

Abstract
Cancer stem cells (CSCs) are believed to be a promising target for cancer therapy because these cells are responsible for tumor development, maintenance and chemotherapy resistance. Finding out the critical factors regulating CSC fate is the key for target therapy of CSCs. Just as normal stem cells are regulated by their microenvironment (niche), CSCs are also regulated by cells in the tumor microenvironment. However, whether various tumor microenvironments can induce CSCs to differentiate into different cancer cells is not clear. Here, we show that single-cell-cloned CSCs, accidentally obtained from a human liver cancer microvascular endothelial cells, express classic stem cell markers, genes associated with self-renewal and pluripotent factors and possess colony-forming ability in vitro and the ability of serial transplantation in vivo. The single-cell-cloned CSCs treated with the different tumor cell/tissue-derived conditioned culture medium, which is a mimic of carcinoma microenvironment, could differentiate into corresponding tumor cells and express specific markers of the respective type of tumor cells at the gene, protein and cell levels, respectively. Interestingly, this multilineage differentiation potential of single-cell-cloned liver CSCs sharply declined after the specific knockdown of octamer-binding transcription factor 4 (Oct4) alone, even though they were under the same induction conditions (carcinoma microenvironments). These data support the hypothesis that single-cell-cloned liver CSCs have the potential of differentiating into different types of tumor cells, and the tumor microenvironment does play a crucial role in deciding differentiation directions. Simultaneously, Oct4 in CSCs is indispensable in this process. These factors are promising targets for liver CSC-specific therapy.
Keywords: CSCs, carcinoma/cancer microenvironments, multilineage differentiation potential, Oct4, microvascular endothelial cells
The cancer stem cells (CSCs) hypothesis suggests that many solid tumors, including liver cancer,1 are composed of a heterogeneous cell population that differ in phenotype and function, of which only a small subpopulation of stem-like cells (CSCs) has high clonogenic potential and can initiate and maintain tumor growth and relapse after inoculation into immunodeficient mice, whereas the majority of the tumor cells have undergone terminal differentiation and lost this potential.2
A new cancer therapy using CSCs as target will be a promising approach in the future. Therefore, an in-depth understanding of the biological characteristics of CSCs and exploring key factors of controlling CSC fates are important for elucidating the mechanisms of tumor occurrence, progression and metastasis and for establishing novel tumor diagnostic and therapeutic strategies.3 The premise of this study is effective isolation of functional CSCs.
Most CSC assays have so far depended on the use of a variety of different surface markers. However, the isolation of these CSCs is still difficult. CD133, as the most representative specific marker for the isolation of many CSCs from ovarian cancer,4 colorectal cancer,5 melanoma,6 endometrial tumors7 and liver carcinoma8 to date, has been recently questioned since CD133 negative cells possess higher tumorigenicity and resistance to chemotherapy than CD133 positive cells.9 Therefore, a definitely unique CSC marker is not available now for various tumors. Even in the same type of tumor, no single marker or combinations of markers unequivocally identifies CSCs. This is because it is currently uncertain whether these markers overlap and define a similar population or designate different populations of cells. These data imply the limitation of using current identification methods and show the need for a better definition of CSCs from the functional perspectives.
Multilineage differentiation potential is the important characteristic of CSCs. At present, most studies have shown that CSCs can differentiate into a wide variety of lineage cell types. But first of all, we must answer a question as to whether the multilineage differentiation potential of CSCs is derived from a single-clonal cell population or depends on multiple different CSCs. Furthermore, whether certain CSCs can differentiate into a variety of distinct types of tumor cells and what the key determinants in this process are is unclear. Therefore, it is necessary for investigating the multilineage differentiation potential to establish a stable and single-cell-cloned CSC line.
There is now increasing evidence that microenvironments (niche) play critical roles in the behavior of stem cells, especially in deciding differentiation directions of stem cells. Several studies have recently demonstrated that the embryonic stem cell microenvironment has a significant influence on the phenotypic characteristics of aggressive cancer cells,10 resulting in the decrease of tumorigenicity. Likewise, tumor microenvironment makes iPS cells turn into malignant cells too.11 All these data imply that the microenvironment is a critical factor for deciding differentiation fate. Based on these studies, we ascertained whether CSCs are able to differentiate into different types of tumor cells after exposure to the corresponding tumor microenvironment. If it is so, the drugs targeted at a certain type of CSCs are also effective for killing other types of CSCs. Therefore, targeted therapy of CSCs would be truly implemented.
In this study, we set out to analyze the multilineage differentiation patterns of liver CSCs by using single-cell-clone screening, combined with cell proliferation assay and xenotransplantation methods of sorting CSCs, and further characterizing these cells by using multiple markers. We show that many currently reported CSC markers are co-expressed on the cells that contain a tumor-initiating capacity. More importantly, we show that single-cell-cloned liver CSC T3A-A3 cells have the multilineage differentiation potential of various types of cancer cells both in vitro and in vivo under the corresponding tumor microenvironment induction. Our in vitro and in vivo experiments furthermore revealed that, besides the tumor microenvironment, octamer-binding transcription factor 4 (Oct4) is a crucial determinant in this cell fate decision.
Results
Accidently obtained cancer stem-like cells from human primary liver cancer microvascular endothelial cells
Recent evidence indicates that endothelial cells interact closely with CSCs, and CSCs are maintained within vascular niches.12, 13, 14 In agreement, in the cultured eighth generation of microvascular endothelial cells (Figure 1a) derived from a human primary liver cancer tissue, we accidently found a cell population morphologically different from endothelial cells, which is small, oval-like or polygonal-like (Figure 1b), and grows fast (Figure 1c). The cells were purified by subcloning and named T3A (Figure 1d). T3A cells express stem cell markers and are able to form a solid tumor after a subcutaneous injection into nude mice (Figure 1e), and the histopathological type appeared to be poorly differentiated adenocarcinoma (Figure 1f).
Figure 1.

