P = 24 and removing the released calcein by passage through a Sepharose CL-6B column.
Atomistic MD simulations
To gain more insight into possible pore-forming properties of MAX peptides, we performed atomistic MD simulations using the CHARMM program (15). Using the coordinates of an idealized β-hairpin, we constructed annular β-sheets in the barrel (Fig. 7 A) and channel (Fig. 7 B) topologies. To construct the barrel structure, the β-hairpin was inclined ∼37° relative to the pore axis and then a 10-fold rotational symmetry operation was performed with respect to the pore axis, creating a decameric MAX barrel (16). To construct the channel structure with the conventional β-strands arrangement, 10 β-hairpins were inserted without inclination, generating a decameric MAX channel (17,18). We modeled MAX barrels/channels with the β-sheet structure. For MAX barrels, our simulations employed the β-barrel morphology by mimicking naturally occurring β-barrels observed in transmembrane proteins that are found frequently in the outer membranes of bacteria, mitochondria, and chloroplasts. The β-barrel motif is a large β-sheet composed of an even number of β-strands. Some known structures of β-barrel membrane proteins have β-strands ranging in size from 8 to 22 (19,20). Our modeled 10-mer MAX barrels contain 20 β-strands enclosing the solvated pore. This number is also in the range of the number of β-strands for natural β-barrels ranging from 8 to 22, which can form a β-barrel motif. For MAX channels, we modeled the β-sheet channel morphology for an extension of the β-barrel morphology. The decameric MAX β-sheet channels also contain 20 β-strands enclosing the solvated pore. Our previous simulations for Aβ channels, which contain the same β-sheet channel morphology, indicate that different numbers of Aβ monomers could produce channels with different outer and pore dimensions (21–25). For the β-sheet channel morphology, we found that Aβ channels obtained a preferred size in the range of 16–24 β-strands lining the pores (23,24). This range was also found to hold for other toxic β-sheet channels; K3 (a fragment of β2-microglobulin) channels with 24 β-strands (26) and protegrin-1 (PG-1) channels with 16–20 β-strands (17,18). In both barrel/channel topologies, the β-hairpin arrangements give rise to two potential β-sheet motifs; turn-next-to-tail β-hairpins in NCCN packing mode and turn-next-to-turn β-hairpins in NCNC packing mode (27), where N and C denote the N- and C-terminal strands, respectively (Fig. 7). In both cases, the β-hairpins form antiparallel β-sheets, positioning the positively charged Lys side chains into the central pore (Fig. S1 in the Supporting Material). For PG-1 β-hairpin, intermolecular packing and alignment in the ordered β-hairpin oligomerization have been determined (28–30). These experiments suggested some possible β-hairpin packings with antiparallel and parallel intermolecular β-strand arrangements. However, for the MAX β-hairpin, no experimental data for the peptide arrangement is currently available. In our simulation, we modeled MAX β-sheet barrels/channels with the antiparallel β-hairpin arrangement. This ensures that all charged Lys side chains point toward the solvated pore. However, in the parallel MAX β-hairpin arrangement, half of the peptides in the barrels/channels unfavorably point their charged Lys side chains toward the lipid hydrophobic core. We constructed 12 independent initial conformations of MAX barrels and channels for explicit solvent MD simulations with a mixed zwitterion/anionic lipid bilayer composed of POPC/POPS (mole ratio 3:1).
Figure 7.
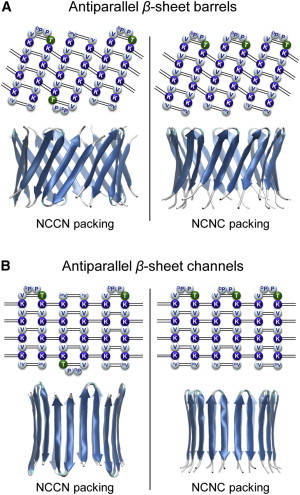
Topological diagrams for the MAX1 β-hairpins in the antiparallel β-sheet arrangement for the MAX1 (A) barrel and (B) channel in the NCCN and NCNC packing modes. Blue and white beads represent the positively charged (Lys) and hydrophobic (Val, DPro, and Pro) residues, respectively, and the polar residue (Thr) is denoted by green beads. Dotted lines indicate the backbone hydrogen bond (H-bond). The first and the last residues for each monomer are indicated by the residue number. The cartoons representing the barrels/channels are in lateral view.
