

Abstract
De novo protein design offers a unique means to test and advance our understanding of how proteins fold. However, most current design methods are native structure eccentric and folding kinetics has rarely been considered in the design process. Here, we show that a de novo designed mini-protein DS119, which folds into a βαβ structure, exhibits unusually slow and concentration-dependent folding kinetics. For example, the folding time for 50 μM of DS119 was estimated to be ∼2 s. Stopped-flow fluorescence resonance energy transfer experiments further suggested that its folding was likely facilitated by a transient dimerization process. Taken together, these results highlight the need for consideration of the entire folding energy landscape in de novo protein design and provide evidence suggesting nonnative interactions can play a key role in protein folding.
Introduction
De novo protein design critically tests our current understanding of protein folding principles (1,2). In particular, it helps delineate the factors that govern the roughness of protein folding energy landscapes (3,4). For example, complex folding kinetics was observed for Top7, a computationally designed protein with a novel ββαβαββ fold (5). At least three distinct phases were required to describe the folding kinetics of Top7, and this kinetic complexity arose from the topology of Top7, which likely imposes a series of folding barriers (6,7). On the other hand, a de novo designed three-helix bundle protein α3D was found to fold with an ultrafast rate (8). Molecular dynamic simulations suggested that the transition state ensemble of α3D was highly heterogeneous and dynamic, allowing fast access to the native state via multiple pathways. In comparison to the folding kinetics of natural proteins, these examples highlight the difficulty in designing specific protein folding energy landscapes or pathways.
To further illustrate this point and to provide new insights into the relationships between protein sequences and folding kinetics, this study focuses on the folding mechanism of DS119, a de novo designed 36-residue protein (9). DS119 folds into a βαβ structure with two parallel β-strands connected by a α-helix (Fig. 1). Because standalone βαβ motifs have not been found in naturally occurring proteins, DS119 was designed rationally with negative design considerations and the introduction of a tryptophan pair. Further NMR studies indicated that DS119 is a well-folded monomeric protein, and its native structural ensemble is in good agreement with the targeted topology (9). In addition, thermal unfolding measurements showed that DS119 has an unusually high thermal melting temperature (Tm >80°C) at neutral pH. A computational study using both all-atom and coarse-grained models has estimated the folding time of DS119 to be 6 to 10 μs (10). A priori estimate of the folding rate based on the Plaxco-Simmons-Baker correlation has led to a similar result (128 μs) (11). Thus, initially we employed an infrared (IR) temperature-jump (T-jump) technique (12) to investigate the folding dynamics of this putative fast folder. In contrast to our expectation and predictions from empirical folding-rate models, there were no detectable population relaxation signals occurring at the 10 ns to 1 ms timescale, suggesting that DS119 is a slow folder compared to natural proteins with similar sizes, and the folding process probably involves either a high free energy barrier or a new mechanism. Based on these initial observations, we present an extensive characterization of the folding mechanisms of DS119 using stopped-flow refolding experiments and mutational studies.
Figure 1.
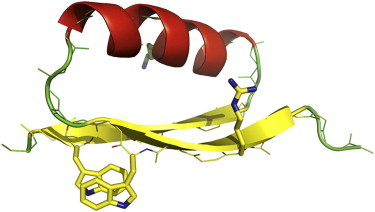
Structure of DS119 (Protein Data Bank identification code: 2KI0). Special design features are shown in sticks, including the tryptophan (Trp) pair (W9 and W34) and the negatively designed residues (R6 and K21).
Materials and Methods
Cloning and site-directed mutagenesis
The genes encoding DS119 and DS103 were synthesized and cloned into the BamHI and XhoI sites of pGEX4T-1 vectors (Invitrogen, Gaithersburg, MD) as reported before (13). Point mutations on DS119 genes (P14A, E23C, W9F, and W9E) were generated with site-directed mutagenesis kits (Saibaisheng, Beijing, China) according to the manufacturer’s instructions. All constructs were confirmed by DNA sequencing.
Protein expression and purification
All proteins were expressed and purified as previously described (13). Briefly, they were expressed in Escherichia coli as a GST-fusion protein (GST: glutathione S-transferase) and purified on a GST-affinity column (GE Healthcare, Piscataway, NJ) and then by reversed-phase HPLC. Lyophilized samples were obtained and their molecular weights were confirmed by high-resolution mass spectrometry.