Origin and characteristics of T3A. (a) Microvascular endothelial cells derived from human liver cancer. (b) A cell subpopulation (T3A) was observed in cultured human liver cancer microvascular endothelial cells in the eighth generation. (c) The T3A cells rapidly proliferated. (d) T3A cells were purified by subcloning. (e, f) T3A cells formed a solid tumor after a subcutaneous injection into nude mice and the pathological type of tumor tissue was poorly differentiated carcinoma
Generation of single-cell-cloned CSCs (T3A-A3)
Single-cell-cloning origin is the premise to study the multilineage differentiation potential of stem cells. To this end, T3A cells were plated in a 96-well plate for single-cell sorting by limiting dilution (Figure 2a). It is important to note that T3A cells consist of heterogeneous populations of cells. The majority of single cells have limited proliferative potential, whereas only a small fraction has the capacity to persist long term and has unlimited proliferative capacity. We eventually have gained 20 clones. The 20 clones are unequal. The MTT assay (Figure 2b) shows that their proliferation rates vary.
Figure 2.
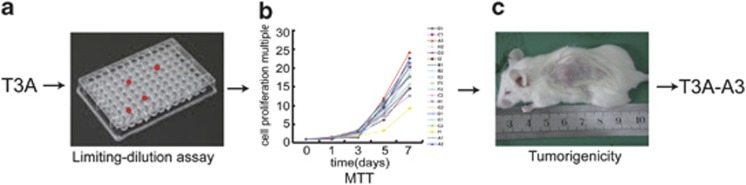
Generation of the T3A-A3 cells. (a) Single-cell clone screening of T3A cells by limiting-dilution assay. (b) Compared with the proliferation rate of 20 clones in vitro by MTT assay. (c) Compared the tumorigenicity of high and low proliferation rate clones in SCID mice
As indicated in a study of papilloma, using a genetic labeling strategy to mark individual tumor cells, Driessens et al.15 demonstrated the presence of two distinct proliferative cell compartments within the papilloma, and that the more persistent population has stem-cell-like characteristics and cycles twice per day, whereas the second represents a slower cycling transient population that gives rise to terminally differentiated tumor cells.
In agreement with their opinion, we also found that clones with high clonogenic potential in single-cell clone sorting had a higher proliferation rate in vitro and higher tumorigenicity in vivo, which were the best population of cancer stem-like cells (Figures 2b and c). Therefore, we selected one of the T3A clones with higher proliferative capacity (A3) as the subject in the following study, and named it T3A-A3. T3A-A3 cells can be passaged over 100 generations, and biological characteristics are stable. Therefore, the clone can become a good cell tool for the study of CSCs. The cell clone was awarded a national patent of the People's Republic of China (Patent No. ZL 2009 1 0177379.2)
Single-cell-cloned T3A-A3 cells express ‘stemness' genes
To further identify the cancer stem-like cell identity of T3A-A3, we detected the expressions of many vital genes and surface markers related to stemness. Using semiquantitative RT-PCR analyses (Figure 3a), we found that single-cell-cloned T3A-A3 cells express a wide variety of ‘stemness' genes such as CD133, SCF, CD117/c-kit, Nestin, CD34 and ABCG216, 17, 18 in different degrees. Moreover, T3A-A3 cells also express those genes associated with proliferation, self-renewal and differentiation of stem cells, such as Wnt/β-catenin, Notch-1, Hedgehog/SMO, Bmi-1 and Oct-4 pathways.19, 20, 21 Additionally, the T3A-A3 cells are positive for pluripotent factors related to induction of induced pluripotent stem (iPS) cells, including Oct-4, Klf4, C-myc, Sox2, Nanog and Lin28.22, 23 In this assay, human embryonic stem cell line (hESC) and human hepatoma cell line (BEL7402) were used as controls. All these markers in T3A-A3 cells were further confirmed by flow cytometry (Figure 3b).
Figure 3.

Identification of T3A-A3 cells. (a) RT-PCR analysis for the expressions of classic stem cell markers and genes associated with the proliferation and self-renewal of stem cells. (b) Flow cytometric analysis for the expressions of classic stem cell markers and genes associated with the proliferation and self-renewal of stem cells. (c) Evaluation of the self-renewing capacity of T3A-A3 cells. Secondary colony formation ability (the first and second panels). Tumor sphere-forming ability (the middle panel). Histopathology of the primary and the secondary grafted tumor (the last two panels). (d) Evaluation of tumor properties of T3A-A3 cells. Comparison of chromosomal karyotype between human fetal liver cells, human liver cancer cells and T3A-A3 cells (upper panels). Evaluation of tumorigenic and metastatic capacities of T3A-A3 cells in SCID mice (bottom panels)
Single-cell-cloned T3A-A3 cells show self-renewing capacity
The self-renewing capacity is an important characteristic of CSCs. To elucidate whether single-cell-cloned T3A-A3 cells have the capacity to self-renew, we initiated a serial of self-renewal experiments. The gold standard test is the in vivo serial transplantation of CSCs.24 The majority of T3A-A3 secondary engrafted mice formed tumors that resembled the histopathological type of the primary xenograft, providing direct evidence for the self-renewal capability of the T3A-A3 cells (Figure 3c). However, recently, many groups have commenced utilizing sphere (serum-free medium) or colony-forming assays as attractive surrogates24 in vitro. In our experiments, we further utilized the tumor sphere assay and secondary colony-forming assay in vitro culture assay for self-renewal capacity. We found T3A-A3 cells were able to form tumor spheres (Figure 3c) when plated in ultra-low attachment plates, and secondary colony-forming experiments were also positive in three-dimensional culture conditions (Figure 3c). All these data indicated that T3A-A3 cells have self-renewal capacity.
Single-cell-cloned T3A-A3 cells possess the characteristics of tumor cells
To verify whether T3A-A3 cells possess the characteristics of tumor cells, the chromosomal karyotype assay showed that human fetal liver cells are diploid, while T3A-A3 cells, akin to human liver cancer cells, are polyploid (Figure 3d). Furthermore, 106 T3A-A3 cells were intrahepatically inoculated into six SCID mice. One month later, the primary tumors were found in the liver (6/6) and metastatic tumors were seen in the lung (3/6), the stomach (2/6) and the colon (4/6) (Figure 3d).
We further detected the histopathological type and differentiated markers, including liver cell-derived differentiated markers HepPar1 and biliary epithelial cell-derived differentiated markers CK19, in various metastatic carcinomas as well as the primary carcinomas of T3A-A3 cells. To exclude metastatic adenocarcinoma, we also detected CK20. The results indicated that the histopathological type in metastatic carcinomas was consistent with the primary carcinoma. CK19 was positive; HepPar1 and CK20 were negative both in primary cancer and in various metastatic carcinomas (Supplementary Figure S1).
To further analyze the tumorigenic potential, different numbers of T3A-A3 cells were inoculated subcutaneously into SCID mice. As few as 1000 T3A-A3 cells were sufficient for tumor formation (Table 1).
