

Abstract
Thyroid paraganglioma is an extremely rare tumor and frequently mistaken for other thyroid neoplasms. Increased awareness of its potential presentation in thyroid and its characteristic features is essential for avoiding diagnostic and therapeutic pitfalls. We describe here three additional cases of primary thyroid paraganglioma and analyze their clinical findings and pathological characteristics. Patients included two women and one man presenting with asymptomatic thyroid nodules. Radiological examinations were nonspecific and none had been diagnosed correctly before surgery. On intraoperative frozen section consultation they were all misdiagnosed as carcinomas, either primary or metastatic. However, the permanent sections showed features consistent with paraganglioma. Of note, two cases displayed extension into adjacent thyroid tissues, one of which exhibited increased mitotic activity, confluent tumor necrosis and vascular invasion. Immunohistochemically, the neoplastic chief cells expressed chromogranin, synaptophysin, neuron-specific enolase and CD56, whereas the sustentacular cells were highlighted by S100 protein. All three patients were well with normal hormone secretion, without local recurrence or distant metastasis at last follow-up (range 10–47 months). We further reviewed the literature to summarize the characteristics of this distinctive entity. Albeit being very rare, paraganglioma should be included in the differential diagnosis of hypervascular thyroidal neoplasms. Accurate diagnosis relies on the histopathogical findings and adjunctive immunohistochemcial studies. To date, all the reported cases have pursued a benign course. Although atypical features seem to have no association with clinical behavior, long time postoperative surveillance with biochemical screening of hormone secretion, cervical ultrasonography and whole-body CT scan is recommended.
Keywords: Thyroid gland, Paraganglioma, Immunohistochemistry, Differential diagnosis, Hormone secretion
Introduction
Paraganglioma is a neuroendocrine tumor that originates from the neural crest-derived paraganglia of the autonomic nervous system, which is located from the skull base to the pelvic floor [1]. Although the tumor can occur in a wide variety of sites, the majority arise in the head and neck region, the superior mediastinum and the abdomen [2]. Occasionally, it may involve unusual sites where paraganglia are not normally distributed. Paraganglioma manifesting as a primary thyroid lesion is extremely rare, comprising less than 0.1 % of all thyroid neoplasms [3]. Due to the unexpected occurrence and overlapping features, thyroid paraganglioma is frequently misdiagnosed as other common types of thyroid neoplasms, which may result in inappropriate treatment. Therefore, increased awareness of its potential occurrence in the thyroid gland and familiarity with its characteristic features are important for both clinicians and pathologists to avoid diagnostic and therapeutic pitfalls. We describe here three additional cases of primary thyroid paraganglioma to enhance the recognition and broaden the morphological spectrum.
Materials and Methods
Three cases of primary thyroid paraganglioma were collected from the archival files of the Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, China. The clinical data and pathological information were obtained from the clinicians and from the medical records. The study was approved by the institutional review board. All available H&E sections were reviewed by two senior pathologists. Each case was assessed for the growth pattern of tumor cells, cell morphology, nuclear atypia, mitotic activity, presence of confluent tumor necrosis, invasion of vessels and involvement of adjacent tissues.
The immunohistochemical study was performed on deparaffinized sections by the standard EnVision technique using a panel of antibodies, including pan-cytokeratin (AE1/AE3, dilution 1:100; DAKO, Glostrup, Denmark), epithelial membrane antigen (EMA) (E29, dilution 1:200; DAKO), thyroglobulin (DAK-TG6, dilution 1:500; DAKO), thyroid transcription factor-1 (TTF1) (SP14, dilution 1:100; DAKO), chromogranin A (DAK-A3, dilution 1:200; DAKO), synaptophysin (SP11, dilution 1:100; Zymed, California, USA), neuron-specific enolase (NSE) (E27, dilution 1:100; Zymed), CD56 (123C3, dilution 1:50; DAKO), S100 protein (polyclonal, dilution 1:300; DAKO), CD34 (QBEnd10, dilution 1:50; DAKO), calcitonin (polyclone, dilution 1:300; DAKO), carcinoembryonic antigen (CEA) (II-7, dilution 1:50; DAKO), parathormone (MRQ-31, dilution 1:50; Zymed) and Ki67 (MIB-1, dilution 1:200; DAKO). Heat-induced epitope retrieval was carried out by using a pressure cooker. For each antibody, appropriate positive and negative controls were employed throughout the study.