Labeling of fluorescent dyes
E23C was modified with the thiol-reactive fluorophores Alexa Fluor 488 C5-maleimide and 5-((((2-iodoacetyl) amino) ethyl) amino) naphthalene-1-sulfonic acid (1, 5-IAEDANS or I14) (Molecular Probes, Eugene, OR). A fivefold molar excess of fluorophore was used for quantitative modification. Labeling reactions were carried out for 12 h at 4°C in 50 mM Tris-HCl, pH 7.5 with a 10-fold molar excess of Tris (2-carboxyethyl) phosphine (TCEP) (Sigma, St. Louis, MO) to prevent oxidation of cysteine. Labeled proteins were purified by reversed-phase HPLC. All labeled proteins showed the expected increase in mass, as determined by high-resolution mass spectrometry.
Chemical cross-linking
Cross-linking experiments with E23C were conducted with thiol-reactive 1, 11-bis (maleimido) triethylene glycol, or BM-PEG3 (Pierce Biotechnology, Rockford, IL) in two different conditions: steady state and refolding process. In steady state, E23C was diluted to 500 μM BM-PEG3 solutions containing 20 mM PB (Na2HPO4/NaH2PO4 pH 7.0) and 200 μM TCEP. The final concentration of E23C was 200 μM. In the refolding process, unfolded E23C dissolved in 6 M guanidine hydrochloride (Gdn-HCl) was diluted to 500 μM BM-PEG3 solutions with the same concentrations of PB and TCEP. The final concentration of Gdn-HCl was 0.6 M and different concentrations of E23C were tested in this case from 20 to 200 μM. In both cases the sample was incubated at 25°C for 5 min before SDS-PAGE analysis.
Equilibrium experiments
Circular dichroism (CD) spectra were measured on a MOS 450 AF/CD device (Bio-Logic, Claix, France) at room temperature, using 1-mm quartz cuvettes for the far-ultraviolet (UV) region (190–250 nm) and 10-mm cuvettes for the near-UV region (260–320 nm). Thermal denaturation curves were obtained with Peltier accessory in10-mm quartz cuvettes. Heating was performed at 1°C/min from 1°C to 97°C. In both experiments, the protein concentrations were kept at 0.2 mg/mL in 50 mM PB buffer (Na2HPO4/NaH2PO4 for pH 7.3 or NaH2PO4/H3PO4 for pH 2.5).
Chemical denaturation experiments were also performed on the MOS 450 AF/CD with a titration accessory in 10-mm quartz cuvettes. The tryptophan fluorescence was monitored by an excitation wavelength of 290 nm and the emission above 320 nm was recorded.
Fluorescence spectra were collected on a FluoroLog 3 spectrofluorometer (HORIBA Jobin Yvon, Longjumeau, France). For fluorescence resonance energy transfer (FRET) measurements, a 1-mm cuvette was used and the buffer was 50 mM PB (pH 7.3). The excitation wavelength was 365 nm, and the spectra between 400 and 650 nm were recorded.
Fourier transform IR spectra were collected on a Magna-IR 860 spectrometer (Nicolet, Madison, WI) with a homemade CaF2 sample cell. After hydrogen-deuterium exchange, protein samples were dissolved in KD2PO4/D3PO4 buffer (pH 2.5) to 0.2 mg/mL. Spectra scanning and thermal denaturation were performed as previously described (8).
Fluorescence correlation spectroscopy was conducted with Alexa Fluor 555-labeled E23C at different concentrations (16 nM and 100 μM). The fluorescence correlation spectroscopy (FCS) setup, data collecting, and fitting procedures were described elsewhere (14).
In gel filtration experiments, protein samples were dissolved in 50 mM PB (pH 7.3) buffers with 100 mM NaCl and loaded onto a Superdex Peptide 10/300 GL column (GE Healthcare). The column was eluted using the same buffer with a flow rate of 0.4 mL/min and the UV absorption at 280 nm was monitored.
Proton 1D NMR and 2D NOESY data were collected on a 600 MHz Bruker AVANCE III spectrometer with TXI probe. NOESY spectra were recorded with the mixing time of 200 μs. 1 mM DS119 samples were prepared in 50 mM sodium phosphate buffer (pH 7.3 and pH 2.5) with 10% D2O. For the samples in pH 7.3 solutions, data were recorded at 20°C and 60°C. For the samples in pH 2.5 solutions, data were recorded at 20°C and 46°C.
