

Abstract
The miR-17/92 cluster is among the best-studied microRNA clusters. Interest in the cluster and its members has been increasing steadily and the number of publications has grown exponentially since its discovery with more than 1000 articles published in 2012 alone. Originally found to be involved in tumorigenesis, research work in recent years has uncovered unexpected roles for its members in a wide variety of settings that include normal development, immune diseases, cardiovascular diseases, neurodegenerative diseases and aging. In light of its ever-increasing importance and ever-widening regulatory roles, we review here the latest body of knowledge on the cluster's involvement in health and disease as well as provide a novel perspective on the full spectrum of protein-coding and non-coding transcripts that are likely regulated by its members.
Keywords: cancer, ‘expanded' model of miRNA targeting, microRNA, miRNA, miR-17/92 cluster, oncomir
Facts
MiR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1 are members of the miR-17/92 cluster.
The miR-17/92 cluster is important in cell cycle, proliferation, apoptosis and other pivotal processes.
The miR-17/92 cluster is important in normal development and also the first group of microRNAs (miRNAs) to be implicated in a human syndrome (Feingold syndrome).
The miR-17/92 cluster is also known as ‘oncomiR-1'.
The miR-17/92 cluster is very often dysregylated in hematopoietic and solid cancers.
The miR-17/92 cluster is often dysregylated in cardiovascular, immune and neurodegenerative diseases.
The miR-17/92 cluster has been implicated in age-related conditions.
There are two models of miRNA targeting: the ‘standard' that has been in use for a decade and the ‘expanded' that is emerging with the help of recent technological advances.
The ‘standard' model assumes Watson–Crick pairing in the ‘seed' region of a miRNA and targets that are primarily in the 3′ untranslated region (3′UTR) and conserved across genomes.
The ‘expanded' model also incorporates Watson–Crick pairing but additionally allows for combinations of unmatched bases and G:U wobbles in the ‘seed' region; moreover, the targets can be anywhere along the messenger RNA (not just the 3′UTR) as well as in the intergenic and intronic genomic space; under this model, miRNA targets need not be conserved.
Open Questions
What currently unsuspected processes and human diseases/conditions are regulated by the miR-17/92 cluster?
Are there any protein-coding genes that are important for human diseases or conditions and are regulated by the miR-17/92 cluster?
Does the miR-17/92 cluster have functionally significant genomic targets in the intergenic and intronic parts of the genome?
Are there additional paralogues of the miR-17/92 cluster that have not yet been reported?
The presence of guanines and thymines in the seed region of the cluster's members suggests great potential for targeting under the ‘expanded' model; what is the relative fraction of the cluster's targets under the ‘expanded' model?
MiRNAs are abundant non-coding RNAs (ncRNAs), ∼22 nucleotides (nts) in length, which have significant roles in regulating gene expression.1, 2 The first animal miRNA, lin-4, was discovered during a genetic screen in Caenorhabditis elegans (C. elegans) and was found to repress the expression of the protein-coding gene lin-14.3, 4 In 2000, a second miRNA, the well-conserved let-7, was discovered and functionally characterized as important for C. elegans development.5 Since then, thousands of miRNAs have been predicted and identified in animals, plants and viruses (see http://www.mirbase.org).6, 7, 8
Herein, we focus on the miR-17/92 cluster of miRNAs and review the current knowledge to date as to the roles of its members in health and disease. In light of recent findings, we also examine and discuss the topic of miRNA target identification in the context of the miR-17/92 cluster.
The Cluster and its Paralogues
In 2004, a novel gene, ‘chromosome 13 open reading frame 25' or C13orf25 for short, was identified.9 Analysis of 70 human B-cell lymphoma cases showed amplification of this region.9 The miR-17/92 cluster as it is now known is located in the locus of the non-protein-coding gene MIR17HG (the miR-17/92 cluster host gene) (also known as C13orf25). The miR-17/92 cluster transcript spans 800 nts10, 11 out of MIR17HG's 7 kb and comprises six miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1 (Figure 1). The miR-17/92 cluster is conserved among vertebrates.12 Soon after its discovery, the ectopic expression of a truncated version of the cluster (lacking miR-92) in B-cell lymphoma revealed its oncogenic character and miR-17/92 was given the distinction of being the first ‘oncomir'.13
Figure 1.

Genomic representation of the human miR-17-92 cluster host gene (MIR17HG) and neighborhood genes on Chr 13q31.1-q33-1. (a) Genomic representation of genes located±10 kb around human MIR17HG. (b) Genomic representation of MIR17HG. Two transcripts are shown in light blue and individual members of the cluster represented as red rectangles. The two panels were created using the UCSC genome browser (http://genome.ucsc.edu/)
The human genome contains two paralogues of the main cluster (Figure 2): the miR-106b/25 and the miR-106a/363 cluster, respectively. MiR-106b/25 is located on chromosome 7 (7q22.1), in the 13th intron of the MCM7 gene. MiR-106a/363 is located on chromosome X (Xq26.2). The miR-106b/25 cluster comprises three miRNAs: miR-106b, miR-93 and miR-25 (Figure 2). The miR-106a/363 cluster comprises six miRNAs: miR-106a, miR-18b, miR-20b, miR-19b-2, miR-92a-2 and miR-363. MiR-17/92 and miR-106b/25 are expressed abundantly in a wide spectrum of tissues but miR-106a/363 is expressed at lower levels.14, 15 Together these three miRNA clusters represent a combined total of 15 miRNAs that form four ‘seed' families: the miR-17 family, the miR-18 family, the miR-19 family and the miR-92 family (Figure 3).
Figure 2.

Members of the miR-17/92 cluster and its two paralogues miR-106a/363 and miR-106b/25 and their chromosomal location. Red: members of the miR-17 family; blue: members of the miR-18 family; green: members of the miR-19 family; orange: members of the miR-92 family
Figure 3.

Sequences of the members of the miR-17/92 cluster (in bold face) and its two paralogues miR-106a/363 and miR-106b/25. The sequences are divided into four families according to the miRNA ‘seed' (the sequence spanning positions 2 through 7 inclusive counting from the 5′ end of the miRNA). The ‘seed' in each case is shown in boldface and is highlighted in blue
Transcriptional Regulation of the Cluster
One of the early findings was C-MYC's involvement in activating MIR17HG transcription through a site that is located 1484 nts upstream of MIR17HG's transcription start site.16, 17 N-MYC also transcriptionally activates MIR17HG18 as well as E2F1 and E2F3.19, 20 The data show close functional interactions between c-Myc/n-Myc and the miR-17/92 cluster. Both c-Myc and n-Myc can directly bind to the promoter of miR-17/92 and initiate transcription.17, 21, 22 Indeed, some patients with an N-MYC mutation have developmental abnormalities similar to those in Feingold syndrome patients (see below).23, 24
Additional information on transcriptional regulation became available through the recent release by the ENCODE (Encyclopedia of DNA Elements) project25 of data from the study of 118 transcription factors (TFs). ENCODE's results revealed 1292 TF:miRNA interactions and 421 miRNA:TF interactions that in turn suggest tightly coupled auto-regulatory loops involving miRNAs and TFs. Of the 118 TFs, 34 pertain to the miR-17/92 cluster: they include the previously known MYC17, 18 and MXI26, 27 and 32 novel TFs (Figure 4). Among the newly identified TFs, BCL3 was found to regulate miR-17/92 and miR-106b/25. Further, BCL3, IRF1, SP1, TAL1 and ZBTB33 are targeted by individual miRNAs of the cluster, in addition to being TFs for the cluster (Figure 4). Moreover, several novel targets for members of miR-17/92 and miR-106b/25 were identified and are also summarized in Figure 4.25, 28 With regard to the miR-106a/363 cluster, it is likely regulated by the microphthalmia-associated transcription factor (MITF) through a binding site at position 133,135,780 (hg19) of chromosome X in the cluster's immediate vicinity.29
Figure 4.