Results
Clinical Summary (Table 1)
Table 1.
The clinical and histopathologic findings of 3 cases with primary thyroid paraganglioma
| Case | Age (years)/sex | Laterality | Size (cm) | Ultrasound scan | Computed tomography (CT) scan | CDFI | Invasion | Lymphadenopathy | Metastasis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 30/F | Left | 3.0 | Inhomogeneous hypoechoic nodule | Mass of low density, with a suspicion of an adenoma | Normal | Yes | No | No |
| 2 | 47/M | Right | 1.8 | Solid nodule with distorted cord-like hypoechoic structure | Solitary mass with indistinct border | Normal | Yes | No | No |
| 3 | 37/F | Right | 3.0 | Hypoechoic nodule | ND | Rich blood flow in the nodule | No | No | No |
| Case | Intraoperative diagnosis | Surgery | Tumor capsule | Cellular atypia | Vascular invasion | Mitot(ic activity | Follow-up (mo) |
|---|---|---|---|---|---|---|---|
| 1 | Medullary thyroid carcinoma | Left thyroid lobectomy + subtotal right thyroid lobectomy | No | Foci of pleomorphism | Yes | Yes | 39 |
| 2 | Metastatic carcinoma | Right thyroid lobectomy | No | Foci of pleomorphism | No | Inconspicuous | 47 |
| 3 | Follicular carcinoma | Right thyroid lobectomy + isthmus and partial left thyroid lobectomy | Yes | Foci of pleomorphism | No | Inconspicuous | 10 |
ND not done, CDFI color doppler flow imaging
Patient 1
A 30-year-old female presented with an asymptomatic neck mass of 2 months’ duration. Physical examination revealed enlargement of the left thyroid lobe. Ultrasonography showed an inhomogeneous nodule of low echogenicity located in the upper portion, measuring 3.0 × 1.5 × 1.5 cm in size. On color doppler flow imaging (CDFI), remarkable hypervascular flow signal was detected within the nodule (Fig. 1). Emission computed tomography (ECT) revealed a cold nodule with increased uptake of iodine. Bilateral cervical computed tomography (CT) demonstrated a mass of low density with a suspicion of an adenoma (Fig. 2). Past histories were negative for thyroid or other endocrine disorders. The laboratory examinations, including the serum high sensitive-thyrotropin (h-TSH), total triiodothyronine (TT3), free triiodothyronine (FT3), total thyroxine (TT4) and free thyroxine (FT4) were all within normal limits. At operation, the mass was ill-circumscribed, showing involvement of the recurrent laryngeal nerve. The right lobe displayed features of nodular goiter. Left thyroid lobectomy and subtotal right thyroid lobectomy were performed. On intraoperative frozen section consultation, the mass of the left thyroid lobe was considered as a medullary thyroid carcinoma (MTC). The patient recovered with uneventful course and was well with no signs of recurrence at 39-months follow up.
Fig. 1.
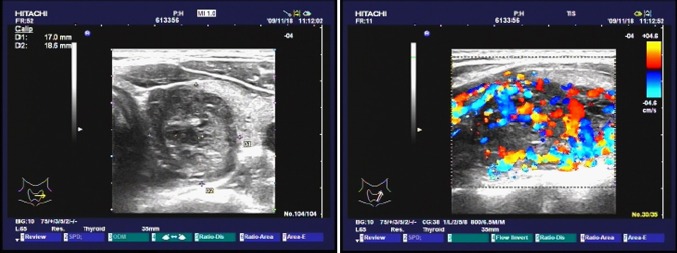
Sonographic image demonstrated an ovoid hypoechoic mass apparently in the left lobe of the thyroid gland. Color dopper evaluation revealed extreme hypervascularity of the mass
Fig. 2.