Table 1. Tumorigenic potential of T3A-A3 in SCID mice.
| Number of cells injected | Tumor incidencea | Latency (days)b |
|---|---|---|
| 500 | 0/5 | — |
| 1000 | 3/5 | 90 |
| 2500 | 3/5 | 60 |
| 5000 | 4/5 | 45 |
| 10 000 | 5/5 | 30 |
Mice with tumor/mice with cell injection
Days from tumor cell injection to appearance of tumor
Single-cell-cloned T3A-A3 cells possess the potential of differentiating into different tumor cell types
Another key property of stem cells is their ability to differentiate into different lineages. To detect whether tumor microenvironments play critical roles in deciding differentiation fate of CSCs, we prepared appropriate tumor microenvironment to induce single-cell-cloned T3A-A3 cells differentiation.
Phase-contrast microscopy analysis revealed changes in the cell morphology of the T3A-A3 cells following melanoma cell-directed differentiation after 72 h induction, from polygonal-like into longitudinal stretched cells, which are similar to melanoma cell line SK-MEL-1, especially in the group of tissue-conditioned medium (Figure 4a). Furthermore, melanoma-specific expressing marker gp100 detected by RT-PCR, western blot, immunofluorescence staining in vitro and immunohistochemistry staining on graft tumor tissue in vivo, respectively at the gene, protein and cell levels became significantly positive in T3A-A3 cells after induction, but T3A-A3 cells did not express gp100 before induction.
Figure 4.

Evaluation of multi-differentiation potential of T3A-A3 cells. The multi-differentiation potential of T3A-A3 cells was analyzed after induction with melanoma, lymphoma and prostate cancer cell- or tissue-derived conditioned culture medium. The morphological changes of T3A-A3 after induction were photographed. The tumor-specific markers were compared between T3A-A3 cells before and after induction by RT-PCR, western blot, immunofluorescence staining in vitro and immunohistochemistry staining on graft tumor tissue in vivo. (a) Morphological changes and expression of gp100 in T3A-A3 after induction by melanoma-derived conditioned culture medium. (b) Morphological changes and expression of CD10 in T3A-A3 after induction by lymphoma-derived conditioned culture medium. (c) Morphological changes and expression of PSA in T3A-A3 after induction by prostate cancer-derived conditioned culture medium. (1: positive control cells (corresponding cancer cell line); 2: T3A-A3 cells; 3: T3A-A3 cells after induction with tumor cell-derived conditioned culture medium; 4: T3A-A3 cells induced with tumor tissue-derived conditioned culture medium)
In vivo differentiation of T3A-A3 cell-derived tumors revealed that gp100-positive cells were present in graft tumor after differentiation by immunohistochemistry staining (Figure 4a).
Similar results were also found in T3A-A3 cells after induction with lymphoma or prostate cancer cell/tissue-derived conditioned culture medium. The expressions of CD10 (a specific marker of lymphoma) and PSA (a specific marker of prostate cancer cell) changed from negative in T3A-A3 cells before differentiation to positive in T3A-A3 cells after differentiation. Both markers were positive in the grafted tumors derived from T3A-A3 cells after differentiation by lymphoma or prostate cancer cell/tissue-derived conditioned culture medium (Figures 4b and c).
Taken together, these data settle an important point in the origin of multiple distinct tumor types within a tumor, suggesting that subsets of single-cell-cloned T3A-A3 cells have the capacity to differentiate into different types of carcinoma cells under the corresponding tumor microenvironment stimuli. In addition, we found that upon the onset of differentiation, related stemness markers, such as CD133, Oct4, SCF, Notch-1 and Nestin, are significantly declined on T3A-A3 cells (Supplementary Figure S2).
To exclude the possibility that the multilineage differentiation potential of A3 clone was cell-type specific, we selected another clone, A2, with high proliferation rate. We found that A2 can also differentiate into different types of tumor cells after induction with melanoma, lymphoma and prostate cancer tissue-derived conditioned culture medium (Supplementary Figure S3), which showed that the phenomenon we observed in A3 clone is a general phenomenon.
The multilineage differentiation potential of T3A-A3 cells can be regulated by Oct4
To study whether expression of Oct4 is required to maintain the multilineage differentiation potential of CSCs, we knocked down Oct4 expression in the T3A-A3 cells with Oct4 shRNA. T3A-A3 cells infected with vector containing scramble shRNA were used as control. As expected, Oct4 shRNA significantly decreased the Oct4 expression of T3A-A3 cells at both mRNA (Figure 5a) and protein (Figure 5b) levels.
Figure 5.

Effect of Oct4 knockdown on the stem cell properties of T3A-A3. Oct4 in T3A-A3 cells was knocked down by infection with shOct4-carring lentivirus. (a) The expression level of Oct4 was evaluated by RT-PCR. (b) The expression level of Oct4 was evaluated by western blot. (c), The self-renewal ability of shOct4-T3A-A3 was detected by colony formation assay in vitro, **P<0.01 versus shNC, n=3. (d) The tumorigenicity of shOct4-T3A-A3 was examined by grafted tumor growth in vivo **P<0.01 versus shNC, n=6. (e) The sphere assay of shOct4-T3A-A3 was performed on ultra-low adherent plates
After Oct4 knockdown, T3A-A3 cells cannot differentiate into different cancer cells, and the specific markers of three kinds of tumors cannot be detected both in vitro and in vivo even if under the corresponding tumor microenvironments as in the previous differentiation experiments (Figure 6).
Figure 6.

Effect of Oct4 knockdown on the multi-differentiation potential of T3A-A3 cells. After Oct4 knockdown in T3A-A3 cells, the committed inductions were repeated using melanoma, lymphoma and prostate cancer tissue-derived conditioned culture medium. The tumor-specific markers were detected by RT-PCR, western blot, immunofluorescence staining in vitro and immunohistochemistry staining on graft tumor tissue in vivo. (a) Detection of the expressions of gp100 in shOct4-T3A-A3 cells induced by melanoma tissue-derived conditioned culture medium. (b) Detection of CD10 in shOct4-T3A-A3 cells induced by lymphoma tissue-derived conditioned culture medium. (c) Detection of PSA in shOct4-T3A-A3 induced by prostate cancer tissue-derived conditioned culture medium. 1: Positive control (corresponding cancer cell line); 2: shNC-T3A-A3 cells; 3: induced shNC-T3A-A3 cells; 4: shOct4-T3A-A3 cells; 5: induced shOct4-T3A-A3 cells)
In addition, we also detected the phenomenon of the recently reported25 expression of Oct4 positively correlated with colony forming ability (Figure 5c) and tumorigenicity in our cultures (Figure 5d). Moreover, we applied Oct4-knockdown T3A-A3 cells to a spheroid cell culture that was subjected to adherent plate differentiation even if on ultra-low adherent plates. Cells were much more flattened, which indicated that differentiation had occurred in Oct4-knockdown T3A-A3 cells (Figure 5e).