The MAX barrels/channels are preassembled initially as an annular shape with 10 β-hairpins. During the simulations, the initial annular conformation is further optimized in the MAX1 and MAX35 barrels preserving the stable β-barrel topology through the formation of the inter- and intramolecular backbone hydrogen bonds (H-bonds) (Fig. 8), although the control barrel composed of MAX8V16E and all channels (composed of MAX1, MAX35, and MAX8V16E) gradually dismiss the annular shape via relaxation of the lipid bilayer yielding discontinuities in the β-sheet network (Fig. S2). All MAX barrels/channels increase their outer diameter, 1–2 nm from the starting points, due to the charge repulsion between the Lys side chains in the solvated pore. Both MAX1 and MAX35 barrels preserve the solvated pore with pore diameter ∼2.0 nm, wide enough for conducting ions or small molecules as calculated by the HOLE program (31). Although the β-sheets are partially broken in the MAX1 and MAX35 channels, they also retain the solvated pore. However, the MAX8V16E barrels/channels generally collapse the pore due to unstable β-hairpins in the lipid bilayer (Fig. S3).
Figure 8.
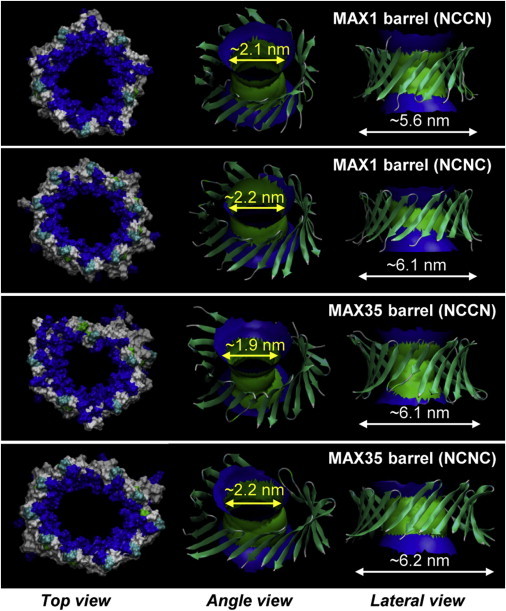
Averaged pore structures calculated by the HOLE program (31) embedded in the averaged barrel conformations during the simulations for the MAX1 and MAX35 barrels in the NCCN and NCNC packing modes. In the top view, MAX barrels are shown in the surface representation with the color representations: hydrophobic residues are shown in white, positively charged residues are shown in blue, and polar residues are shown in green. In the angle and lateral views of the pore structure, barrel structures are shown with the ribbon representation. For the pore structures in the surface representation, red denotes pore radius of r < 0.8 nm, green denotes pore radius in the range, 0.8 nm ≤ r ≤ 1.2 nm, and blue denotes pore radius of r > 1.2 nm.
The interaction of individual MAX β-hairpins with surrounding solvent including lipids, water, and ions is inhomogeneous. Well-balanced β-hairpin interactions with solvent could support stable barrel/channel conformation in the complex lipid environment. We calculated the interaction energy for each MAX β-hairpin with lipids, water, and anions (Cl−). The β-hairpin interaction energy was then averaged over time and the number of peptides in the barrel/channel. The percentage of peptide-solvent interactions for the three components, i.e., lipid, water, and anion, represents the relative strength of peptide interactions made with the surrounding environment (Fig. S4). For the MAX1 and MAX35 barrels, the lipid and water interactions are relatively weak, whereas anion interaction is relatively strong. In our simulations, the initial locations of ions were in the bulk with |z| > 3 nm from the bilayer center. No ions were initially placed in the water pore. However, during the course of preequilibration, these ions rapidly migrated into the water pore. Especially, the anion, Cl− penetrated into the pore and screened the charged Lys-Lys side-chains interactions. After the initial transient t > 30 ns, we found that the average number of anions in the pore is ∼40 Cl−. Three peaks in the probability distribution curve for Cl− denote the anion locations in between four Lys side chains lined along the pore axis, indicating that anions effectively screen the positively charged Lys side chains in the solvated pore (Fig. 9). The MAX1 and MAX35 channels exhibit similar interaction patterns as the barrel motif, suggesting that the channel topology is in the metastable state toward more stable β-barrel conformation. In the starting conformations of MAX1 and MAX35 channels, β-hairpins are inserted conventionally without any tilt with respect to the membrane normal. However, we observed that the β-strand tilt angle increases, close to the values of inclined β-hairpins in the MAX barrel topology during the simulations (Fig. S5). The MAX8V16E is unstable in the lipid bilayer and partitions back into solution.