The transcriptional regulation and main targets of the miR-17/92 cluster and its paralogues. The transcriptional factors (TFs) in the left upper corner have been functionally validated; dark blue arrows indicate upregulation; black lines indicate repression. TFs in the blue ‘cloud' were identified by the ENCODE project and the relationship of most of them to the miR-17/92 cluster and its paralogues is putative. Blue TFs were validated previously and confirmed by ENCODE; red TFs putatively regulate the miR-17/92 cluster and at the same time are known to be targeted by cluster members; green TFs putatively regulate the miR-17/92 cluster and at the same time are known to be targeted by paralogue miR-106b/25. If the specific gene that is targeted by a miRNA is known, the repressor line ends at the gene; otherwise, it ends at the box boundary of the respective cell process
Among TFs, the E2F family (E2F1, E2F2 and E2F3) have a central role in the regulation of G1 to S phase progression.30 All E2Fs,17, 19 especially E2F3,20 have been shown to occupy miR-17/92's promoter region. E2Fs are also known to be targeted by miR-17/92, forming an auto-regulatory loop (Figure 4).19, 20 Finally, recent studies indicate that TP53 targets the miR-17/92 cluster31 while also being targeted by miR-25 through regulation of the latter by Myc and E2F1.32
Despite these significant advances, knowledge about the transcriptional control of miR-17/92 and its paralogues remains largely fragmentary. It is also important to note that the specific processing of individual miRNAs adds a new level of complexity; that is, it is conceivable that there is a cell-type-dependent and context-dependent dimension to post-transcriptional silencing.14, 15, 33, 34
Main Targets of the miR-17/92 Cluster
Phosphatase and tensin homolog (PTEN) and E2Fs were among the first validated miR-17/92 targets.15, 17, 19 Reporter assays revealed targets for miR-19a and miR-19b-1 in PTEN's 3′UTR, and the introduction of miR-19a and miR-19b-1, or of the full cluster, in miR-17/92-deficient cells sufficed to restore PTEN expression levels.15 In addition, miR-17 and miR-20a modulate the expression of E2F1.17, 19 Lastly, miR-20a targets the 3′UTRs of both E2F2 and E2F3 (Figure 4).19
The ability of the cluster's members to cooperate is evident in the context of TGF-β signaling. In particular, miR-17 and miR-20a directly target the TGF-β receptor II (TGFBRII), whereas miR-18a targets Smad2 and Smad4, two members of the TGF-β signaling pathway.35, 36, 37 TGF-β activation exerts an effect mediated in part by the cyclin-dependent kinase inhibitor (p21) and the apoptosis facilitator BCL2L11 (BIM), both of which are targeted by miR-17/92.35, 38 In addition, BCL2L11 is targeted by miR-20a, miR-92, miR-19a and miR-19b-115 and also by miR-106b/25.39 During the endoplasmic reticulum related stress, unfolded protein response TFs, activating TFs, activating transcription factor 4 (Atf4) and nuclear factor-erythroid-2-related factor 2 (Nrf2) are activated and downregulate Mcm7, the host gene for the miR-106a/25 cluster. Downregulation of miR-106b/25 and repression of BCL2L11 consequently trigger apoptosis.39
Lastly, miR-18a and miR-19 directly repress the anti-angiogenic factors thrombospondin-1 (TSP-1) and connective tissue growth factor (CTGF).40 In addition, miR-17 and miR-20a participate in the regulation of the insulin gene enhancer protein (Isl-1) and the T-box 1 protein (Tbx1) (Figure 4).41
MiR-17/92 and Normal Development
The miR-17/92 cluster is highly expressed in embryonic cells15 and has an important role in development.
MiR-17/92 was the first group of miRNAs to be implicated in a developmental syndrome in humans. Indeed, studies of patients with Feingold syndrome revealed an important role for the miR-17/92 cluster in normal skeletal development.24 Human patients with heterozygous microdeletions in the MIR17HG locus have autosomal dominant Feingold syndrome, characterized by multiple skeletal abnormalities in the fingers and toes, short stature and microcephaly. Some patients also show various degrees of learning and developmental disabilities.24
Subsequent mouse studies showed that deletion of the miR-17/92 cluster is perinatal lethal.15 MiR-17/92−/− embryos exhibit severe skeletal abnormalities and recapitulate the phenotype observed in patients with Feingold syndrome. The mice are also smaller in size than normal embryos, and die at birth from cardiac defects and lung hypoplasia.15, 22 The miR-17/92 cluster is involved also in normal lung morphogenesis, epithelial proliferation and branching through the targeting of signal transducer and activator of transcription 3 (STAT3) and mitogen-activated protein kinase 14 (MAPK14).42 The overexpression of the miR-17/92 cluster leads to lung epithelium hyper-proliferation and suggests a role in lung cancer.43
Analogously to the miR-17/92 studies that indicated a role in B-cell differentiation, the normal process of B-cell maturation in miR-17/92-/-mice is blocked during the progression from pro-B to pre-B cells. Mice with a deleted miR-17/92 cluster have a reduced number of pre-B cells at E18.5.15 In experiments with adult mice whose hematopoietic system is reconstituted with fetal liver cells from a miR-17/92Δneo/Δneo embryo at E14.5, the number of circulating lymphocytes, circulating B cells, splenic B cells and pre-B-cells bone marrow cells is significantly reduced compared with mice with reconstituted fetal liver cells from wild-type embryos at 8–10 weeks post transplant.15 Mice that were irradiated and had their hematopoietic system reconstituted with hematopoietic stem cells (HSC) derived from fetal liver of Eμ-myc transgenic mice expressing miR-17/19 under the control of murine stem cell virus (MSCV) show a massive enlargement of lymph nodes, splenic hyperplasia, infiltration of the thymus by lymphoma cells and leukemias.13 Moreover, almost half of the animals in the test group exhibited hind leg paralysis as the result of tumors at the lumbar node. These results suggest the importance of miR-17/92 in normal B-cell development and survival. On the other hand, overexpression of the cluster was also shown to cause lymphoproliferative diseases.44
Parallel studies have also implicated miR-17/92 in normal lymphocyte development.45 In mouse knockout models, miR-19b-1 and miR-17 were shown to promote T-cell expansion; the mice display reduced lymphocyte proliferation that was attributed to the promotion of interferon-γ (IFNγ) production by miR-19b-1 and the promotion of a Th1 response by miR-17 and miR-19b-1.45 Indeed, miR-17 and miR-19b-1 have an important role in promoting B-cell proliferation, protecting B-cells from death, supporting IFNγ production and suppressing T-cell differentiation.45
The Cluster as an Oncogene
We next review the increasing body of literature on the cluster's oncogenic role (Table 1).46, 47
Table 1. Relative expression of individual miRNAs from the miR-17/92 cluster or its paralogues in normal development, cancer, other diseases and age-related conditions.