These data suggest that Oct4 expression is required for maintaining the CSC pluripotency.
Discussion
In this study, we provide evidence to support an important aspect of the cancer stem cell hypothesis, which claims that single-cell-cloned liver CSCs can self-renew and initiate a carcinoma. Importantly, we now formally confirm that the single-cell-cloned liver CSCs have the multilineage potential to differentiate into a variety of cancer cells under various tumor microenvironments, which is, in part, dictated by Oct4 and is intrinsic to CSCs.
Multi-differentiation potential is an essential feature of stem cells. In theory, CSCs have differentiating plasticity under epigenetic control as they express many pluripotent factors. The liver CSC T3A-A3 cells used in our study express a variety of pluripotent factors associated with iPS, including Oct4, Sox2, Nanog, C-myc, Lin28 and Klf4, so they have the possibility of multilineage differentiation.
Although it has been demonstrated that CSCs could differentiate into a wide variety of lineage cell types, there is no evidence that CSCs also have the potential to differente into different types of tumor cells. Can various cancer cells be derived from certain common CSCs? The answer to this question will provide vital experimental data for targeting CSCs in anti-tumor therapy.
As we all know, tumor is a heterogeneity population. This heterogeneity has been attributed to the genetically distinct clones present in a tumor. There are different subpopulations in CSCs, and they may exhibit different responses to different inducers. Moreover, since the CSCs used in the present study of multi-differentiation potential come from different tumors, the comparison and evaluation between these results are difficult. Therefore, obtaining a single-cell-cloned CSC cell line is important in investigating the multi-differentiation potential of CSCs.
We address the question whether one single population of CSCs is indeed responsible for various tumor phenotypes, or several competing clones can co-exist in one tumor to produce the diversity of phenotypes. We utilized a series of methods to generate single-cell-derived CSC cultures by combining limiting-dilution assay with cell proliferation assay and the xenotransplantation method.
There is now increasing evidence that the microenvironment (niche) can exert profound epigenetic effects on stem cells through interactions between cells or through cell-derived factors originating from the surrounding cells within the niche.
In our study, we prepared cell-derived conditioned culture medium and tissue-derived conditioned culture medium, as described by several teams, to induce stem cell differentiation.26, 27 Given that hypoxia and reoxygenation of tumor progression cooperate to provide growth advantages essential for the progressive development of tumors,28 the difference in our study is that the tumor cell lines used in cell-derived conditioned culture medium were prepared by repeated rounds of hypoxia-reoxygenation stimulations, which is different from other teams. In addition, the tumor microenvironment, composed of non-epithelial matrix cells, such as vascular cells, fibroblasts, inflammatory cells and extra-cellular matrix, is essential for maintaining the proliferating and differentiating capacity of CSCs.29 Thus, the tumor tissue-derived conditioned culture medium used here was prepared from the grafted tumors of three types of tumor cells. Therefore, the cell-/tissue-derived conditioned culture medium in our study can better imitate tumor microenvironments.
The multipotent phenotype of CSCs permits them to respond to various microenvironments. Our data show that T3A-A3 cells can express specific tumor cell markers after the induction of the respective tumor microenvironment at gene, protein and cell levels, especially in tumor tissue-derived conditioned culture medium (Figure 4). It indicates that a suitable microenvironment is critical for CSCs in deciding differentiation directions. This result is consistent with the data reported on mouse iPS cells, where the conditioned culture medium of cancer cell lines can make iPS cells develop into CSCs.11
Whether there are other factors in deciding CSC differentiation directions besides the tumor microenvironment is unclear. Oct-4 is the most critical mediator for reprogramming somatic cells into induced pluripotent stem (iPS) cells as a single factor,30 as well as an important transcription factor regulating the pluripotency of embryonic stem cells.31
Recently, the process of reprogramming and tumorigenesis has been linked for sharing common mechanisms,32 which makes us speculate that Oct4 may be playing a critical role in pluripotency of CSCs as well. Moreover, Oct4 is detectable in a variety of cancer types33 and CSC-like cells.34, 35 It has been shown that Oct4 plays crucial roles in self-renewal and tumorigenicity of CSCs.34, 35
Nevertheless, it is not known whether Oct4 has similar functions with regard to pluripotency in CSCs as in normal cells. It has only been shown that Oct4 expression is associated with the differentiation state of cancer cells.35 Our study showed that after the specific knockdown of Oct4 in T3A-A3 cells, the tumorigenicity sharply declined in an orthotopic mouse model concomitant with decreasing colony formation and tumor-sphere in vitro (Figure 5). It was surprising that T3A-A3 cells were not able to differentiate into different cancer cells, and the specific markers of three kinds of tumors cannot be detected both in vitro and in vivo, although they were induced under various tumor microenvironments.
To our knowledge, this is the first study to show that Oct4 alone is sufficient to eliminate multilineage differentiation of liver CSCs. The loss of pluripotency in CSCs is likely resulted from their endogenous expression repression of Sox2, Lin28, Nanog, C-myc and Klf4. Endogenous expression of reprogramming factors has been attributed to the Oct4-induced reprogramming of neural stem cell to iPS cells.30
Moreover, it has been shown that expression of endogenous Oct4 in tumor cells is regulated by its microenvironment, especially under hypoxia condition.36 All these data indicate that the pluripotency or plasticity of CSCs is associated not only with the tumor microenvironment but also with the expression of Oct4 in CSCs, and this interaction plays a critical role in deciding tumor cell differentiation fates.