Figure 9.
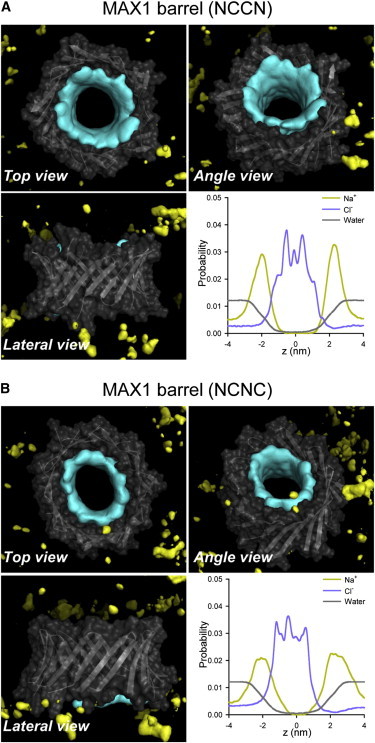
Three-dimensional density maps of Na+ (yellow surface) and Cl− (cyan surface) for the MAX1 barrels in the (A) NCCN and (B) NCNC packing modes. In the maps, the averaged barrel structures are shown as cartoons in gray. Probability distribution functions for Na+ (orange line), Cl− (blue line), and water (gray line) as a function of the pore axis are shown.
Discussion
Peptide-induced leakage of various molecules through liposome membranes studied here involves binding of the peptide to the membrane surface, followed by conformational changes, peptide aggregation, insertion, and formation of a pore (1). We have been able to follow these events using a variety of biophysical techniques. The peptides MAX1 and MAX35 bind to negatively charged membranes as shown by Zeta potential measurements but not to liposomes composed of phospholipids containing only zwitterionic headgroups. Therefore, peptide binding occurs through electrostatic interactions between positively charged amino acids on the peptide and negatively charged headgroups on phospholipids. Although in this study we have focused on phosphatidylserine as the negatively charged lipid because of its presumed overexpression in cancer cells (5,32), incorporation of alternative negatively charged lipids such as phosphatidylglycerol showed similar leakage kinetics (Fig. S6).
MAX1 and MAX35 are capable of folding on POPC/POPS membranes as shown by CD measurements. By contrast MAX8V16E does not fold at the surface of those membranes presumably due to the presence of the two sequential glutamate residues. In contrast to MAX8V16E, both MAX1 and MAX35 induce rapid content release when added to POPS-containing liposomes. We ruled out leaky fusion (12) as a mechanism of release by incorporating lipid-anchored PEG (PEG-PE) into the liposome membrane, which inhibits aggregation/fusion. Such leaky fusion can be demonstrated in the case of Ca2+-mediated fusion of POPS liposomes (Fig. S7).
At high peptide/lipid ratios release of contents of all liposomes can be achieved, but at lower ratios only a given percentage of vesicles release their contents in an all-or-none fashion. Because there are nine positively charged residues on MAX1 and MAX35, nine POPS molecules on the liposome surface would constitute a binding site that satisfies all the charge-charge interactions between lipids and peptide. POPS:POPC (1:1) liposomes would then have 1553 binding sites on their surface and sufficient peptide is available to bind to these sites (see Document S2 in the Supporting Material for calculations). Therefore, under these conditions a large excess of peptide is available to form a 10 peptide-containing pore that should release small molecules in all liposomes. The apparent contradiction between high amounts of peptide required to produce quantitative leakage and low amounts of peptide to produce a pore can be reconciled by considering studies showing that certain AMPs initially bind to the membrane surface causing an increase in outer monolayer tension that allows peptide insertion to form a pore (33,34). Accordingly, sufficient binding of MAX peptides to POPS-containing liposomes is required to produce the outer monolayer expansion required for MAX peptide insertion. For instance, at a peptide/lipid ratio of 1/24 only about half of the population of POPS:POPC (1:1) liposomes release calcein and ∼15% of that population release TRD-40 k (Fig. 5). From these experiments we surmise that in 50% of the liposomes insufficient outer monolayer tension is produced to allow peptide insertion and no leakage is observed. We have entertained the possibility that the leakage of TRD-40 k is due to the formation of very large (>10 nm) lipidic or toroidal pores (33,34). However, based on the theoretical analysis and MD simulations presented here we do favor a barrel stave model in the case of the MAX peptides. Final resolution of the pore structure will require solid-state NMR (35) or x-ray diffraction (36).