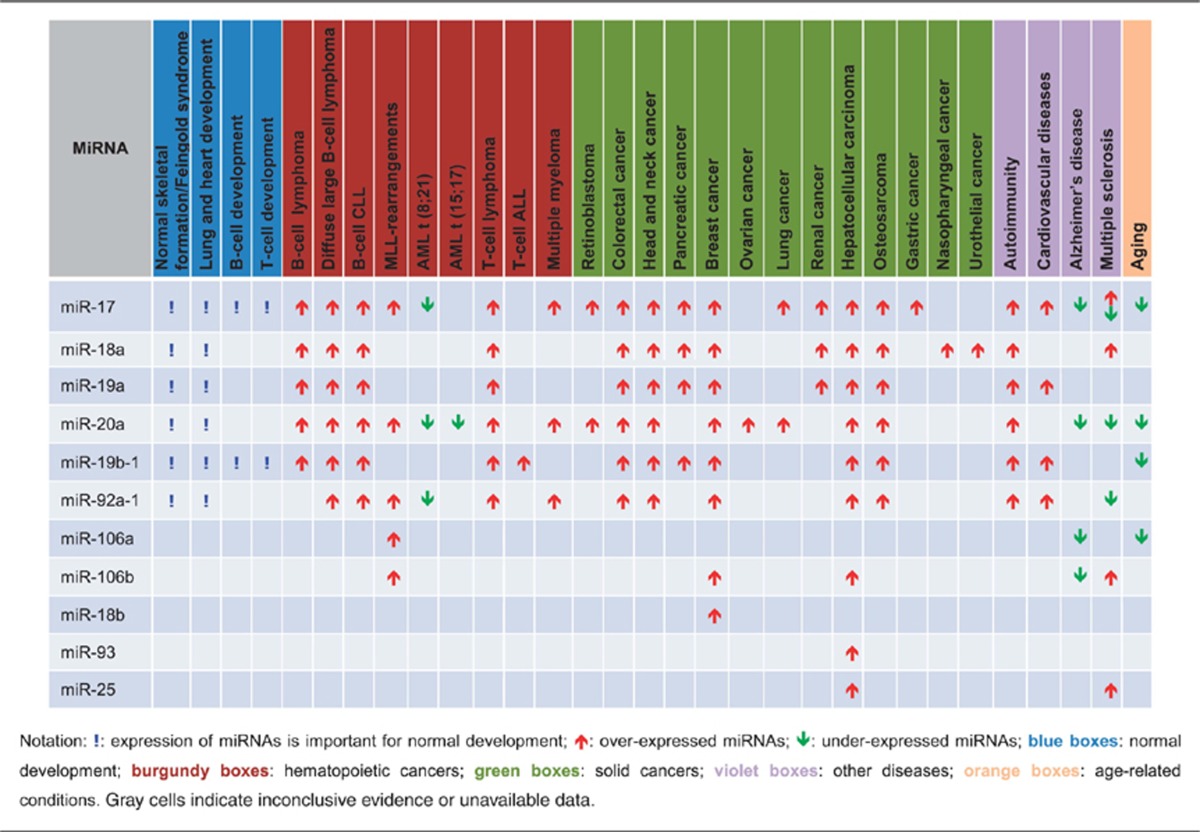
B-cell Lymphomas
The miR-17/92 cluster was initially found amplified in diffuse cell lymphomas.9 Later, in B-cell lymphoma, an ectopically overexpressed truncated version that lacked miR-92 showed the cluster's role as an oncogene.13 Moreover, as already discussed, c-Myc was shown to transcribe the truncated cluster in mouse models of B-cell lymphoma.13 These findings represent early evidence that miR-17/92 can act as an oncogene by suppressing apoptosis.13 MiR-18a levels in diffuse large B-cell lymphoma correlate strongly and negatively with survival (higher expression-shorter survival).48 As mentioned already, miR-19a and miR-19b-1 are necessary and sufficient to promote tumorigenesis B-cell lymphoma.49, 50, 51 In addition, the conditional knockout of miR-17/92 in Myc-driven lymphomas was shown to increase apoptosis and to reduce tumorigenicity and tumor progression.49
B-cell Chronic Lymphocytic Leukemia
MiR-20a was found to correlate with diagnosis to treatment time in B-cell chronic lymphocytic leukemia (CLL) and thus can potentially serve as a blood biomarker.52 The cluster members miR-17 and miR-19b-1 are highly overexpressed in CLL cultures with fibroblast expressing human CD40 ligand (CD154) with IL-4. Another study found miR-20, miR-18a, miR-19a and miR-92a to be overexpressed in CLL cultures, but at much lower levels than those of miR-17 and miR-19b-1.53
Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a heterogeneous group of diseases with different genetic rearrangements, different prognosis and required treatment options. MiR-17-5p, miR-17-3p, miR-20a and miR-92 are upregulated in myeloid/lymphoid, or mixed-lineage leukemia (MLL), display rearrangements in AML and are downregulated in AML with the translocation t(8;21). On the other hand, miR-17-5p and miR-20a are downregulated in t(15;17). AML with MLL-rearrangements is considered to have intermediate or poor prognosis; moreover, it requires a different treatment from AML with t(8;21)/t(15;17) that usually carries a favorable prognosis.54 In mouse studies, the levels of miR-106 were found to be upregulated in AML and to target Sequestosome 1 (SQSTM1).55 In AML characterized by the translocation t(8;16)(p11;p13), miR-17/92 is downregulated.56 However, there is no significant difference in the expression of MYC between t(8;16)(p11;p13) AML and other types of AML suggesting that other mechanisms of downregulation, for example, methylation, may be at work.56
T-cell Lymphoma
In an experiment with the SL3-3 murine leukemia virus, 2545 BALB/c newborn mice were infected and nearly all developed T-cell lymphoma. Quantitative RT-PCR analysis showed elevated expression of miR-17/92 after virus integration.57 The miR-17/92 integration sites were found to cluster together at three distinct regions; the integration sites within each such region were ∼1 kb apart.57
Retinoblastoma
Overexpression and genomic amplification of miR-17/92 were shown in retinoblastoma.58 In particular, in Rb−/− and p107−/− retinoblastomas, ectopic expression of miR-17/92 induces rapid proliferation and disease onset. This increase in proliferation is linked to the miR-17 sub-family, which target the cell cycle inhibitors p21Cip1 and p57Kip2.58, 59
Colorectal Cancer
MiR-17/92 was also found overexpressed in colon cancer.40 In a tumor engraftment model, upregulation of the cluster by Myc in colonocytes increased tumorigenesis by promoting angiogenesis through direct repression of TSP-1 and CTGF by miR-18a and miR-19, respectively.40 MiR-18a and miR-20a are significantly overexpressed in colorectal cancer (CRC); in fact, miR-18a is a marker of poor prognosis.60 In addition, miR-92 levels in colon adenocarcinoma have been shown to correlate negatively with BCL2L11 expression and, thus, with reduced apoptosis.61 High miR-17 expression correlates with low overall survival in patients with CRC.62 Another study that comprised 90 patients with CRC, 90 patients with inflammatory bowel diseases (IBD), 20 patients with gastric cancer and 50 healthy controls also confirmed high expression level of miR-17 and miR-92 in tumors and serum from patients with CRC.63 Patients with CRC had higher miR-92 levels compared with healthy controls or patients with IBD or gastric cancer.63 Interestingly, results revealed a correlation between miR-18a expression and Adenomatous Polyposis Coli (APC) mutation in CRC samples.64
Head and Neck Cancers
MiR-17/92 is often overexpressed in meduloblastomas, especially those with an active Sonic hedgehog signaling pathway. The cluster is overexpressed in mouse models in cerebellar granule neuron progenitors, where the tumor arises.65, 66 Ectopic expression of miR-17/92 increases tumor formation through the suppression of TGF-β signaling upon orthotopic transplantation into immunocompromised mice.65, 66 These studies suggest a tissue-specific function for members of the miR-17/92 cluster. The miR-17/92 cluster amplification was also reported in neuroblastomas and is linked to poor prognosis.67 Lastly, miR-17 has been shown to promote the growth of neuroblastoma cell lines.68
Pancreatic Cancer
In pancreatic cancer, miR-17, miR-18a, miR-19a and miR-19b-1 expression levels are increased.69, 70 Another study also showed that the level of miR-18a in the blood is significantly higher before surgery in patients with pancreatic cancer compared with after surgery, suggesting the possibility that blood levels of miR-18 can potentially be used as a biomarker.70
Breast Cancer
Deep sequencing of triple negative breast cancer (TNBC) samples revealed a threefold increase of miR-17/92 levels.71 In estrogen receptor (ER)-positive breast cancer, it was shown that miR-18a/-18b directly target the 3′UTR of the ERα.72 In addition, miR-17 and miR-20 are overexpressed in metastatic breast cancer73 and have been shown to directly suppress the 3′UTR of IL-8 and to inhibit cytokeratin 8 through cyclin D1.73 Another study has shown that miR-106b positively correlates with homeotic TF Six1 expression levels in breast cancer:74 Six1 depends on the upregulation of the TGF-β pathway to induce epithelial–mesenchymal transition. In addition, high levels of miR-106b are indicative of shorter time to relapse.74
Ovarian Cancer
Studies have implicated the overexpression of miR-20a in proliferation and invasion in the OVCAR3 cell line, whereas the downregulation of miR-20a has been shown to lead to the suppression of proliferation and invasion. A possible mechanism is through binding to the amyloid protein precursor (APP), a gene of central importance in Alzheimer's disease (AD).75
Lung Cancer
In lung cancer, miR-17-5p and miR-20a are overexpressed.76 Their targets include HIF-1α, PTEN, BCL2L11, CDKNA and TSP-1.76 A study of 221 lung cancer patients and 54 matching controls showed a significant increase of miR-17-5p expression in tumor and serum and a negative correlation with patient survival.77 However, the blood of non-small cell lung cancer patients had a low level of miR-17-5p.78
Renal Cancer