Defining CSCs within a tumor currently depends on expression of cell surface markers, which has recently been described, and cannot completely represent true CSCs. Here, we have reported the isolation and characteristics of the CSC population from human liver cancer based on function sorting, but not their cell surface marker expression. We have shown that a single-cell clone (T3A-A3) obtained by limiting dilution assay, with higher proliferation ability in vitro and higher tumorigenicity in vivo, expresses a variety of CSCs markers. This provides strong support for the power of this sorting method. Our findings reveal that the characteristics of the single-cell cloned T3A-A3 subpopulation were consistent with the predicted behavior of both primitive stem and cancer cells, which expresses a variety of surface markers associated with stemness, possess self-renewal and multilineage differentiation potential and even can initiate a tumor with as few as 1000 T3A-A3 cells. As tumor is a heterogeneity population, single-cell-clone origin ensures the credibility to study the multilineage differentiation potential of CSCs.
Additionally, particularly noteworthy are T3A cells existing in cultured microvascular endothelial cells of liver cancer, which is consistent with recent findings that endothelial cells interact closely with CSCs, and CSCs are maintained within vascular niches.12, 13, 14 Some studies showed that CSCs are most likely to be quiescent, slow cycling dormant cells,37 whereas addition of certain angiogenic factors or co-culture of tumor cells together with angiogenic cells was sufficient to revert cancer dormancy by triggering the angiogenic switch.38, 39 Once dormant CSCs were activated, they resulted in rapid cell proliferation and progression of the disease.40 So we speculated that dormant T3A cells exist in the tumor microvascular endothelial cells and underwent rapid proliferation upon activation by addition of endothelial cell growth supplement (ECGS) and interaction with tumor microvascular endothelial cells. The discovery of T3A cells provided another important clue for studying the mechanism of CSC dormancy and the relationship between CSCs and tumor microvascular endothelials.
Combined, our data reveal that liver cancer is a heterogeneity population. A single-cell clone of T3A cells obtained by combining tumorigenicity in vivo and clonogenic potential and proliferation ability in vitro screening, T3A-A3, has the characteristics of CSCs, which exhibit high tumorigenic and metastatic potentials and express various classic stem cell markers, important genes associated with the self-renewal capacity and pluripotent factors related to iPS cell induction. Under the corresponding tumor environment, CSCs can differentiate into various cancer cells, which indicate that microenvironment is of vital importance in deciding differentiation directions. In addition, specific knockdown of Oct4 alone is sufficient to eliminate this potential, which suggests that Oct4 is a positive regulator of multilineage differentiation at least in liver CSCs. Nevertheless, more studies are needed to elucidate the regulation mechanism between tumor microenvironment and Oct4 in the multilineage differentiation of CSCs. Appreciation of the importance of the interaction between pivotal regulator of CSCs and its microenvironment has led to the development of novel anti-cancer therapeutic agents.
Materials and Methods
Human sample
The human liver cancer sample was collected by the China-Japan Friendship Hospital with written informed consent from a hepatoma patient.
Animals
SCID CB17/Icr-Prkdcscid/IcrlcoCrlVr mice and nude CAnN.Cg-Foxn1nu/CrlVr mice were purchased by Vital River Laboratories (VRL, Beijing, China), a leading company in commercial production of laboratory animals in China.
Cell culture
Human melanoma cell line SK-MEL-1, human lymphoma cell line Daudi and human prostate cancer cell line LNCaP were obtained from the Cell Culture Center of Chinese Academy of Medical Sciences. SK-MEL-1 was maintained in alpha modified Eagle's Minimal Essential Media (MEM) supplemented with 10% fetal bovine serum (FBS), and Daudi and LNCaP were maintained in RPMI-1640 Media supplemented with 10% FBS.
T3A-A3 was maintained in DMEM-F12 supplemented with 1% FBS, B27 (1 : 50; Invitrogen, Shanghai, China), 20 ng/ml human epidermal growth factor (hEGF; Invitrogen), 10 ng/ml basic fibroblast growth factor (bFGF; R&D Systems, Shanghai, China), 100 μg/ml endothelial cell growth supplement (ECGS), 2 μg/ml heparin (Sigma, Shanghai, China), 2 mM glutamine (Sigma), 100 units/ml penicillin (Sigma), 100 μg/ml streptomycin (Sigma), 5 μg/ml insulin (Sigma) and 0.5 μg/ml hydrocortisone (Sigma).
Isolation of CSCs-like cells emerged in microvascular endothelial cells of human liver cancer
In the culture of the 8th generation of microvascular endothelial cells of human liver cancer tissue, a cell subpopulation with rapid proliferation was accidently observed and was isolated by subcloning. This subpopulation was named T3A, which is morphologically different from the endothelial cells when they are adherent growth.
Limiting dilution assay
T3A cells were washed with phosphate-buffered saline (PBS) and tryspinized. The cells were then re-suspended at 100 cells per ml in DMEM media. Cells were then dispensed at 5 ml of cell suspension into 96-well plates containing 95 ml of DMEM media. A volume of 200 μl was plated into each well of a 96-well culture plate (approximately 1 cell per well). Four hours after plating, each well was checked with an inverted microscope (Nikon TS100). Wells containing only a single cell were marked; wells with no cells or with more than one cell were excluded. Wells were checked daily and a further 100 μl of cloning medium added after 7 days. After 10–14 days in culture, the colonies were passaged into 24-well culture plates, and then T25 culture bottles. In the culture of the colonies, some of the colonies could not proliferate and died out.
Generation of single-cell-clone CSCs (T3A-A3)
T3A cells were performed by limiting the dilution assay, and gained 20 single-cell clones. By combining the clonogenic potential by limiting dilution assay and proliferation ability in vitro by MTT with the tumor-initiating ability in vivo by inoculating subcutaneously into SCID mice, the single-cell clone A3 with the highest proliferation ability, clonogenic potential and the highest tumorigenic potential was selected and named as T3A-A3. Also, T3A-A3 could be passaged over 100 generations.