Although no significant differences in the leakage-inducing capabilities between MAX1 and MAX35 could be detected at POPS ≥25%, at 10% POPS, MAX35 clearly induced greater leakage than MAX1 (Fig. 2). Presumably, in a significant portion of the 10% POPS containing liposome population, an insufficient amount of surface POPS is available to provide the underlying matrix required for peptide binding and outer monolayer expansion. However, at a given outer monolayer tension the more hydrophobic MAX35 appears to have a larger probability of insertion than MAX1. The POPS distribution on the liposome surface and aggregation of peptides required to form a pore are complex issues still under investigation. Because liposome contents are emptied out within ∼10 ms (see Document S2 in the Supporting Material for calculations) following pore formation the rate-limiting steps in the overall processes are presumably aggregation/membrane expansion/insertion.
Previous studies on AMP PG-1 have also shown that membrane perturbation activity was dependent on their β-hairpin forming nature (17,18,27,37,38). PG-1 is a small β-hairpin peptide consisting of 18 amino acids with a high content of positively charged Arg residue, and two disulfide S-S bonds between Cys side chains that constrain the hairpin conformation (39,40). PG-1 is able to alter the permeability of bilayers, forming β-sheet channels in the membrane (30,35,41,42), and electrical recordings show multiple single channel conductances (18). PG-1 and MAX peptides are very similar: i), Both share the same β-hairpin secondary structure. ii), They are highly positively charged. iii), Conformational β-hairpin constraints depend on their sequences, i.e., disulfide S-S bonds versus tetrapeptide turn sequence (-VDPPT-). However, the charged side chains of the peptides play a critical role in the formation of channels in the membrane. In the PG-1 peptide, positively charged side chains are located at the β-hairpin’s turn and termini. Thus, when assembled in a channel, both ends of the cylindrical channel are charged. This bifurcated charge distribution in the PG-1 channel aligns the channel in the lipid bilayer. In contrast, our modeled MAX barrels/channels contain their positively charged side chains in the interior of a solvated pore. The asymmetric charge distribution across the β-sheet plane induces strong repulsive forces at the clustered Lys side chains. This suggests that small MAX barrels/channels may not be stable due to closely packed charged side chains and larger barrels, greater than the 10-mer barrel in our simulations, can be populated.
Conclusions
CD, liposome release, and sizing experiments, as well as modeling suggest a mechanism of membrane permeation consistent with surface-induced peptide folding followed by outer monolayer expansion, peptide insertion, and barrel formation. CD shows that MAX1 and MAX35 are capable of folding at the surface of negatively charged liposomes forming amphiphilic hairpins. The lysine-rich face of the hairpin interacts with the negatively charged headgroup region of the liposome and it is this interaction that drives the folding event. Once folded at the liposome surface, the hydrophobic face of the hairpin would be largely exposed to bulk water, an energetically unfavorable proposition. In response, the hairpin partitions into the membrane to release ordered water and solubilize its hydrophobic side chains in the lipid. Modeling and release studies indicate that once associated with the liposome, the hairpin rapidly assembles to form barrel-like structures with pore diameters of at least 3 nm. The data also indicate that these barrels induce leakage in an all-or-none fashion where the liposomes that do undergo disruption rapidly leak all of their content. It should be noted that the mechanism leading to liposomal disruption undercover in this study is distinct from the mechanism of antibacterial action enjoyed by MAX1 and MAX35 in their gelled state. Once self-assembled into the hydrogel’s fibrillar network, there is no free peptide available for barrel formation. Thus, MAX1 and MAX35 are capable of disrupting membranes in both their self-assembled, gel-state and their freely soluble state by distinct mechanisms.