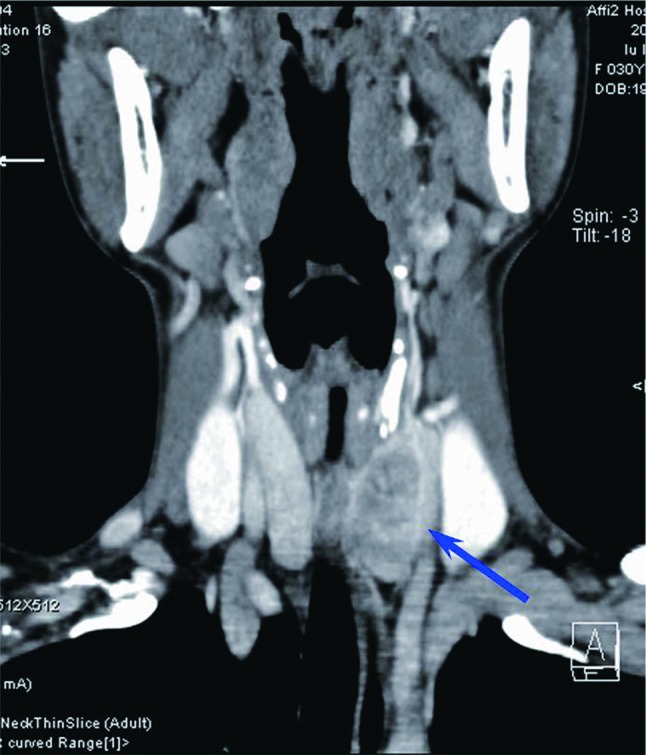
CT scan showed an oval-shaped noncalcifying mass in low density located in the left thyroid lobe (arrow)
Patient 2
A 47-year-old male was referred to the hospital because of a right thyroid nodule that incidentally discovered during a health check. The non-tender nodule was movable with swallowing. At ultrasound assessment, it was solid and hypervascular with distorted cord-like hypoechoic structure. No cervical lymphadenopathy was detected. CT scan revealed a solitary mass with indistinct border, measuring 1.8 × 1.2 × 1.2 cm in size. The patient had no past history of thyroid disease. Serum TSH, T3, T4, and calcitonin were all within normal limits. A diagnosis of thyroid carcinoma was supposed. Fine needle aspiration biopsy (FNAB) material was insufficient to make a diagnosis. The intraoperative frozen section was reported as thyroid carcinoma with a suspicion of being metastatic. The patient underwent right thyroid lobectomy. No adjunctive therapy was administrated after surgery. He was alive and free of disease at 47-months follow up.
Patient 3
A 37-year-old female was admitted to the hospital because of a mass situated in the right anterior neck with 4-year duration. The mass was neither painful nor tender, however, it showed enlargement in recent months. The patient had no past history of thyroid diseases or other endocrine disorders and the blood pressure remained normal. Physical examination revealed a solid mass in the right lobe of thyroid. Ultrasonography showed a hypoechoic nodule in the right lobe with rich intra-nodular blood flow. The left thyroid lobe displayed features suggestive of Hashimoto’s thyroiditis. Thyroid function testing and serum calcium were all within normal ranges. At operation, a well circumscribed mass was found in the upper portion of the right thyroid lobe, measuring 3.0 × 2.5 × 1.8 cm. Intraoperative pathologic consultation suggested a follicular carcinoma. Right thyroid lobectomy together with isthmus and partial left thyroid lobectomy was performed. The patient was well with no evidence of recurrent disease 10 months after surgery.
Pathological Findings
Grossly, all three tumors arose in the thyroid gland. They were ill demarcated in case 1 and case 2, but well circumscribed in case 3. On cut section, they were red-brown to tan-gray in color and soft to firm in consistency. Tumor size ranged from 1.8 to 3.0 cm (mean 2.6 cm).