Sphere assay
T3A-A3 cells were suspended in serum-free DMEM-F12 supplemented with B27 (1 : 50; Invitrogen), 20 ng/ml human epidermal growth factor (hEGF; Invitrogen), 10 ng/ml basic fibroblast growth factor (bFGF; R&D Systems), 2 μg/ml heparin (Sigma), 2 mM glutamine (Sigma), 100 U/ml penicillin (Sigma), 100 μg/mlstreptomycin (Sigma), 5 μg/ml insulin (Sigma) and 0.5 μg/ml hydrocortisone (Sigma) and plated into ultra-low attachment plates (Corning, Shanghai, China) as described previously.41
Reverse transcription-PCR
Total RNA was isolated from T3A-A3 cells using the RNeasy mini kit (Qiagen, Valencia, CA, USA) following the instructions of the manufacturer. One microgram RNA was reverse-transcribed by Reverse Transcription System (Promega, Madison, WI, USA). The expressions of SCF, c-kit, Nestin, CD34, ABCG2, CD133, β-catenin, Notch-1, SMO, Bmi-1, Oct-4, Klf4, C-myc, Sox2, Nanog and Lin28 were detected by PCR. The gene-specific primer sequences are available on request. The PCR products were analyzed by gel imaging system AlphaImager 2200 (AlphaIInnotech, San Leandro, CA, USA). The specific primers for RT-PCR were the following:
β-actin: forward, 5′-TGGCACCACACCTTCTACAATGAGC-3′, reverse, 5′-GCACAGCTTCTCCTTAATGTCACGC-3′
Oct-4: forward, 5′-CGACCATCTGCCGCTTTGAG-3′, reverse, 5′-CCCCCTGTCCCCCATTCCTA-3′
SCF: forward, 5′-CTCCTATTTAATCCTCTCGTC-3′, reverse, 5′-TACTACCATCTCGCTTATCCA-3′
C-kit: forward, 5′-GGCTCTTCTCAACCATCTGTG-3′, reverse, 5′-ATTTGCCGGTGTTGGTGGCTT-3′
CD34: forward, 5′-ACAACCTTGAAGCCTAGCCTG-3′, reverse, 5′-CAAGACCAGCAGTAGACACTG-3′
ABCG2: forward, 5′-GGCCTCAGGAAGACTTATGT-3′, reverse, 5′-AAGGAGGTGGTGTAGCTGAT-3′
Nestin: forward, 5′-AGAGGGGAATTCCTGGAG-3′, reverse, 5′-CTGAGGACCAGGACTCTCTA-3′
CD133: forward, 5′-CCTTGTGGCAAAGCTCAACC-3′, reverse, 5′-TCACCTCCTCTCTCACCCAG-3′
SMO: forward, 5′-ATCTCCACAGGAGAGACTGGTTCGG-3′, reverse, 5′-AAAGTGGGGCCTTGGGAACATG-3′
Notch-1: forward, 5′-ATCGGGCACTGAACGTGGCG-3′, reverse, 5′-CACGTCTGCCTGGCTCGGCTC-3′
Bmi-1: forward, 5′-GGAGACCAGCAAGTATTGTCCTTTTG-3′, reverse, 5′-CATTGCGCTGGGCATCGTAAG-3′
β-catenin: forward, 5′-ACTGGCAGCAACAGTCTTACC-3′, reverse, 5′-TTTGAAGGCAGTCTGTCGTAAT-3′
Sox2: forward, 5′-GGCAGCTACAGCATGATGCAGGAGC-3′, reverse, 5′-CTGGTCATGGAGTTGTACTGCAGG-3′
Nanog: forward, 5′-CAATGGTGTGACGCAGAAGG-3′, reverse, 5′-GCAGAGATTCCTCTCCACAGT-3′
Lin28: forward, 5′-GGAGGCCAAGAAAGGGAATA-3′, reverse, 5′-CCGCCCCATAAATTCAAGAT-3′
C-myc: forward, 5′-TCTTGACATTCTCCTCGGTG-3′, reverse, 5′-TACCCTCTCAACGACAGCAG-3′
Klf4: forward, 5′-CCAGCCAGAAAGCACTAC-3′, reverse, 5′-GACTCACCAAGCACCATC-3′
gp100: forward, 5′-AGTTCTAGGGGGCCCAGTGTCT-3′, reverse, 5′-GGGCCAGGCTCCAGGTAAGTAT-3′
CD10: forward, 5′-TGGAGATTCATAATGGATCTTGTAAGC-3′, reverse, 5′-CATCCAAGTGAGGTCATCTAAAGTCTG-3′
PSA: forward, 5′-GGTGATGACTCCAGCCACGA-3′, reverse, 5′-GCGCACACACGTCATTGGAA-3′.
Fluorescence-activated cell sorting analysis
To detect the variety of stem cell marker expression, T3A-A3 cells were labeled with CD34 (mouse anti-human CD34-FITC, 130-081-001, Miltenyi Biotech, Shanghai, China), ABCG2 (mouse anti-human ABCG2, ab3380, Abcam, Cambridge, UK), Oct4 (rabbit anti-human Oct3/4, Sc5279, Santa Cruz, Santa Cruz, CA, USA), CD133 (mouse anti-human CD133-PE, 130-080-801, Miltenyi Biotech), Bmi-1 (rabbit anti-human Bmi-1, ab126783, Abcam), C-myc (mouse anti-human C-myc, MCA2200GA, AbD Serotec, Oxford, UK), β-catenin (mouse anti-human Notch-1, ab22656, Abcam), SCF (rabbit anti-human SCF, ab52603, Abcam), C-kit (rabbit anti-human C-kit, ab32362, Abcam), Sox2 (rabbit anti-human Sox2, 3579, Cell Signaling, Shanghai, China), Nanog (rabbit anti-human Nanog, ab109250, Abcam), Klf4 (mouse anti-human Klf4, ab130243, Abcam), Lin28 (rabbit anti-human Lin28A, ab124765, Abcam), Notch-1 (rabbit anti-human Notch-1, ab65297, Abcam), SMO (rabbit anti-human SMO, ab72130, Abcam) and Nestin (mouse anti-human Nestin, MAB5236, Chemicon, Temecula, CA, USA) followed by fluorescence-activated cell sorter (FACS) analysis. Flow cytometric analysis was performed using a FACS Canto flow cytometer (Becton Dickinson, San Jose, CA, USA).
Secondary colony-forming assay
Briefly, T3A-A3 cells were suspended in 0.9% methylcellulose-based semisolid medium MethoCult H4100 (StemCell, Beijing, China). After 14 days, individual primary clones (>50 cells) were trypsinized and re-plated in the same conditions to examine the secondary colony-forming ability for self-renewal.
Serial transplantation assay
We performed serial transplantation experiments from T3A-A3 cell-derived tumors that grew from an initial injection of 50 000 T3A-A3 cells. After 8 weeks, the xenograft tumor was excised from the primary mouse recipient, dissociated into single-cell suspension, grown in culture for approximately 4 weeks, resorted for rapid proliferative single-cell clone using limited dilution assay and then re-injected into secondary mouse recipients.