The miR-17/92 cluster is regulated by the von Hippel-Lindau (VHL) tumor suppressor: in the absence of VHL, miR-17/92 levels increase.79 Other studies have shown miR-17, miR-18a and miR-20a to be overexpressed in renal cancer; however, overexpression of these miRNAs did not correlate with survival.80, 81
Hepatocellular Carcinoma
All six members of the miR-17/92 cluster are often overexpressed in hepatocellular carcinoma (HC).82 The use of antisense nts specific to all six members of the miR-17/92 cluster caused a 50% reduction in proliferation and anchorage-independent growth.82 In addition, several members of miR-17/92 and its paralogues (miR-92,83 miR-18a, miR-106b, miR-93 and miR-2584) are highly expressed in HC cells compared with paired non-tumor samples. Another study showed that miR-18, and the miR-106b/25 paralogue, was overexpressed in 50% of clinical samples used in the study.84 Cell culture studies have also shown that the knockdown of miR-106b/25 leads to decreased cell proliferation and anchorage-independent growth in three different cell lines: HepG2, HeLa and HuH7.84
Osteosarcoma
MiR-17/92 is overexpressed in osteosarcoma as demonstrated by luciferase assays.85 In particular, miR-17 and miR-20a are overexpressed in metastasized cells compared with parental cells.86 In addition, mouse studies with anti-miR-20a showed significant increase in lung metastases, possibly through the repression of FasL in the lung tissues.86
The Cluster in Other Diseases
Beyond cancer, the miR-17/92 cluster has been shown to have important roles in other human conditions including immune, cardiovascular and neurodegenerative diseases.87, 88, 89, 90
Immune Diseases
The miR-17/92 cluster has a role in the innate and acquired immune response.88 In human cord blood, CD34+ hematopoietic progenitor cells differentiate into monocytes upon exposure to macrophage-colony stimulating factor (M-CSF) and the miR-17/92 cluster.91 MiR-17/92 also has a role in the acquired immune response.88 Another study has shown that autoimmunity, characterized by increased proliferation and survival of CD4+ T cell, could be caused by overexpression of the miR-17/92 cluster in the DN1 stage.88
Cardiovascular Diseases
MiR-92a is highly expressed in endothelial cells but overexpression of miR-92a in those cells under ischemic conditions was shown to inhibit angiogenesis.92 In a mouse model with leg ischemia, the administration of antagomiR-92a led to inhibition of miR-92a with consequent growth of new blood vessels and recovery from ischemia.92 In another study, endothelial cells from patients with coronary artery disease (CAD) exhibited higher levels of miR-17 and miR-92a compared with endothelial cells from healthy controls.93, 94 In another study, the miRNA profile of patients with acute coronary syndrome showed an increase of miR-19 compared with patients with CAD.95 And a study of miRNA levels measured by quantitative RT-PCR in whole blood and serum showed reduced miR-19a levels in patients with CAD compared with healthy controls.
Neurodegenerative Diseases
The amyloid precursor protein APP generates the amyloid-β, Aβ, peptide through the ‘amyloidogenic' pathway with the help of β- and γ-secretases. Aβ accumulates in extracellular spaces forming Aβ plaques. Members of the miR-17 family (i.e., miR-17, miR-20a, miR-106a and miR-106b) were shown to directly suppress APP in vitro.96, 97 In the AD brain, miR-106b was shown to be downregulated in vivo.97
In relapsing patients with multiple sclerosis (MS), miR-18 was found to be overexpressed compared with controls.98 Another study of CD4+ T cells and B cells of relapsing and remitting MS patients shows the upregulation of miR-17-5p in CD4+ T cells and downregulation of miR-92 in B cells.99 Another study has shown under-expression of miR-17 and miR-20a in whole-blood samples from 59 MS patients compared with 37 healthy controls.100 The MS patients represented different disease types (primary progressive, secondary progressive and remitting-relapsing) and for the last 3 months before the study had not received any treatment. In addition, miR-106b and miR-25 were found to be upregulated in 12 relapsing-remitting MS patients and in 14 healthy controls.101 A recent analysis used an integrative approach to study miRNAs dysregulated in MS revealed that miR-20a and miR-20b target ∼500 genes each.102
On a related note, profiling of miRNA expression in the brain of zebrafish (Danio rerio) showed that miR-92 is expressed in periventricular cells and in proliferative zones of larva and adult brain and down-regulated in mature neurons.103
The Cluster and Age-Related Conditions
Considering the importance of miR-17/92 in tumorigenesis, it was not long before the relation between dysregulation of these miRNAs and aging was discovered.104, 105 Studies of different tissue types representing aging revealed downregulation of miR-17, miR-19b, miR-20a and miR-106a.106 This suggests yet another role for these miRNAs, one that transcends cell cycle regulation and tumorigenesis. However, the mechanistic connection between downregulation of the cluster's members and aging has yet to be elucidated.105
The Potentially Wide Spectrum of Coding and Non-coding Sequences Targeted by the Cluster
It should be clear by now that the cluster's members are involved in very diverse settings and a wide range of post-transcriptional regulatory events. The experimental findings to date about the cluster's targetome have been driven primarily by the analyses of gene-expression array data. The exact mechanistic aspects of those interactions have been elucidated in only relatively few contexts. To better appreciate the range of possibilities and to place the cluster's known (and unknown) targets in the appropriate light, it is important to first review briefly how miRNA target prediction has been practiced.
A miRNA effects the post-transcriptional regulation of its targets in a sequence-dependent manner. The ‘seed' in particular, that is, the sequence spanning positions 2–7 inclusive from the 5′ end of a miRNA, is especially important for target determination and coupling.1, 5, 107, 108, 109, 110, 111, 112, 113 Early genetic studies and subsequent efforts demonstrated that miRNAs can function in the presence of inexact matches and/or bulges in the seed region.108, 109, 111, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125 Despite the potential diversity of targets, there has been a strong adherence throughout to a ‘standard' model that anticipates exact base-pairing, that is, Watson–Crick, and the absence of any bulges in the seed region. Consequently, the majority of the designed algorithms treated these criteria as necessary and sufficient attributes of bona fide targets and combined them with the enforcement of cross-genome conservation. However, algorithms were also proposed that neither enforced nor used these constraints, for example, rna22.126 As a matter of fact, rna22 permits Watson–Crick and G:U pairs in the seed region as well as bulges; moreover, a candidate target does not have to be conserved across organisms and can be located anywhere.