Kinetic experiments
The stopped-flow experiments were performed on the MOS 450 AF/CD with a SFM300 module and a 0.8-mm cuvette. The temperature was kept at 25°C by a water bath. Refolding was initiated by an eightfold dilution of protein samples in 6 M Gdn-HCl and 50 mM Na2HPO4/NaH2PO4 buffer (pH 7.3). The dead time was 0.3 to 0.4 ms and the kinetic traces were recorded 10 ms before the mixing finished. The resulting kinetic curve was an average of 6 to 15 independent measurements. In each measurement the HT value of the detector (photomultiplier tube) was automatically adjusted to obtain the best signal/noise ratio. In stopped-flow fluorescence experiments, the excitation wavelength was 290 nm, and emission above 320 nm was recorded. In stopped-flow FRET experiments, the excitation wavelength was 375 nm, and a filter of 515–555 nm (Chroma, Bellows Falls, VT) was used for detection. For stopped-flow CD, the final concentration of DS119 was 174 μM. For stopped-flow fluorescence, the final concentrations were illustrated (see Fig. 3). Based on the expected amplitude in the equilibrium experiments, the observed signal amplitude in the stopped-flow CD experiments corresponded to ∼30% of the total change from unfolded to folded state (−66 deg to −82 deg), and in the stopped-flow fluorescence experiments the observed signal amplitude corresponded to ∼90% of the total change (7.5 volts to 4.2 volts) at the highest concentration (99 μM). The details of the T-jump IR setup were described elsewhere (12), except that in the current study a continuous wave quantum cascade laser (Daylight Solutions, San Diego, CA) served as the IR probe.
Figure 3.
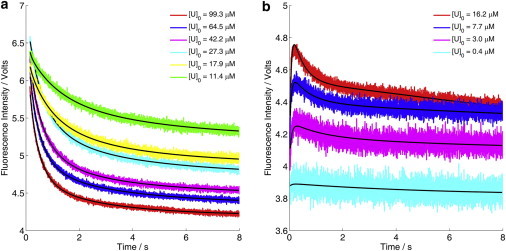
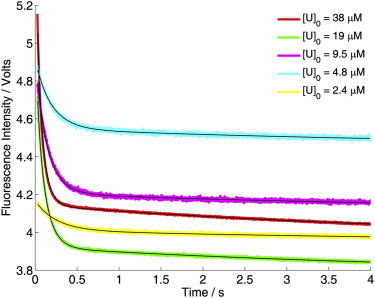
(a) Refolding kinetics of DS119 measured by stopped-flow fluorescence at different protein concentrations at pH 7.3. (b) Stopped-flow refolding kinetics obtained using an E23C-I14 and E23C-488 mixture (1:1). Global fitting to a bimolecular model (Fig. 4a) is shown in black lines.
Data analysis
The kinetic data obtained from stopped-flow experiments were fitted to the bimolecular model described in the main text. The burst phase in each kinetic curve was truncated and only the smooth part was used for the model fitting. Therefore, the start time for the stopped-flow FRET was 22 ms, and for the stopped-flow Trp fluorescence it was 148 ms (see Fig. 3). The changes of protein concentrations with time in the refolding process were described by ordinary differential equations. And the fluorescent emission parameters E (V/μM) were defined for each species. The signal of a specific species, for example U, at a time t was determined by [U](t) × QY(U) × R, where [U](t) is the concentration of U at time t, QY is the quantum yield, and R is the response of the instrument. QY (U) × R is the emission parameter E.
The curve fitting toolbox of MATLAB (The MathWorks, Natick, MA) was used to estimate the microscopic rate constants (k1, k−1, and so on) and emission parameters by modified simulated annealing, an optimization algorithm for nonlinear squared fitting using local Hessian to accelerate convergence. Because the experimental data can be represented by a linear combination of concentrations of species in the ordinary differential equations model, a solver for linear least squared error problems was applied to determine the combinational coefficients. In the cases that different curves share part of the combinational coefficients, the shared and unshared coefficients were iteratively computed by two different linear least squared error solvers.
The asymptotic approximation of 100(1-α) % confidence region was used to evaluate the 95% confidence region:
where p stands for the number of parameters and n for the number of data points. We used the Metropolis sampling method to search for parameter sets in the confidence region (15).
Results
Equilibrium unfolding measurements of DS119
The conformational stability of DS119 at neutral pH was assessed by guanidine hydrochloride (Gdn-HCl)-induced denaturation, using both CD and tryptophan (Trp) fluorescence as conformational probes. As shown (Fig. 2 a), the original Trp fluorescence intensity was low, and it progressively increased along with the Gdn-HCl concentration, indicating that Gdn-HCl-induced protein unfolding alleviated the Trp fluorescence quenching in the native DS119 structure. Furthermore, the CD unfolding curve showed significant deviation from that of Trp fluorescence, suggesting that the secondary structures (i.e., the CD signal) and the tertiary structure (i.e., the fluorescence signal) may not be formed in a cooperative manner, or that the underlying folding/unfolding process involves stable intermediates.