Microscopically, the ill-demarcated tumor in case 1 and case 2 showed focal extension into the adjacent thyroid tissues (Fig. 3a). By comparison, the tumor in case 3 was well separated from the thyroid tissue by a thick fibrous capsule. Under high power magnification, all three tumors showed similar features. They were composed of epithelioid to polygonal cells with eosinophilic to finely granular cytoplasm, forming nesting, organoid or trabecular patterns of growth. A rich network of delicate vessels surrounded the clusters of tumor cells, creating the characteristic ‘Zellballen’ structure (Fig. 3b). Tumor cells were generally uniform in shape with relatively bland cytomorphology, albeit scattered cells with enlarged hyperchromatic nuclei were noted in focal areas. Whereas mitotic figures were rare or absent in case 2 and case 3, they were readily encountered in case 1 (Fig. 3c). Besides, coagulative necrosis and vascular invasion were also seen in case 1 (Fig. 3d, e). The stroma in case 1 and case 3 was unremarkable; however, it showed extensive hyalinization in case 2, creasing a sclerosing appearance (Fig. 3f). The thyroid tissues in the contralateral lobes of case 1 and case 3 showed nodular goiter and Hashimoto’s thyroiditis respectively.
Fig. 3.

Microscopic appearance of thyroid paraganglioma with hematoxylin and eosin staining. a The tumor was partially surrounded by a thin fibrous capsule. In some area, the tumor protruded into the thyroid tissue as a tongue. b The tumor was arranged in typical well-defined “Zellballen pattern”. c Scattered enlarged tumor cells with hyperchromatin were present. Mitotic figures can be seen in some foci in case 1 (arrow). d Coagulative necrosis can be seen in case 1. e Thyroid paraganglioma with vascular invasion. f The prominent hyalinized stroma overwhelmed the cellular component, showing a “sclerosing” appearance in some area
Immunohistochemical Findings
The polygonal chief cells showed strong positivity for chromogranin A, synaptophysin (Fig. 4a), NSE and CD56. They were negative for AE1/AE3, EMA, thyroglobulin, TTF1, calcitonin, CEA and parathormone. Ki-67 index was low with <1 % in case 2 and case 3, and showing about 3 % in case 1. In each case, the staining of S100 protein highlighted the presence of elongated sustentacular cells that located at the periphery of the tumor nests (Fig. 4b). The endothelial marker of CD34 outlined the rich vascular network.
Fig. 4.
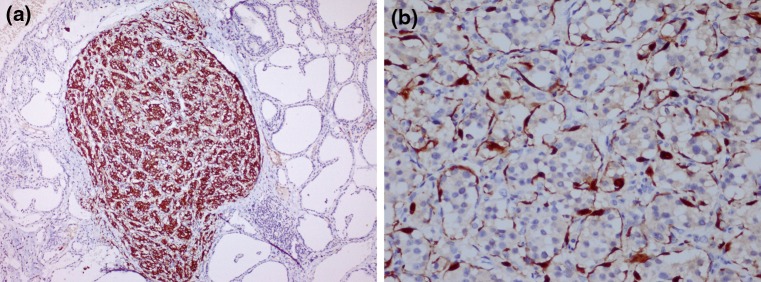
a Immunohistochemical analysis showed the neoplastic chief cells were strong positive for synaptophysin (×100). b S100 protein staining highlighted the spindle sustentacular cells at the periphery of the cellular nests (×100)
Discussion
Primary thyroid paraganglioma is an uncommon neuroendocrine tumor supposed to originate from the inferior laryngeal paraganglia. In our personal experience with more than 300 cases of extraadrenal paragangliomas, we have seen only 3 cases (<1 %) of paraganglioma originating within the thyroid gland. Possibly due to the rarity and unexpected occurrence in the thyroid gland, this tumor type has remained underrecognized. A comprehensive search of the PubMed showed that only 35 cases have been reported in the English literature [1–5, 7–29]. To better understand this rare entity, we undertook a brief review of the existing literature. For analysis, the current study was also included. Apart from one case arising in a 9-year-old girl which was described in Italian [30], all the other cases have been described to occur in adult patients with age ranging from 24 to 78 years (median, 48.5 years; mean, 48.2 years). There is a marked predilection for female patients, with a male to female ratio of 1:5.3. Clinically, most patients presented with an asymptomatic neck mass. In 2 cases, thyroid paraganglioma was associated with synchronous carotid body paraganglioma [3, 23, 29]. In another case, the tumor was accompanied with papillary thyroid carcinoma, parathyroid adenoma and bilateral carotid body paragangliomas [6].