Tumorigenic potential for cell transplantation into SCID mice
T3A-A3 cells were harvested and suspended in Matrigel (diluted 1 : 1 with DMEM), and then injected subcutaneously into 4-week-old SCID mice at cell numbers ranging from 500 to 10 000 cells per site. Mice were assessed regularly for tumor formation for up to 24 weeks.
Preparation of tumor-derived conditioned culture medium
Three types of tumor cell lines including melanoma cell line SK-MEL-1, lymphoma cell line Daudi and prostate cancer cell line LNCaP, with the respective specific marker and all negative in T3A-A3 cells, were selected and prepared in cell/tissue conditioned medium.
Tumor cell-derived conditioned culture medium was prepared as described previously.26 Briefly, the tumor cells were cultured under hypoxic conditions (95% N2 and 5% CO2) for 24 h followed by reoxygenation (95% O2 and 5% CO2) for another 24 h. Then the process was repeated thrice. The supernatant was collected and sterilized by 0.22-μm filters. The tumor cell-derived conditioned culture medium was prepared by mixing the supernatant 1 : 1 with DMEM containing 20% FBS.
Tumor tissue-derived conditioned culture medium was prepared using grafted tumors derived from SK-MEL-1 cells, Daudi cells or prostate cancer cells on SCID mice as described previously.27 Briefly, the grafted tumor tissues were crushed in liquid nitrogen and then lysed in buffer with freshly added protease inhibitor cocktail. After centrifugation (12k r.p.m.), the supernatant was sterilized by 0.22-μm filters, and the protein concentration was determined. The tumor tissue-derived conditioned culture medium was prepared by diluting the tumor-tissue derived supernatant 1 : 10 in DMEM containing 20% FBS.
Multilineage differentiation potential assay
Single-cell-cloned T3A-A3 cells were cultured in different tumor cell/tissue-derived conditioned culture medium for 3 weeks. Then the melanoma-specific marker gp100, lymphoma-specific marker CD10 and prostate cancer-specific marker PSA were detected by RT-PCR, western blot and immunofluorescence staining in T3A-A3 cells before differentiation and T3A-A3 cells after differentiation, respectively. In addition, T3A-A3 cells before differentiation and T3A-A3 cells after differentiation were inoculated subcutaneously into SCID mice. Expressions of gp100 (mouse anti-human gp100, ab15228, Abcam), CD10 (mouse anti-human CD10, ab951, Abcam) and PSA (rabbit anti-human KLK3, HPA000764, Sigma) were detected on the grafted tumor tissue by immunohistochemistry.
Knockdown of Oct4 by lentivirus in T3A-A3 cells
Lentivirus production and tittering were carried out according to the following protocols from Trono Lab (http://tronolab.epfl.ch). In HEK293T cells, the vector pLVTHM-shOct4 contains a previously validated Oct4 shRNA sequence.42 The vector pLVTHM-shOct4 and a control vector pLVTHM-scramble (kindly provided by Prof. Kosik)42 were co-transfected with the envelope plasmid pMD2 and the packing vector psPAX2 via calcium phosphate precipitation. The virus-containing medium was collected 48 h after transfection, and the viruses were concentrated by ultracentrifugation at 28 000 r.p.m. for 2 h. Concentrated viruses were reconstituted in phosphate-buffered saline (PBS).
Immunofluorescence staining and immunohistochemistry
After induction by conditioned culture medium, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS containing 0.5% BSA (PBS-BSA) for 30 min. The cells were subsequently incubated with mouse anti-human gp100(ab15228, Abcam), mouse anti-human CD10 (ab951, Abcam) and rabbit anti-human KLK3 (PSA) (HPA000764, Sigma) for 30 min, followed by labeling with Alexa Fluor 488-conjugated rabbit anti-mouse or goat anti-rabbit IgG antibody (Invitrogen). The cells were viewed under a fluorescent microscope (Olympus IX71, Japan).
For immunohistochemistry, tissues were fixed in 10% formalin, paraffin embedded, sectioned and deparaffinized. Microwave antigen retrieval was performed. Antibodies were: mouse anti-gp100 (ab15228, Abcam), mouse anti-CD10 (ab951, Abcam), rabbit anti-KLK3 (PSA) (HPA000764, Sigma), mouse-HepPar1 (IMG-80318, Imgenex, Ste E San Diego, CA, USA), rabbit anti-CK19 (ab76539, Abcam) and rabbit anti-CK20 (ab97511, Abcam). Peroxidase-conjugated secondary rabbit anti- mouse or goat anti-rabbit antibodies were used from Sigma.
Western blot
Induced cells were harvested and lysed in RIPA lysis buffer (50 mM Tris-HCl (pH 8.0), 150 mM Nacl, 0.1% SDS, 1% NP-40, 0.25% sodium deoxycholate and 1 mM EDTA) with freshly added protease inhibitor cocktail (Calbiochem, Shanghai, China) for 30 min on ice, and subsequently centrifuged at 13 000 r.p.m. for 10 min. The total protein concentration of whole-cell extracts was measured with BCA Protein Assay Kit (Pierce, Pittsburgh, PA, USA). Total cellular proteins were fractionated by 10% SDS-PAGE. Then the proteins were electro-transferred to polyvinylidene fluoride membranes (Millipore, Shanghai, China), blocked with PBS-T containing 5% non-fat dry milk, and the primary antibodies were diluted in PBS-T containing 5% non-fat dry milk: mouse anti-human gp100 (ab15228, Abcam), mouse anti-human CD10 (ab951, Abcam) and rabbit anti-human KLK3 (HPA000764, Sigma) followed by horseradish peroxidase (HRP)-conjugated secondary antibody (Sigma). Binding was detected by enhanced chemiluminescence (Millipore).
Ethics statement
The use of human liver cancer tissues and the waiver of patient consent in the present study were approved by the Clinical Research Ethics Board of China-Japan Friendship Hospital. The study was conducted according to the principles of the Declaration of Helsinki. The animal studies were approved by the Animal Ethics Committee of China-Japan Friendship Hospital.
Statistical analysis
The results are expressed as mean±SD. Two-tailed t-tests were employed to analyze the in vitro and in vivo data using the software (SPSS 16.0; SPSS Inc., Chicago, IL, USA). Statistical significance was set at P<0.05, while P<0.01 indicated a highly significant difference.
Acknowledgments
This work was supported by the grants from National Basic Research Program of China (2012 CB966402), Key New Drug Creation & Manufacture Program (No.2011ZX09102-010-03) and National Basic Research Program of China (2009 CB521804) to LOU J.