Figure 2.
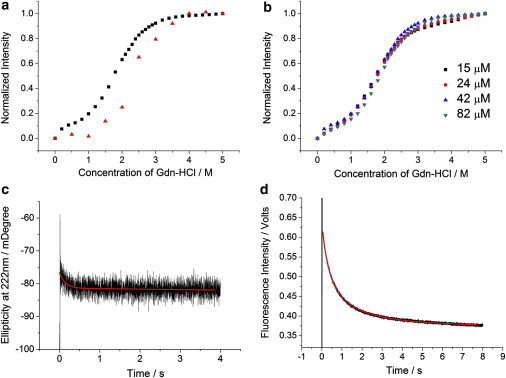
Chemical denaturation and refolding kinetics of DS119. (a) Chemical denaturation curves measured by CD (red) and Trp fluorescence (black). (b) Chemical denaturation curves of different concentrations of DS119 measured by Trp fluorescence. (c) Stopped-flow refolding kinetics measured by CD. (d) Stopped-flow refolding kinetics measured by Trp fluorescence. In the kinetic experiments, the final concentration of Gdn-HCl was 0.75 M. Red lines show the fitting to a single-exponential function.
DS119 exhibits slow and concentration-dependent folding kinetics
Because the initial attempt of using time-resolved IR spectroscopy to measure the population redistribution kinetics of DS119 in response to a nanosecond T-jump pulse did not reveal any relaxation events at the submillisecond timescale (Fig. S1 in the Supporting Material), we employed stopped-flow CD and fluorescence to measure the folding kinetics of DS119, via rapid dilution of the protein samples dissolved in a 6 M Gdn-HCl solution (50 mM Na2HPO4/NaH2PO4 buffer, pH 7.3). Both the stopped-flow CD and fluorescence kinetics indicated that DS119 folded extremely slowly (Fig. 2, c and d). By fitting the data to a single-exponential function, we roughly estimated the folding time of the secondary and tertiary structures to be 136 ms (CD signal) and 517 ms (fluorescence signal), respectively. More importantly, the folding kinetics of DS119 showed strong concentration dependence in the range of 11 and 99 μM (Fig. 3 a). In contrast, the equilibrium unfolding transitions measured at different DS119 concentrations (15–82 μM) overlapped with each other within our experimental uncertainties (Fig. 2 b), suggesting that both the folded and unfolded states are monomeric in the concentration range studied.
We further confirmed that DS119 is a monomer in the steady states using FRET, FCS, and analytical gel filtration. We labeled a mutant of DS119 (E23C) with either a fluorescent donor (I14) or an acceptor (Alexa Fluor 488). Far-UV CD and thermal unfolding experiments suggested that the dye labeling did not introduce any significant changes to the structure or thermodynamics properties of the protein (Fig. S2). In equilibrium measurements, no detectable FRET signal was observed for E23C-I14 and E23C-488 mixtures in the unfolded or the folded state (Fig. S3). FCS analysis showed that the dye-labeled DS119 mutant had similar diffusion times at 16 nM and 100 μM (Fig. S4). In the gel-filtration experiments, the elution peaks of DS119 did not show concentration dependence in the range of 5 to 200 μM, and based on the apparent molecular weight calculated from the standard curve, DS119 was determined to be a monomer (Fig. S5). These results are consistent with our previous NMR studies (9).
Next, we employed chemical cross-linking to detect possible oligomeric states of the mutant E23C both in the folded state and in the refolding process (Fig. S6). In the refolding process, a small amount of dimers were trapped for 50 to 200 μM of E23C. For the same concentration of E23C in the steady folded state, however, only the band of monomeric proteins was observed after cross-linking. In the cross-linking experiments, simultaneously linking of three or more proteins is difficult, so we cannot rule out the possibility that other oligomeric forms are involved. Taken together, these results suggest that the folding process of DS119, from a monomeric unfolded state to a monomeric folded state, probably involves a transient dimer formation step.
Stopped-flow FRET kinetics corroborate the transient dimer formation model
To substantiate the previous notion that a dimeric intermediate is populated on the folding pathway of DS119, we carried out stopped-flow FRET experiments with a mixture of E23C-I14 and E23C-488 to detect whether molecular association occurs via detection of intermolecular FRET signals. A transient FRET signal was observed in the kinetic analysis, and the amplitude of this FRET signal showed a strong dependence of the total protein concentration (Fig. 3 b). Moreover, within the time window of the stopped-flow experiments, this FRET signal first increased and then decreased. These results provide evidence for the hypothesis that the folding of DS119 involves a partially folded dimeric species, which dissociates to yield the monomeric and folded protein.