Acknowledgments
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Supporting Material
References
- 1.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 2.Shai Y., Makovitzky A., Avrahami D. Host defense peptides and lipopeptides: modes of action and potential candidates for the treatment of bacterial and fungal infections. Curr. Protein Pept. Sci. 2006;7:479–486. doi: 10.2174/138920306779025620. [DOI] [PubMed] [Google Scholar]
- 3.van der Weerden N.L., Bleackley M.R., Anderson M.A. Properties and mechanisms of action of naturally occurring antifungal peptides. Cell. Mol. Life Sci. 2013;70:3545–3570. doi: 10.1007/s00018-013-1260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mader J.S., Hoskin D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs. 2006;15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 5.Sinthuvanich C., Veiga A.S., Schneider J.P. Anticancer β-hairpin peptides: membrane-induced folding triggers activity. J. Am. Chem. Soc. 2012;134:6210–6217. doi: 10.1021/ja210569f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoskin D.W., Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salick D.A., Kretsinger J.K., Schneider J.P. Inherent antibacterial activity of a peptide-based beta-hairpin hydrogel. J. Am. Chem. Soc. 2007;129:14793–14799. doi: 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozbas B., Kretsinger J., Pochan D.J. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules. 2004;37:7331–7337. [Google Scholar]
- 9.Rajagopal K., Lamm M.S., Schneider J.P. Tuning the pH responsiveness of beta-hairpin peptide folding, self-assembly, and hydrogel material formation. Biomacromolecules. 2009;10:2619–2625. doi: 10.1021/bm900544e. [DOI] [PubMed] [Google Scholar]
- 10.Schneider J.P., Pochan D.J., Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002;124:15030–15037. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 11.Wilschut J., Düzgüneş N., Papahadjopoulos D. Studies on the mechanism of membrane fusion: kinetics of calcium ion induced fusion of phosphatidylserine vesicles followed by a new assay for mixing of aqueous vesicle contents. Biochemistry. 1980;19:6011–6021. doi: 10.1021/bi00567a011. [DOI] [PubMed] [Google Scholar]
- 12.Yang S.T., Zaitseva E., Melikov K. Cell-penetrating peptide induces leaky fusion of liposomes containing late endosome-specific anionic lipid. Biophys. J. 2010;99:2525–2533. doi: 10.1016/j.bpj.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee D.K., Brender J.R., Ramamoorthy A. Lipid composition-dependent membrane fragmentation and pore-forming mechanisms of membrane disruption by pexiganan (MSI-78) Biochemistry. 2013;52:3254–3263. doi: 10.1021/bi400087n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinstein J.N., Klausner R.D., Blumenthal R. Phase transition release, a new approach to the interaction of proteins with lipid vesicles. Application to lipoproteins. Biochim. Biophys. Acta. 1981;647:270–284. doi: 10.1016/0005-2736(81)90255-8. [DOI] [PubMed] [Google Scholar]
- 15.Brooks B.R., Bruccoleri R.E., Karplus M. Charmm - a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 16.Jang H., Arce F.T., Nussinov R. β-Barrel topology of Alzheimer’s β-amyloid ion channels. J. Mol. Biol. 2010;404:917–934. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang H., Ma B., Nussinov R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic alzheimer β-amyloid ion channels. Biophys. J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capone R., Mustata M., Lal R. Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 2010;98:2644–2652. doi: 10.1016/j.bpj.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz G.E. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta. 2002;1565:308–317. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- 20.Sansom M.S., Kerr I.D. Transbilayer pores formed by beta-barrels: molecular modeling of pore structures and properties. Biophys. J. 1995;69:1334–1343. doi: 10.1016/S0006-3495(95)80000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang H., Zheng J., Nussinov R. Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang H., Zheng J., Nussinov R. New structures help the modeling of toxic amyloidbeta ion channels. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Jang H., Arce F.T., Nussinov R. Misfolded amyloid ion channels present mobile β-sheet subunits in contrast to conventional ion channels. Biophys. J. 2009;97:3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang H., Teran Arce F., Nussinov R. Structural convergence among diverse, toxic β-sheet ion channels. J. Phys. Chem. B. 2010;114:9445–9451. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang H., Arce F.T., Lal R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:6538–6543. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mustata M., Capone R., Nussinov R. K3 fragment of amyloidogenic β(2)-microglobulin forms ion channels: implication for dialysis related amyloidosis. J. Am. Chem. Soc. 2009;131:14938–14945. doi: 10.1021/ja9049299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jang H., Ma B., Nussinov R. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007;7:21. doi: 10.1186/1472-6807-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buffy J.J., Waring A.J., Hong M. Determination of peptide oligomerization in lipid bilayers using 19F spin diffusion NMR. J. Am. Chem. Soc. 2005;127:4477–4483. doi: 10.1021/ja043621r. [DOI] [PubMed] [Google Scholar]