An additional element, likely guided by the early C. elegans studies with the 3′UTR of the lin-14 and lin-41 genes, relates to an adherence to seek miRNA targets primarily in the 3′UTR of the candidate mRNAs. Nevertheless, the use of artificial constructs demonstrated the absence of any mechanistic constraints that would prevent the targeting of other mRNA regions by miRNAs: let-7 target sites embedded in the coding sequence (CDS) were shown to be functional.127 Analogously, the introduction of miRNA target sites into the 5′UTR of luciferase reporter mRNAs containing internal ribosome entry sites (IRESs) revealed that miRNAs could regulate the abundance of a target by binding to its 5′UTR.128
Further support for these findings came more recently through a number of efforts by our group and others that reported the discovery and validation of extensive and naturally occurring miRNA targets in the CDS of an mRNA as well as its 5′UTR.129, 130, 131, 132, 133, 134 In earlier work, we estimated that as many as 90% of the known protein-coding human genes could be targeted by miRNAs.126 On a related note, miRNAs were recently shown to also target intergenic and intronic regions, pseudogenes and long ncRNAs.119, 121, 123, 135, 136
Another dimension of the miRNA target prediction problem relates to the observation of conservation of miRNA sequences across animals and plants (and viruses). At the sequence level, most miRNAs are evolutionarily conserved among distant species.137 However, not every known miRNA is conserved: indeed, there are reports of miRNAs that are species- or genus-specific.138, 139 Moreover, it is known that conserved miRNAs do not have the same functional behavior in different species: let-7, which is conserved between C. elegans and Drosophila melanogaster, is a characteristic example. In the worm, let-7 is a component of the heterochronic pathway; it is expressed at a late stage of larva development, regulates the transition from larva to adult5 and is embryonic lethal.140 In the fruit fly, let-7-knockout flies are externally normal but exhibit behavioral defects and juvenile features in their neuromusculature.141
In recent work113 we used molecular dynamics to analyze the crystal structure of the Argonaute (Ago) silencing complex and demonstrated in a very general way the existence of many admissible targets that transcend the ‘standard' model that has been in use for more than a decade already. The molecular dynamics findings were further corroborated by publicly available Ago-immunoprecipitated119 and sequenced miRNA targets.113 These results provide strong evidence in support of an expanded model of miRNA targeting and are very relevant for the members of the miR-17/92 cluster and for our estimates of this cluster's targetome (Figure 5). Glimpses of evidence supporting the ‘expanded' model were also observed experimentally in earlier work109, 111, 116, 117, 118 as well as discussed in the literature.110, 112, 124, 130, 142
Figure 5.

Differences between the ‘standard' and ‘expanded' model of miRNA targeting and the corresponding targetome. The seed of miR-18 is used as an example (bases at positions 2 through 7 inclusive). For the expanded model's G:U wobble examples and bulge examples, only a few representative cases are shown
The members of miR-17/92 clusters are ideal miRNAs for which to explore ‘unexpected targets' under the ‘expanded' model. Indeed, as all of the members have at least two G/U bases in their seed region, (Figure 3) they could potentially base pair with U and G, respectively, on the side of the target to create wobbles and additional targets (Figure 5). In addition, potential incorporation of bulges on either the miRNA or the target side would lead to an even higher number of non-standard targets.
There is also increasing evidence with regard to non-protein-coding transcripts that could be targeted by miRNAs. The evidence comes both from studies of individual miRNA:target pairs as well as global analyses. In particular, it has already been shown for several miRNAs, some belonging to the miR-17/92 cluster, that they target and suppress the expression of PTENP1, the PTEN pseudogene.143, 144 The same study also showed targeting and regulation by miRNAs of the KRAS pseudogene KRASP1 as well as of several of the pseudogenes of OCT4. More recently,136 it was shown that miR-133 and miR-135 target a long non-coding RNA, linc-MD1, thereby regulating the expression of MAML1 and MEF2C, two TFs that activate muscle-specific gene expression. In addition, the advent of the crosslinking and immunoprecipitation followed by high-throughput sequencing (CLIP-seq) technology has enabled global studies of miRNA targeting preferences in a variety of contexts.119, 123, 145, 146, 147 Initial analyses of the available data have provided additional evidence that miRNA-targeted transcripts include numerous transcripts that are not protein-coding.113
These findings indicate that the miR-17/92 targetome may be larger and further ranging than originally anticipated. As we saw above, several members of the miR-17/92 cluster have been implicated in the regulation of non-protein-coding transcripts. In addition, the sequence composition of the seed region of the cluster's members and in particular the presence of G/U's in the seed provides them with an expanded base-pairing ability. Consequently, there is great potential that a very rich set of currently unrecognized heteroduplexes comprising miRNAs of the miR-17/92 cluster awaits discovery.
Conclusion
MiR-17/92 is one of the best-known miRNA clusters. The cluster's members have pivotal roles in normal development, and dysregulation of their expression leads to a wide array of diseases including hematopoietic and solid cancers, and immune, neurodegenerative and cardiovascular diseases. The cluster is also important because its members are the first described in the context of a developmental syndrome in humans. Related to this, other recent work uncovered novel important connections between the miR-17/92 cluster and aging.
Despite great progress in understanding the cluster's roles, several key questions remain unanswered. For example, until the recently reported findings by the ENCODE project very little was known about the transcriptional control of the cluster by TFs as well as about the targeting of TFs by members of the cluster and its paralogues. Considering the ENCODE project's findings, it is reasonable to conjecture that the actual transcription control of the cluster is significantly more complex than research to date has managed to reveal.
A parallel and very important question is that of elucidating the cluster's targetome. The currently known set of validated protein-coding targets is small. In light of the many miRNAs that the cluster and its paralogues comprise and the recent evidence obtained through molecular dynamics studies and CLIP-seq data analyses, it is increasingly apparent that the true spectrum of targets is potentially very large. Additional research effort will be needed before the full complement of the cluster's targets can be elucidated. It is also important to note that an increased target set opens up new opportunities and new avenues for therapeutic intervention in those settings, where one or more of the cluster's members are dysregulated.
Acknowledgments
We thank Eric Londin for feedback on an earlier version of the manuscript, Peter Clark for suggestions with some figures and the other members of the Rigoutsos laboratory for helpful discussions. This research was supported in part by the William M Keck Foundation (IR), the Hirshberg Foundation for Pancreatic Cancer Research (IR) and by institutional funds.