Based on the results of the stopped-flow fluorescence and FRET measurements, we proposed a bimolecular folding model to elucidate the refolding mechanism of DS119, which involves a dimeric intermediate species I2 (Fig. 4 a). We used this model to globally fit the stopped-flow kinetic traces by numerically solving the differential rate equations (Fig. S7). The fits were satisfactory and the resulting microscopic rate constants indicate that the rate-limiting step is the formation of the transient dimer (Tables 1, S1 and S2). The time courses of the refolding process were simulated based on the rate constants at different initial concentrations (Fig. 5). At the protein concentration of 50 μM, I2 reached its maximum concentration at ∼150 ms and then dissociated to the folded state F rapidly. At ∼2 s, >90% of the unfolded protein U changed to F (Fig. 4 c). The timescales for the formation of I2 at different initial concentrations are in the range of 100–200 ms (Fig S8 b), which is consistent with the folding time observed in the stopped-flow CD (136 ms), indicating that I2 probably has a significant amount of secondary structures.
Figure 4.
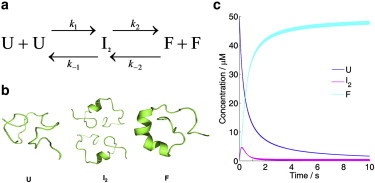
(a) The bimolecular folding model. (b) A cartoon representation of different species in the model. The structural model of I2 is an illustration of one possible configuration. (c) A simulation for the concentration changes of U, I2, and F in the folding process. The total protein concentration is 50 μM.
Table 1.
Microscopic rate constants derived from the global fitting of the stopped-flow kinetics data
| k1/μM-1s−1 | 2.97 × 10−2 | [2.89 × 10−2 2.98 × 10−2]a |
| k-1/s−1 | 2.94 × 10−9 | (0 6.36 × 10−2] |
| k2/s−1 | 6.57 | [6.50 6.91] |
| k-2/μM-1s−1 | 1.01 × 10−4 | (0 1.76 × 10−3] |
The 95% confidence intervals are shown in brackets.
Figure 5.
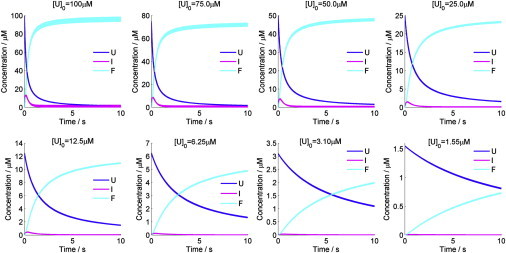
Simulated time course of the concentration of each species (U, I2, and F) in the bimolecular folding model of DS119. The total protein concentrations are labeled as [U]0.
We also calculated the 95% confidence intervals of each microscopic rate constant (Table 1, k1, k-1, k2, and k-2). The results revealed that k1 was more accurately determined than k-1, k2, and k-2.This is because the strong folding conditions (Gdn-HCl was 0.75 M after mixing) was applied in our experiment and the formation of the transient dimer is the rate-limiting step in the folding kinetics. We further applied different final concentrations of Gdn-HCl in the stopped-flow experiments and we found the resulting rate constant k1 had a linear relationship with the concentration of Gdn-HCl in the semilogarithmic plot (Fig. S9 and Table S3).
The refolding process of DS119 at pH 2.5
We also studied the refolding process of DS119 in acidic solutions. The T-jump IR experiments were performed at pH 2.5 because the Tm of DS119 (46°C) at this pH was lower than that at neutral pH. The stopped-flow results indicated that at pH 2.5 DS119 also had slow and concentration-dependent folding kinetics (Fig. S10). The kinetic curves were well fitted by our bimolecular folding model and the resulting rate constants indicated that the disaggregation of I2 to form monomeric F happened much slower in the acidic condition (the underlying rate constant k2 was ∼170 times smaller than that at pH 7.3).
We further applied NMR to probe the structure of DS119 at the temperatures around Tm in both pH 7.3 and pH 2.5 solutions. At pH 7.3 we compared the 1D-NMR and NOSEY spectra of DS119 at 20°C and 60°C, respectively (Fig. S11 and Fig. S12). Multiple peaks were observed at both temperatures, indicating the transition between the folded and unfolded states was a slow process. At pH 2.5 the exact Tm could be reached in our NMR measurement and the results are similar as that in pH 7.3 (Fig. S13). Taken together, these results provided further evidence supporting the proposed slow folding model.