Glossary
- CSCs
cancer stem cells
- hEGF
human epidermal growth factor
- RT-PCR
reverse transcriptase-polymerase chain reaction
- DMEM
Dulbecco's, modified Eagle's medium
- bFGF
basic fibroblast growth factor
- MTT
3-(4,5-dimethylthioazol-2-yl)-2,5-diphenyl tetrazolium bromide
- iPS cells
induced pluripotent stem cells
- ECGS
endothelial cell growth supplement
- HE
hematoxylin and eosin stain
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Disease website (http://www.nature.com/cddis)
Author Contributions
LOU J designed and supervised this study, wrote and edited the manuscript; LIU H and ZHANG W performed experiments, analyzed data and wrote the manuscript; JIA Y and PENG L provided technical support for some critical experiments; GRAU G, YU Q and DENG H participated in analysis and discussion of results and assisted in the manuscript; Ran Y and YANG Z contributed to the establishment and identification of T3A-A3 cell line.
Edited by Y Shi
Supplementary Material
References
- Rountree CB, Mishra L, Willenbring H. Stem cells in liver diseases and cancer: recent advances on the path to new therapies. Hepatology. 2012;55:298–306. doi: 10.1002/hep.24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. International journal of cancer Journal international du cancer. 2012;130:29–39. doi: 10.1002/ijc.25967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Huber J, Hadaschik B, Siegers GM, Fiebig HH, Schuler J. Characterization of colon cancer cells: a functional approach characterizing CD133 as a potential stem cell marker. BMC cancer. 2012;12:96. doi: 10.1186/1471-2407-12-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CY, Schwartz BE, Hsu MY. CD133+ melanoma subpopulations contribute to perivascular niche morphogenesis and tumorigenicity through vasculogenic mimicry. Cancer research. 2012;72:5111–5118. doi: 10.1158/0008-5472.CAN-12-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutella S, Bonanno G, Procoli A, Mariotti A, Corallo M, Prisco MG, et al. Cells with characteristics of cancer stem/progenitor cells express the CD133 antigen in human endometrial tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:4299–4311. doi: 10.1158/1078-0432.CCR-08-1883. [DOI] [PubMed] [Google Scholar]
- Rountree CB, Ding W, He L, Stiles B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem cells. 2009;27:290–299. doi: 10.1634/stemcells.2008-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. The Journal of clinical investigation. 2008;118:2111–2120. doi: 10.1172/JCI34401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4329–4334. doi: 10.1073/pnas.0800467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Kasai T, Li Y, Sugii Y, Jin G, Okada M, et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One. 2012;7:e33544. doi: 10.1371/journal.pone.0033544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- Bautch VL. Cancer: tumour stem cells switch sides. Nature. 2010;468:770–771. doi: 10.1038/468770a. [DOI] [PubMed] [Google Scholar]
- Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–530. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levina V, Marrangoni A, Wang T, Parikh S, Su Y, Herberman R, et al. Elimination of human lung cancer stem cells through targeting of the stem cell factor-c-kit autocrine signaling loop. Cancer research. 2010;70:338–346. doi: 10.1158/0008-5472.CAN-09-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem cells. 2009;27:2405–2413. doi: 10.1002/stem.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672–2680. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- Perryman SV, Sylvester KG. Repair and regeneration: opportunities for carcinogenesis from tissue stem cells. Journal of cellular and molecular medicine. 2006;10:292–308. doi: 10.1111/j.1582-4934.2006.tb00400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeb BG, Lacerda L, Xu W, Larson R, Solley T, Atkinson R, et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem cells. 2012;30:2366–2377. doi: 10.1002/stem.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:869–881. doi: 10.1158/1078-0432.CCR-11-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer research. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA, et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012;31:4898–4911. doi: 10.1038/onc.2011.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Koppen A, Joles JA, van Balkom BW, Lim SK, de Kleijn D, Giles RH, et al. Human embryonic mesenchymal stem cell-derived conditioned medium rescues kidney function in rats with established chronic kidney disease. PLoS One. 2012;7:e38746. doi: 10.1371/journal.pone.0038746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu B, Liu Y, Yao H, Lu N, Li B, et al. The ovarian cancer-derived secretory/releasing proteome: a repertoire of tumor markers. Proteomics. 2012;12:1883–1891. doi: 10.1002/pmic.201100654. [DOI] [PubMed] [Google Scholar]
- Louie E, Nik S, Chen JS, Schmidt M, Song B, Pacson C, et al. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast cancer research: BCR. 2010;12:R94. doi: 10.1186/bcr2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Sebastiano V, Wu G, Arauzo-Bravo MJ, Sasse P, Gentile L, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Munoz Descalzo S, Rue P, Garcia-Ojalvo J, Arias AM. Correlations between the levels of oct4 and nanog as a signature for naive pluripotency in mouse embryonic stem cells. Stem cells. 2012;30:2683–2691. doi: 10.1002/stem.1230. [DOI] [PubMed] [Google Scholar]
- Daley GQ. Common themes of dedifferentiation in somatic cell reprogramming and cancer. Cold Spring Harbor symposia on quantitative biology. 2008;73:171–174. doi: 10.1101/sqb.2008.73.041. [DOI] [PubMed] [Google Scholar]
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer research. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010;29:2153–2159. doi: 10.1038/onc.2009.500. [DOI] [PubMed] [Google Scholar]
- Zhang X, Han B, Huang J, Zheng B, Geng Q, Aziz F, et al. Prognostic significance of OCT4 expression in adenocarcinoma of the lung. Japanese journal of clinical oncology. 2010;40:961–966. doi: 10.1093/jjco/hyq066. [DOI] [PubMed] [Google Scholar]
- Li Z, Rich JN. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Current topics in microbiology and immunology. 2010;345:21–30. doi: 10.1007/82_2010_75. [DOI] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Indraccolo S, Stievano L, Minuzzo S, Tosello V, Esposito G, Piovan E, et al. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4216–4221. doi: 10.1073/pnas.0506200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nature reviews Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadley KW, Hunn MK, Farrand KJ, Price KM, Grasso C, Miller RJ, et al. Side population is not necessary or sufficient for a cancer stem cell phenotype in glioblastoma multiforme. Stem cells. 2011;29:452–461. doi: 10.1002/stem.582. [DOI] [PubMed] [Google Scholar]
- Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2 and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.