- 29.Mani R., Tang M., Hong M. Membrane-bound dimer structure of a β-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–8349. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- 30.Tang M., Waring A.J., Hong M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007;129:11438–11446. doi: 10.1021/ja072511s. [DOI] [PubMed] [Google Scholar]
- 31.Smart O.S., Goodfellow J.M., Wallace B.A. The pore dimensions of gramicidin A. Biophys. J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenis H., Reutelingsperger C. Targeting phosphatidylserine in anti-cancer therapy. Curr. Pharm. Des. 2009;15:2719–2723. doi: 10.2174/138161209788923903. [DOI] [PubMed] [Google Scholar]
- 33.Lee M.T., Hung W.C., Huang H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA. 2008;105:5087–5092. doi: 10.1073/pnas.0710625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamba Y., Ariyama H., Yamazaki M. Kinetic pathway of antimicrobial peptide magainin 2-induced pore formation in lipid membranes. J. Phys. Chem. B. 2010;114:12018–12026. doi: 10.1021/jp104527y. [DOI] [PubMed] [Google Scholar]
- 35.Mani R., Cady S.D., Hong M. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee M.T., Sun T.L., Huang H.W. Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. USA. 2013;110:14243–14248. doi: 10.1073/pnas.1307010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang H., Ma B., Nussinov R. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 2006;91:2848–2859. doi: 10.1529/biophysj.106.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang H., Arce F.T., Lal R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011;100:1775–1783. doi: 10.1016/j.bpj.2011.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fahrner R.L., Dieckmann T., Feigon J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996;3:543–550. doi: 10.1016/s1074-5521(96)90145-3. [DOI] [PubMed] [Google Scholar]
- 40.Miyasaki K.T., Lehrer R.I. β-sheet antibiotic peptides as potential dental therapeutics. Int. J. Antimicrob. Agents. 1998;9:269–280. doi: 10.1016/s0924-8579(98)00006-5. [DOI] [PubMed] [Google Scholar]
- 41.Sokolov Y., Mirzabekov T., Kagan B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta. 1999;1420:23–29. doi: 10.1016/s0005-2736(99)00086-3. [DOI] [PubMed] [Google Scholar]
- 42.Gottler L.M., de la Salud Bea R., Marsh E.N. Using fluorous amino acids to probe the effects of changing hydrophobicity on the physical and biological properties of the beta-hairpin antimicrobial peptide protegrin-1. Biochemistry. 2008;47:9243–9250. doi: 10.1021/bi801045n. [DOI] [PubMed] [Google Scholar]
- 43.Szoka F., Jr., Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes) Annu. Rev. Biophys. Bioeng. 1980;9:467–508. doi: 10.1146/annurev.bb.09.060180.002343. [DOI] [PubMed] [Google Scholar]
- 44.Düzgüneş N., Wilschut J., Papahadjopoulos D. Physicochemical characterization of large unilamellar phospholipid vesicles prepared by reverse-phase evaporation. Biochim. Biophys. Acta. 1983;732:289–299. doi: 10.1016/0005-2736(83)90214-6. [DOI] [PubMed] [Google Scholar]
- 45.Ames B.N., Dubin D.T. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J. Biol. Chem. 1960;235:769–775. [PubMed] [Google Scholar]
- 46.Gupta K., Singh V.P., Maiti S. Nanoparticles of cationic chimeric peptide and sodium polyacrylate exhibit striking antinociception activity at lower dose. J. Control. Release. 2009;134:47–54. doi: 10.1016/j.jconrel.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Düzgüneş N., Faneca H., Lima M.C. Methods to monitor liposome fusion, permeability, and interaction with cells. Methods Mol. Biol. 2010;606:209–232. doi: 10.1007/978-1-60761-447-0_16. [DOI] [PubMed] [Google Scholar]
- 48.Kucerka N., Tristram-Nagle S., Nagle J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 49.Mukhopadhyay P., Monticelli L., Tieleman D.P. Molecular dynamics simulation of a palmitoyl-oleoyl phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys. J. 2004;86:1601–1609. doi: 10.1016/S0006-3495(04)74227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durell S.R., Brooks B.R., Bennaim A. Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 1994;98:2198–2202. [Google Scholar]
- 52.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 53.Connelly L., Jang H., Lal R. Atomic force microscopy and MD simulations reveal pore-like structures of all-D-enantiomer of Alzheimer’s β-amyloid peptide: relevance to the ion channel mechanism of AD pathology. J. Phys. Chem. B. 2012;116:1728–1735. doi: 10.1021/jp2108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capone R., Jang H., Lal R. All-d-enantiomer of β-amyloid peptide forms ion channels in lipid bilayers. J. Chem. Theory Comput. 2012;8:1143–1152. doi: 10.1021/ct200885r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.