The folding kinetics of the mutants of DS119
We have studied the thermodynamic properties and folding mechanisms of several mutants of DS119, including DS103 (Table 2), W9F, W9E, and P14A. DS103 is an intermediate sequence in the de novo design process of DS119. Compared to DS119, the major differences in DS103 residues are V5E, T13D, and K21L. Gel filtration experiments demonstrated that DS103 folded into a stable dimer (Fig. S14). The CD spectra and thermal denaturation experiments indicated that DS103 has a well-folded structure, and the stopped-flow fluorescence showed that the folding process of DS103 was also slow and concentration-dependent, which was well fitted to a different bimolecular model (Fig. 6 and Fig. S15). For DS103, both the intermediate and native states (I2 and F2) are dimers, and the step of forming I2 was approximately seven times faster than that of DS119 (Table S4), suggesting that the mutated residues accelerate the formation of the transient dimers.
Table 2.
Sequences of DS119 and DS103
| DS119 | GSGQV RTIWV GGTPE ELKKL KEEAK KANIR VTFWG D |
| DS103 | GSGQE RTIWV GGDPE ELKKL LEEAK KANIR VTFWG D GG |
Figure 6.
Stopped-flow refolding kinetics of DS103 measured by Trp fluorescence at pH 7.3. The fitting curves are shown in black lines.
Of the three mutations in DS103 (compared with DS119), K21L was the only one representing a change from a charged residue to a highly hydrophobic residue. It is reasonable to assume that Leu-21 in DS103 plays a significant role in stabilizing the dimer both in the folding process and in the folded structure, whereas the Lys-21 in DS119 destabilizes the transient dimer and leads to a monomeric folded structure. We speculate one possible configuration of the intermediate is that the interfaces of the transient dimers consist of a partially folded hydrophobic core (I29, V31, F33, etc.) and probably the tryptophan residues.
For the other two mutants W9E and W9F, the near-UV CD spectra showed that only W9F preserved a stable tertiary structure (Fig. S16 and Fig. S17). The thermal unfolding process and kinetic refolding results of W9F are similar to those of DS119. These results highlight the importance of π-π stacking of aromatic residues in stabilizing the βαβ structure and in shaping the folding process.
Slow kinetic events observed in the protein folding studies usually arise from proline isomerization, which occurs on a timescale of hundreds of milliseconds (16). To exclude this possibility, we mutated the single proline residue in the N-cap region of DS119 to alanine and measured the folding thermodynamics and kinetics of the resulting mutant P14A. Comparison of the CD spectra of P14A and DS119 suggests that this mutation has not altered the characteristics of the folded structure (Fig. S18). The stopped-flow fluorescence kinetics further showed that just like the native sequence, P14A also folded with an extremely slow rate, indicating that the slow folding behavior of DS119 does not stem from proline isomerization, but rather reflects the uniqueness of the underlying folding energy landscape of DS119.
Discussion
Our results are the first, to our knowledge, to demonstrate the formation of a transient dimer in the folding process of a designed protein. A transient dimer has been observed in the refolding process of cytochrome c using small-angle x-ray scattering (17). Furthermore, transient aggregation or partially unfolded oligomers have been reported for maltose-binding protein (18), the SH3 domain of α-spectrin (19), the human spliceosomal protein U1A (20), and most interestingly, a water-soluble protein with an introduced Alzheimer sequence (21). In most studies, the self-associated intermediates were verified by equilibrium experiments, such as gel filtration (17), differential scanning calorimetry (19), or observable precipitates (18). Therefore, they were more stable than the transient dimer we observed and were usually considered to be kinetic traps. In the case of DS119, the lifetime of the transient dimer was much shorter and could not be detected by these techniques. We observed the transient dimer by chemical cross-linking and FRET experiments. However, the structural details of the transient dimer need further investigation.
An interesting question is whether the dimeric intermediate pathway is obligatory and whether the dimeric structure is unique or not. To demonstrate the monomeric folding pathway is kinetically disfavored, we have roughly fit the stopped-flow refolding kinetics to a unimolecular reaction model (Fig. S19 b). Although the refolding process cannot be rigorously described by the single-exponential function, this approach nevertheless offers the easiest means to estimate the folding rate of DS119 at nearly zero protein concentration. By extrapolating the rate constants to zero concentration, we estimated that the folding time for monomeric proteins was ∼1.8 s (Fig. S20). Comparing this value to the rate constants in Table 1, we think the bimolecular folding pathway is more favored and has a lower free energy barrier. We have also tried to globally fit the kinetic curves in Fig. 3 a with a bimolecular reaction model and the fitting was more reasonable (Fig. S19 a), indicating the main kinetic process is a bimolecular process.