Glossary
- 3′UTR
3′ untranslated region
- 5′UTR
5′ untranslated region
- CLIP-seq
crosslinking and immunoprecipitation followed by high-throughput sequencing
- Ago
Argonaute protein
- AD
Alzheimer's disease
- AML
acute myeloid leukemia
- APC
Adenomatous Polyposis Coli
- APP
amyloid protein precursor
- Atf4
activating transcription factor 4
- C13orf25
chromosome 13 open reading frame 25
- C. elegans
Caenorhabditis elegans
- CAD
coronary artery diseases
- CDS
coding sequence
- CLL
chronic lymphocytic leukemia
- CRC
colorectal cancer
- CTGF
connective tissue growth factor
- D. melanogaster
Drosophila melanogaster
- ENCODE
the Encyclopedia of DNA Elements
- ER
estrogen receptor
- IBD
inflammatory bowel diseases
- IRES
internal ribosome entry site
- IFNγ
interferon-γ
- Isl-1
insulin gene enhancer protein
- HC
hepatocellular carcinoma
- HSC
hematopoietic stem cells
- MAPK 14
mitogen-activated protein kinase 14
- M-CSF
macrophage-colony stimulating factor
- MIR17HG
the miR-17/92 cluster host gene (non-protein coding)
- miRNAs
microRNAs
- MITF
microphthalmia-associated transcription factor
- MLL
mixed-lineage leukemia
- MS
multiple sclerosis
- MSCV
murine stem cell virus
- ncRNAs
non-coding RNAs
- Nrf2
nuclear factor-erythroid-2-related factor 2
- Nts
nucleotides
- PTEN
phosphatase and tensin homolog
- STAT3
signal transducer and activator of transcription 3
- Tbx1
T-box 1 protein
- TFs
transcription factors
- TNBC
triple negative breast cancer
- TSP-1
thrombospondin-1
- VHL
von Hippel-Lindau tumor suppressor
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by G Melino
Supplementary Material
References
- Bartel DP. Micrornas: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. Micrornas: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57. doi: 10.1016/0092-8674(89)90171-2. [DOI] [PubMed] [Google Scholar]
- Ruvkun G, Giusto J. The Caenorhabditis elegans heterochronic gene lin-14 encodes a nuclear protein that forms a temporal developmental switch. Nature. 1989;338:313–319. doi: 10.1038/338313a0. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 rna regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Chang S, Johnston RJ, Jr, Frokjaer-Jensen C, Lockery S, Hobert O. Micrornas act sequentially and asymmetrically to control chemosensory laterality in the nematode. Nature. 2004;430:785–789. doi: 10.1038/nature02752. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Hobert O. A microrna controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426:845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Cohen SM. Towards a complete description of the microrna complement of animal genomes. Genome Biol. 2003;4:228. doi: 10.1186/gb-2003-4-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, et al. Identification and characterization of a novel gene, c13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- Mendell JT. Miriad roles for the mir-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diosdado B, van de Wiel MA, Terhaar Sive Droste JS, Mongera S, Postma C, Meijerink WJ, et al. Mir-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br J Cancer. 2009;101:707–714. doi: 10.1038/sj.bjc.6605037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion CP, Bonetti C, Ventura A. The microrna-17-92 family of microrna clusters in development and disease. Cancer J. 2012;18:262–267. doi: 10.1097/PPO.0b013e318258b60a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microrna polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell-specific micrornas. Developmental Cell. 2003;5:351–358. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the mir-17 through 92 family of mirna clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Jothi R, Cuddapah S, Cui K, Roh TY, Schones DE, et al. Chromatin poises mirna- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. C-myc-regulated micrornas modulate e2f1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, et al. Mycn regulates oncogenic micrornas in neuroblastoma. Int J Cancer. 2008;122:699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, et al. An e2f/mir-20a autoregulatory feedback loop. The Journal of biological chemistry. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-rna cluster by e2f transcription factors. J Biol Chem. 2007;282:2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- Loven J, Zinin N, Wahlstrom T, Muller I, Brodin P, Fredlund E, et al. Mycn-regulated micrornas repress estrogen receptor-alpha (esr1) expression and neuronal differentiation in human neuroblastoma. Proc Natl Acad Sci USA. 2010;107:1553–1558. doi: 10.1073/pnas.0913517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, et al. Germline deletion of the mir-17 approximately 92 cluster causes skeletal and growth defects in humans. Nature Genet. 2011;43:1026–1030. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, van Beusekom E, et al. Mycn haploinsufficiency is associated with reduced brain size and intestinal atresias in feingold syndrome. Nature Genet. 2005;37:465–467. doi: 10.1038/ng1546. [DOI] [PubMed] [Google Scholar]
- Marcelis CL, Hol FA, Graham GE, Rieu PN, Kellermayer R, Meijer RP, et al. Genotype-phenotype correlations in mycn-related feingold syndrome. Hum Mut. 2008;29:1125–1132. doi: 10.1002/humu.20750. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, et al. Architecture of the human regulatory network derived from encode data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M, Rao E, Ramachandrareddy H, Shen Y, Jiang C, Chen J, et al. The mir-17-92 microrna cluster is regulated by multiple mechanisms in b-cell malignancies. Am J Pathol. 2011;179:1645–1656. doi: 10.1016/j.ajpath.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA, et al. Identification of oncogenic microrna-17-92/zbtb4/specificity protein axis in breast cancer. Oncogene. 2012;31:1034–1044. doi: 10.1038/onc.2011.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, et al. Dicer ablation affects antibody diversity and cell survival in the b lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, et al. Chromatin structure analyses identify mirna promoters. Genes Dev. 2008;22:3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Helin K. The e2f transcription factors: Key regulators of cell proliferation. Biochim Biophys Acta. 2000;1470:M1–12. doi: 10.1016/s0304-419x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF, et al. Repression of the mir-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009;28:2719–2732. doi: 10.1038/emboj.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SS, Yoo JY, Nuovo GJ, Jeon YJ, Kim S, Lee TJ, et al. Micrornas/tp53 feedback circuitry in glioblastoma multiforme. Proc Natl Acad Sci USA. 2012;109:5316–5321. doi: 10.1073/pnas.1202465109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, et al. Human embryonic stem cells express a unique set of micrornas. Dev Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microrna gene expression. Nature Met. 2004;1:47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- Datto MB, Yu Y, Wang XF. Functional analysis of the transforming growth factor beta responsive elements in the waf1/cip1/p21 promoter. J Biol Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- Ohgushi M, Kuroki S, Fukamachi H, O'Reilly LA, Kuida K, Strasser A, et al. Transforming growth factor beta-dependent sequential activation of smad, bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol Cell Biol. 2005;25:10017–10028. doi: 10.1128/MCB.25.22.10017-10028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, et al. The myc-mir-17∼92 axis blunts tgf{beta} signaling and production of multiple tgf{beta}-dependent antiangiogenic factors. Cancer Res. 2010;70:8233–8246. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyashiki M, Ohyashiki JH, Hirota A, Kobayashi C, Ohyashiki K. Age-related decrease of mirna-92a levels in human cd8+ t-cells correlates with a reduction of naive t lymphocytes. Immunity Ageing. 2011;8:11. doi: 10.1186/1742-4933-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, et al. Perk-dependent repression of mir-106b-25 cluster is required for er stress-induced apoptosis. Cell Death Dis. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, et al. Augmentation of tumor angiogenesis by a myc-activated microrna cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, et al. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a microrna-mediated mechanism. Dev Cell. 2010;19:903–912. doi: 10.1016/j.devcel.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro G, El-Hashash A, Guidolin D, Tiozzo C, Turcatel G, Young BM, et al. Mir-17 family of micrornas controls fgf10-mediated embryonic lung epithelial branching morphogenesis through mapk14 and stat3 regulation of e-cadherin distribution. Dev Biol. 2009;333:238–250. doi: 10.1016/j.ydbio.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microrna mir-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–453. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased mir-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, et al. Molecular dissection of the mir-17-92 cluster's critical dual roles in promoting th1 responses and preventing inducible treg differentiation. Blood. 2011;118:5487–5497. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Galasso M, Costinean S, Tagliavini L, Gamberoni G, Drusco A, et al. Reprogramming of mirna networks in cancer and leukemia. Genome Res. 2010;20:589–599. doi: 10.1101/gr.098046.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alencar AJ, Malumbres R, Kozloski GA, Advani R, Talreja N, Chinichian S, et al. Micrornas are independent predictors of outcome in diffuse large b-cell lymphoma patients treated with r-chop. Clin Can Res. 2011;17:4125–4135. doi: 10.1158/1078-0432.CCR-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the mir-17∼92 cluster of micrornas in myc-induced b-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. Mir-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23:2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haaften G, Agami R. Tumorigenicity of the mir-17-92 cluster distilled. Genes Dev. 2010;24:1–4. doi: 10.1101/gad.1887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussay E, Wang K, Cho JH, van Moer K, Pierson S, Paggetti J, et al. Microrna as biomarkers and regulators in B-cell chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2011;108:6573–6578. doi: 10.1073/pnas.1019557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willimott S, Wagner SD. Stromal cells and cd40 ligand (cd154) alter the mirnome and induce mirna clusters including, mir-125b/mir-99a/let-7c and mir-17-92 in chronic lymphocytic leukaemia. Leukemia. 2012;26:1113–1116. doi: 10.1038/leu.2011.299. [DOI] [PubMed] [Google Scholar]
- Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, et al. Distinct microrna expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM, et al. Mir-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood. 2011;118:916–925. doi: 10.1182/blood-2011-02-336487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Beya M, Navarro A, Ferrer G, Diaz T, Gel B, Camos M, et al. Acute myeloid leukemia with translocation (8;16)(p11;p13) and myst3-crebbp rearrangement harbors a distinctive microrna signature targeting ret proto-oncogene. Leukemia. 2012;27:595–603. doi: 10.1038/leu.2012.278. [DOI] [PubMed] [Google Scholar]
- Wang CL, Wang BB, Bartha G, Li L, Channa N, Klinger M, et al. Activation of an oncogenic microrna cistron by provirus integration. Proc Natl Acad Sci USA. 2006;103:18680–18684. doi: 10.1073/pnas.0609030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J, Ventura A. Mir than meets the eye. Genes Dev. 2011;25:1663–1667. doi: 10.1101/gad.17454011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, et al. Mir-17∼92 cooperates with rb pathway mutations to promote retinoblastoma. Genes Dev. 2011;25:1734–1745. doi: 10.1101/gad.17027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama K, Inoue H, Takatsuno Y, Tanaka F, Mimori K, Uetake H, et al. Over- and under-expressed micrornas in human colorectal cancer. Int J Oncol. 2009;34:1069–1075. doi: 10.3892/ijo_00000233. [DOI] [PubMed] [Google Scholar]
- Tsuchida A, Ohno S, Wu W, Borjigin N, Fujita K, Aoki T, et al. Mir-92 is a key oncogenic component of the mir-17-92 cluster in colon cancer. Cancer Sci. 2011;102:2264–2271. doi: 10.1111/j.1349-7006.2011.02081.x. [DOI] [PubMed] [Google Scholar]
- Yu G, Tang JQ, Tian ML, Li H, Wang X, Wu T, et al. Prognostic values of the mir-17-92 cluster and its paralogs in colon cancer. J Surg Oncol. 2012;106:232–237. doi: 10.1002/jso.22138. [DOI] [PubMed] [Google Scholar]
- Ng EK, Chong WW, Jin H, Lam EK, Shin VY, Yu J, et al. Differential expression of micrornas in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut. 2009;58:1375–1381. doi: 10.1136/gut.2008.167817. [DOI] [PubMed] [Google Scholar]
- Reid JF, Sokolova V, Zoni E, Lampis A, Pizzamiglio S, Bertan C, et al. Mirna profiling in colorectal cancer highlights mir-1 involvement in met-dependent proliferation. Mol Can Res. 2012;10:504–515. doi: 10.1158/1541-7786.MCR-11-0342. [DOI] [PubMed] [Google Scholar]
- Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Ann Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A, et al. The mir-17∼92 cluster collaborates with the sonic hedgehog pathway in medulloblastoma. Proc Natl Acad Sci USA. 2009;106:2812–2817. doi: 10.1073/pnas.0809579106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, Marschall T, Martin M, Rosenstiel P, Mestdagh P, Schlierf S, et al. Deep sequencing reveals differential expression of micrornas in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010;38:5919–5928. doi: 10.1093/nar/gkq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and bim. PLoS One. 2008;3:e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, et al. Microrna expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442–4452. doi: 10.1038/sj.onc.1210228. [DOI] [PubMed] [Google Scholar]
- Morimura R, Komatsu S, Ichikawa D, Takeshita H, Tsujiura M, Nagata H, et al. Novel diagnostic value of circulating mir-18a in plasma of patients with pancreatic cancer. Br J Cancer. 2011;105:1733–1740. doi: 10.1038/bjc.2011.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi TA, Horlings HM, Ten Hoeve JJ, Mihailovic A, Halfwerk H, Morozov P, et al. Microrna sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011;71:4443–4453. doi: 10.1158/0008-5472.CAN-11-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leivonen SK, Makela R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K, et al. Protein lysate microarray analysis to identify micrornas regulating estrogen receptor signaling in breast cancer cell lines. Oncogene. 2009;28:3926–3936. doi: 10.1038/onc.2009.241. [DOI] [PubMed] [Google Scholar]
- Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y, et al. Microrna 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci USA. 2010;107:8231–8236. doi: 10.1073/pnas.1002080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, et al. The mir-106b-25 cluster targets smad7, activates tgf-beta signaling, and induces emt and tumor initiating cell characteristics downstream of six1 in human breast cancer. Oncogene. 2012;31:5162–5171. doi: 10.1038/onc.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Liu Y, Jiang J, Ma Z, Wu H, Liu T, et al. Mir-20a promotes proliferation and invasion by targeting app in human ovarian cancer cells. Acta Biochim Biophys Sin. 2010;42:318–324. doi: 10.1093/abbs/gmq026. [DOI] [PubMed] [Google Scholar]
- Osada H, Takahashi T. Let-7 and mir-17-92: Small-sized major players in lung cancer development. Cancer Sci. 2011;102:9–17. doi: 10.1111/j.1349-7006.2010.01707.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Si Q, Xiao S, Xie Q, Lin J, Wang C, et al. Prognostic significance of serum mir-17-5p in lung cancer. Medical Oncol. 2013;30:353. doi: 10.1007/s12032-012-0353-2. [DOI] [PubMed] [Google Scholar]
- Heegaard NH, Schetter AJ, Welsh JA, Yoneda M, Bowman ED, Harris CC. Circulating micro-rna expression profiles in early stage nonsmall cell lung cancer. Int J Cancer. 2012;130:1378–1386. doi: 10.1002/ijc.26153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal CS, Michael MZ, Rawlings LH, Van der Hoek MB, Gleadle JM. The vhl-dependent regulation of micrornas in renal cancer. BMC Med. 2010;8:64. doi: 10.1186/1741-7015-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Mollenkopf HJ, Grimm C, Wagner I, Albrecht M, Waller T, et al. Microrna profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med. 2009;13:3918–3928. doi: 10.1111/j.1582-4934.2009.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow TF, Mankaruos M, Scorilas A, Youssef Y, Girgis A, Mossad S, et al. The mir-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol. 2010;183:743–751. doi: 10.1016/j.juro.2009.09.086. [DOI] [PubMed] [Google Scholar]
- Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, Slagle BL, et al. Elevated expression of the mir-17-92 polycistron and mir-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol. 2008;173:856–864. doi: 10.2353/ajpath.2008.080096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigoka M, Tsuchida A, Matsudo T, Nagakawa Y, Saito H, Suzuki Y, et al. Deregulation of mir-92a expression is implicated in hepatocellular carcinoma development. Pathol Int. 2010;60:351–357. doi: 10.1111/j.1440-1827.2010.02526.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Tan W, Neo TW, Aung MO, Wasser S, Lim SG, et al. Role of the mir-106b-25 microrna cluster in hepatocellular carcinoma. Cancer Sci. 2009;100:1234–1242. doi: 10.1111/j.1349-7006.2009.01164.x. [DOI] [PubMed] [Google Scholar]
- Thayanithy V, Sarver AL, Kartha RV, Li L, Angstadt AY, Breen M, et al. Perturbation of 14q32 mirnas-cmyc gene network in osteosarcoma. Bone. 2012;50:171–181. doi: 10.1016/j.bone.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Nishimoto K, Zhou Z, Hughes D, Kleinerman ES. Mir-20a encoded by the mir-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating fas expression. Cancer Res. 2012;72:908–916. doi: 10.1158/0008-5472.CAN-11-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Baltimore D. Micrornas and immunity: Tiny players in a big field. Immunity. 2007;26:133–137. doi: 10.1016/j.immuni.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Tsitsiou E, Lindsay MA. Micrornas and the immune response. Curr Opin Pharmacol. 2009;9:514–520. doi: 10.1016/j.coph.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Frost RJ, Olson EN. Micrornas add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Olson EN. Pervasive roles of micrornas in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. Micrornas 17-5p-20a-106a control monocytopoiesis through aml1 targeting and m-csf receptor upregulation. Nature Cell Biol. 2007;9:775–787. doi: 10.1038/ncb1613. [DOI] [PubMed] [Google Scholar]
- Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. Microrna-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, et al. Circulating micrornas in patients with coronary artery disease. Circulation Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- Tijsen AJ, Pinto YM, Creemers EE. Circulating micrornas as diagnostic biomarkers for cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2012;303:H1085–H1095. doi: 10.1152/ajpheart.00191.2012. [DOI] [PubMed] [Google Scholar]
- Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, et al. Microparticles: major transport vehicles for distinct micrornas in circulation. Cardiovascular Res. 2012;93:633–644. doi: 10.1093/cvr/cvs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SS, De Strooper B. Alterations of the microrna network cause neurodegenerative disease. Trends Neurosci. 2009;32:199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Schonrock N, Matamales M, Ittner LM, Gotz J. Microrna networks surrounding app and amyloid-beta metabolism--implications for alzheimer's disease. Exp Neurol. 2012;235:447–454. doi: 10.1016/j.expneurol.2011.11.013. [DOI] [PubMed] [Google Scholar]
- Otaegui D, Baranzini SE, Armañanzas R, Calvo B, Muñoz-Culla M, Khankhanian P, et al. Differential micro rna expression in pbmc from multiple sclerosis patients. PLoS One. 2009;4:e6309. doi: 10.1371/journal.pone.0006309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. Altered expression of mir-17-5p in cd4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol. 2010;40:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- Cox MB, Cairns MJ, Gandhi KS, Carroll AP, Moscovis S, Stewart GJ, et al. Consortium ANMSG. Micrornas mir-17 and mir-20a inhibit t cell activation genes and are under-expressed in ms whole blood. PLoS One. 2010;5:e12132. doi: 10.1371/journal.pone.0012132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis G, Ferracin M, Biondani A, Caniatti L, Rosaria Tola M, Castellazzi M, et al. Altered mirna expression in t regulatory cells in course of multiple sclerosis. J Neuroimmunol. 2010;226:165–171. doi: 10.1016/j.jneuroim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Angerstein C, Hecker M, Paap BK, Koczan D, Thamilarasan M, Thiesen HJ, et al. Integration of microrna databases to study micrornas associated with multiple sclerosis. Mol Neurobiol. 2012;45:520–535. doi: 10.1007/s12035-012-8270-0. [DOI] [PubMed] [Google Scholar]
- Kapsimali M, Kloosterman WP, de Bruijn E, Rosa F, Plasterk RH, Wilson SW. Micrornas show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007;8:R173. doi: 10.1186/gb-2007-8-8-r173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DJ, Liang R, Li N, Wang E. The impact of noncoding rna on the biochemical and molecular mechanisms of aging. Biochim et Biophys Acta. 2009;1790:970–979. doi: 10.1016/j.bbagen.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Grillari J, Hackl M, Grillari-Voglauer R. Mir-17-92 cluster: Ups and downs in cancer and aging. Biogerontology. 2010;11:501–506. doi: 10.1007/s10522-010-9272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Muck C, et al. mir-19b, mir-20a, and mir-106a are down-regulated in human aging. Aging Cell. 2010;9:291–296. doi: 10.1111/j.1474-9726.2010.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. Micrornas to nanog, oct4 and sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- Brodersen P, Voinnet O. Revisiting the principles of microrna target recognition and mode of action. Nat Rev Mol Cell Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O'Day E, et al. Mir-24 inhibits cell proliferation by targeting e2f2, myc, and other cell-cycle genes via binding to ‘seedless' 3'utr microrna recognition elements. Mol Cell. 2009;35:610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoutsos I, Tsirigos A.Micrornas in Development and Cancer in Molecular Medicine and Medicinal Chemistry, Slack f(eds)Imperial College Press: Danvers, MA, USA; 237–273.2010 [Google Scholar]
- Xia Z, Clark P, Huynh T, Loher P, Zhao Y, Chen HW, et al. Molecular dynamics simulations of ago silencing complexes reveal a large repertoire of admissible 'seed-less' targets. Sci Rep. 2012;2:569. doi: 10.1038/srep00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha I, Wightman B, Ruvkun G. A bulged lin-4/lin-14 rna duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes Dev. 1996;10:3041–3050. doi: 10.1101/gad.10.23.3041. [DOI] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ. The C. elegans microrna let-7 binds to imperfect let-7 complementary sites from the lin-41 3'utr. Genes Dev. 2004;18:132–137. doi: 10.1101/gad.1165404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easow G, Teleman AA, Cohen SM. Isolation of microrna targets by mirnp immunopurification. RNA. 2007;13:1198–1204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of micrornas on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by micrornas. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute hits-clip decodes microrna-mrna interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mrna translation and stability by micrornas. Ann Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, et al. Transcriptome-wide identification of rna-binding protein and microrna target sites by par-clip. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Lieberman J, Lal A. Desperately seeking microrna targets. Nat Struc Mol Biol. 2010;17:1169–1174. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, et al. Comprehensive discovery of endogenous argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17:173–179. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microrna target recognition. Nat Struc Mol Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, et al. The viral and cellular microrna targetome in lymphoblastoid cell lines. PLoS Pathogens. 2012;8:e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of microrna binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microrna targets dicer within its coding sequence. Proc Natl Acad Sci USA. 2008;105:14879–14884. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Wienholds E, Ketting RF, Plasterk RH. Substrate requirements for let-7 function in the developing zebrafish embryo. Nucleic Acids Res. 2004;32:6284–6291. doi: 10.1093/nar/gkh968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. Mir-519 reduces cell proliferation by lowering rna-binding protein hur levels. Proc Natl Acad Sci USA. 2008;105:20297–20302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoutsos I. New tricks for animal micrornas: Targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res. 2009;69:3245–3248. doi: 10.1158/0008-5472.CAN-09-0352. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Wang WX, Mao G, Wilfred BR, Xie K, Jennings MH, et al. Specific sequence determinants of mir-15/107 microrna gene group targets. Nucleic Acids Res. 2011;39:8163–8172. doi: 10.1093/nar/gkr532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surdziel E, Cabanski M, Dallmann I, Lyszkiewicz M, Krueger A, Ganser A, et al. Enforced expression of mir-125b affects myelopoiesis by targeting multiple signaling pathways. Blood. 2011;117:4338–4348. doi: 10.1182/blood-2010-06-289058. [DOI] [PubMed] [Google Scholar]
- Adilakshmi T, Sudol I, Tapinos N. Combinatorial action of mirnas regulates transcriptional and post-transcriptional gene silencing following in vivo pns injury. PLoS One. 2012;7:e39674. doi: 10.1371/journal.pone.0039674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui YH, Xiao L, Rao JN, Zou T, Liu L, Chen Y, et al. Mir-503 represses cug-binding protein 1 translation by recruiting cugbp1 mrna to processing bodies. Mol Biol Cell. 2012;23:151–162. doi: 10.1091/mbc.E11-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the mir-106b∼25 microrna cluster as a proto-oncogenic pten-targeting intron that cooperates with its host gene mcm7 in transformation. Sci Signaling. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding rna controls muscle differentiation by functioning as a competing endogenous rna. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. Micrornas and other tiny endogenous rnas in C. elegans. Currt Biol. 2003;13:807–818. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- Stark A, Kheradpour P, Parts L, Brennecke J, Hodges E, Hannon GJ, et al. Systematic discovery and characterization of fly micrornas using 12 Drosophila genomes. Genome Res. 2007;17:1865–1879. doi: 10.1101/gr.6593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty JR, Wang WX, Hebert SS, Wilfred BR, Mao G, Nelson PT. The mir-15/107 group of microrna genes: Evolutionary biology, cellular functions, and roles in human diseases. J Mol Biol. 2010;402:491–509. doi: 10.1016/j.jmb.2010.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, et al. Most Caenorhabditis elegans micrornas are individually not essential for development or viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol NS, Xu P, Jan Y-N, Ambros V. Drosophila let-7 microrna is required for remodeling of the neuromusculature during metamorphosis. Genes Dev. 2008;22:1591–1596. doi: 10.1101/gad.1671708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Levin AA. Developing microrna therapeutics: Approaching the unique complexities. Nucleic Acid Ther. 2012;22:213–225. doi: 10.1089/nat.2012.0356. [DOI] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mrnas regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoutsos I, Furnari F. Gene-expression forum: Decoy for micrornas. Nature. 2010;465:1016–1017. doi: 10.1038/4651016a. [DOI] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. A quantitative analysis of clip methods for identifying binding sites of rna-binding proteins. Nat Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, et al. Genome-wide identification of ago2 binding sites from mouse embryonic stem cells with and without mature micrornas. Nat Struc Mol Biol. 2011;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, et al. Transcriptome-wide mir-155 binding map reveals widespread noncanonical microrna targeting. Mol Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.