To probe the possible structure of the intermediate, we have compared the folding mechanisms of DS119 and the mutant DS103. For DS103 (V5E, T13D, and K21L) there are several possible interactions that can accelerate the formation of the transient dimer: 1), nonspecific hydrophobic stacking between K21L and I29, V31, F33; 2), quadrupole-like interactions among 5E, 6R on one monomer, and the same residues on the other monomer, or an intermolecular salt bridge formed between 5E and 6R. The intermolecular β-sheet, which is facilitated by the W-W interactions, is also a possible configuration. Furthermore, it has been reported that nonnative interactions participate in the folding process of several proteins, such as the de novo designed Top7 and the knotted proteins (5,6,22). In our case, the nonnative interactions observed are mainly those at the dimeric interface in the intermediate. The chevron rollover caused by transient nonnative contacts as for Top7 has not been observed in our studies (Fig. S9). Further molecular dynamics simulations and NMR studies are helpful on elucidating the structure of the intermediate.
As a de novo designed protein, DS119 is unique in that it consists of a standalone βαβ motif, in which the two β-strands are parallel to each other. Although the βαβ motif has been predicted to possess high designability (23,24), a tryptophan pair was introduced to stabilize the native fold of DS119. Thus, it is reasonable to assume that the slow folding behavior of DS119 arises from the difficulty of such parallel β-strand arrangement to spontaneously develop within a single protein molecule. In other words, a catalytic event is needed to facilitate the folding of DS119. Our stopped-flow and mutation studies suggest that this catalytic event corresponds to a transient dimerization process. We speculate that the transient dimer of DS119 possesses some secondary structure and the α-helix is probably formed, which means that several charged residues in the helix region are on the protein surface, leading to a relatively rapid disassociation process. At pH 2.5 the repulsive interactions between the charged residues are weakened and consequently the disaggregation is much slower than that at pH 7.3, as suggested by the folding results at pH 2.5. No aggregation was observed for the native state, indicating that the reverse process is rather slow, which is mainly due to the negatively designed lysine and arginine residues.
De novo designed proteins are not subjected to the process of natural selection, which makes most natural proteins fold rapidly and cooperatively. In the living cells, slow-folding kinetics is a drawback because the unfolded region in newly synthesized proteins can easily be digested by various proteases. Our experiments showed that although the thermodynamic properties of designed proteins are optimized to obtain structures with high stability, their kinetic properties are not. This is one possible reason that the designed proteins usually have folding mechanisms distinct from those of natural proteins. De novo designed proteins also provide clues to understand the evolutionary process of protein structures and folding mechanisms. The process of designing DS103 and DS119 can be regarded as an artificial structural evolution. Our results indicate that the differences between DS103 and DS119 originate from the differences in the stabilities of the transient dimers. Thus, in future protein design of novel structures, not only the native states, but also the kinetic properties of proteins need to be considered explicitly.
Conclusion
In this study, we used various kinetic techniques including T-jump IR, stopped-flow CD, and stopped-flow fluorescence to probe the folding process of a de novo designed protein DS119. As the first, to our knowledge, designed small protein with a parallel β structure, DS119 provides a model system for understanding the folding mechanisms of the βαβ structures. Our results and analysis showed that the slow folding kinetics of DS119 originates from a transient dimer formation process and intermolecular interactions participate in the folding process of DS119, though the protein is monomeric after being folded. In summary, folding mechanism studies of de novo designed proteins may shed light on the evolutionary process of protein sequence, structure, and folding kinetics.
Acknowledgments
We thank Professors Zhirong Liu and Zhuqing Zhang for helpful discussions. We acknowledge the help from Dr. Xiaogang Niu and Beijing Nuclear Magnetic Resonance Center on the NMR measurements. We also acknowledge the technical assistance of Arnaldo L. Serrano in the laser T-jump setup and Chun-Wei Lin in the FCS experiments.
This work was supported in part by the Ministry of Science and Technology of China (2009CB918500) and the National Natural Science Foundation of China (11021463, 21173013).
Contributor Information
Feng Gai, Email: gai@sas.upenn.edu.
Luhua Lai, Email: lhlai@pku.edu.cn.
Supporting Material
References
- 1.Samish I., MacDermaid C.M., Saven J.G. Theoretical and computational protein design. Annu. Rev. Phys. Chem. 2011;62:129–149. doi: 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- 2.Dill K.A., MacCallum J.L. The protein-folding problem, 50 years on. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 3.Pitera J.W., Swope W. Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc. Natl. Acad. Sci. USA. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koga N., Tatsumi-Koga R., Baker D. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watters A.L., Deka P., Baker D. The highly cooperative folding of small naturally occurring proteins is likely the result of natural selection. Cell. 2007;128:613–624. doi: 10.1016/j.cell.2006.12.042. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z., Chan H.S. Competition between native topology and nonnative interactions in simple and complex folding kinetics of natural and designed proteins. Proc. Natl. Acad. Sci. USA. 2010;107:2920–2925. doi: 10.1073/pnas.0911844107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z., Chan H.S. Native topology of the designed protein Top7 is not conducive to cooperative folding. Biophys. J. 2009;96:L25–L27. doi: 10.1016/j.bpj.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y., Alonso D.O.V., Gai F. Ultrafast folding of alpha3D: a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. USA. 2003;100:15486–15491. doi: 10.1073/pnas.2136623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang H., Chen H., Lai L. De novo design of a beta alpha beta motif. Angew. Chem. Int. Ed. Engl. 2009;48:3301–3303. doi: 10.1002/anie.200805476. [DOI] [PubMed] [Google Scholar]
- 10.Qi Y.F., Huang Y.Q., Lai L.H. Folding simulations of a de novo designed protein with a betaalphabeta fold. Biophys. J. 2010;98:321–329. doi: 10.1016/j.bpj.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capriotti E., Casadio R. K-Fold: a tool for the prediction of the protein folding kinetic order and rate. Bioinformatics. 2007;23:385–386. doi: 10.1093/bioinformatics/btl610. [DOI] [PubMed] [Google Scholar]
- 12.Huang C.Y., Getahun Z., Gai F. Helix formation via conformation diffusion search. Proc. Natl. Acad. Sci. USA. 2002;99:2788–2793. doi: 10.1073/pnas.052700099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu C., Zhang C., Lai L. Engineering a zinc binding site into the de novo designed protein DS119 with a βαβ structure. Protein Cell. 2011;2:1006–1013. doi: 10.1007/s13238-011-1121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo L., Chowdhury P., Gai F. Denaturant-induced expansion and compaction of a multi-domain protein: IgG. J. Mol. Biol. 2008;384:1029–1036. doi: 10.1016/j.jmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinath S., Gunawan R. Parameter identifiability of power-law biochemical system models. J. Biotechnol. 2010;149:132–140. doi: 10.1016/j.jbiotec.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 16.Jackson S.E., Fersht A.R. Folding of chymotrypsin inhibitor 2. 2. Influence of proline isomerization on the folding kinetics and thermodynamic characterization of the transition state of folding. Biochemistry. 1991;30:10436–10443. doi: 10.1021/bi00107a011. [DOI] [PubMed] [Google Scholar]
- 17.Segel D.J., Eliezer D., Doniach S. Transient dimer in the refolding kinetics of cytochrome c characterized by small-angle X-ray scattering. Biochemistry. 1999;38:15352–15359. doi: 10.1021/bi991337k. [DOI] [PubMed] [Google Scholar]
- 18.Ganesh C., Zaidi F.N., Varadarajan R. Reversible formation of on-pathway macroscopic aggregates during the folding of maltose binding protein. Protein Sci. 2001;10:1635–1644. doi: 10.1110/ps.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casares S., Sadqi M., van Nuland N.A.J. Detection and characterization of partially unfolded oligomers of the SH3 domain of alpha-spectrin. Biophys. J. 2004;86:2403–2413. doi: 10.1016/S0006-3495(04)74297-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silow M., Oliveberg M. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. USA. 1997;94:6084–6086. doi: 10.1073/pnas.94.12.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otzen D.E., Miron S., Oliveberg M. Transient aggregation and stable dimerization induced by introducing an Alzheimer sequence into a water-soluble protein. Biochemistry. 2004;43:12964–12978. doi: 10.1021/bi048509k. [DOI] [PubMed] [Google Scholar]
- 22.Chan H.S., Zhang Z., Liu Z. Cooperativity, local-nonlocal coupling, and nonnative interactions: principles of protein folding from coarse-grained models. Annu. Rev. Phys. Chem. 2011;62:301–326. doi: 10.1146/annurev-physchem-032210-103405. [DOI] [PubMed] [Google Scholar]
- 23.Li H., Helling R., Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 24.Miller J., Zeng C., Tang C. Emergence of highly designable protein-backbone conformations in an off-lattice model. Proteins. 2002;47:506–512. doi: 10.1002/prot.10107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.