To date, none of the reported cases had been diagnosed correctly by radiological examinations before surgery. Ultrasonography typically reveals a solid hypoechoic nodule in the thyroid gland with no significant difference from other relatively common types of thyroid neoplasms. By thyroid scintiscan, the tumor frequently appears as a ‘cold nodule’ [4], although a ‘hot nodule’ was reported in one case [10]. CT scan displays a thyroid mass of low density, usually being considered benign.
Fine needle aspiration biopsy is considered to be a highly sensitive and specific assessment in the work up of thyroid lesions. However, thyroid paraganglioma is rarely suspected by FNAB. Of 17 cases with FNAB, only 1 case was interpreted as a carotid body tumor, whereas 4 cases were mistaken for MTC, 3 cases with suspicion for malignancy, 2 cases as follicular carcinoma, 1 case as papillary thyroid carcinoma, 2 cases described as atypical cells not helpful for diagnosis, and the other 4 cases were not diagnostic due to excessive blood.
At surgery, except for one case in which the tumor was immediately adjacent to the gland [4], all the other tumors were described to develop in the thyroid parenchyma. Of these 37 cases, 13 cases were described to have a nodule located in the left lobe, 14 in the right lobe, 4 in the isthmus, and 1 involved both right and left lobes [5]. The exact location of the tumor in the remaining 5 cases remained unknown. The tumor was encapsulated in 22 cases, partially encapsulated in 6 cases, 2 of which showed focal capsular invasion. Eight cases were nonencapsulated and the remaining 2 cases were unknown. Most tumors were confined within the thyroid capsule. However, in 10 cases the tumors showed extrathyroid invasion, extending into the adjacent organs of trachea, subglottic larynx, esophagus and mediastinum, or involving the recurrent laryngeal nerve [2, 7, 9, 12, 13, 17–20]. In the latter instance, resection of the tumor was very difficult which might result in temporary vocal cord paralysis [2, 12].
Thyroid paraganglioma was frequently misdiagnosed on intraoperative consultation. Among the 22 cases with frozen sections, only 1 case was initially considered to be a paraganglioma [7]. The other cases were misinterpreted as a variety of thyroid diseases, including MTC (n = 10), follicular carcinoma (n = 3), and 1 case each as metastatic carcinoma, anaplastic carcinoma, malignant small cell tumor, Hürthle cell carcinoma, follicular pattern tumor, benign neoplasm, and being inconclusive or deferred for permanent sections.
Histologically, thyroid paragangliomas are quite similar to extra-adrenal paragangliomas at other sites. They are composed of two types of cells, namely polygonal chief cells and elongated sustentacular cells. Tumor cells formed distinctive nesting or organoid growth pattern (Zellballen), recapitulating the structure of normal paraganglion. Cytologically, 17 cases (45 %) showed bland appearance, 16 (42 %) exhibited focal nuclear pleomorphism, and 5 (13 %) displayed moderate to severe nuclear atypia. Mitotic figures were generally rare in most cases. Of note, 3 cases showed vascular invasion [7, 19], 1 case in the current study displayed infiltration of vascular spaces within the tumor capsule. Confluent tumor necrosis was only documented in 1 case [18]. In addition, prominent stromal sclerosis and hyalinization was described in 2 cases [12]. Despite the atypical features suggestive of malignancy, none of the reported cases showed aggressive behavior [16]. In the current study, 1 case displayed extension into adjacent thyroid tissues, increased mitotic activity, confluent tumor necrosis and vascular invasion. The patient was well and free of disease at 39-months follow up.
With regard to the malignancy in paraganglioma, the diagnostic criteria rely on the evidence of metastasis. No histopathological parameters have been shown to be predictive of clinical behavior. Although a few tumors displayed worrisome morphological features, malignant transformation has not been described in a thyroid paraganglioma thus far. Nevertheless, close follow up with radiological surveillance is warranted.
Due to architectural similarities, thyroid paraganglioma may be misdiagnosed as other types of thyroid neoplasms. In particular, the tumor is frequently misdiagnosed as MTC. Both tumors consist of nesting and organoid cell clusters. The coexpression of neuroendocrine differentiation makes the distinction further complicated. As the management of the disease and follow-up strategy vary greatly, it is essential to make a distinction between these two entities. In contrast to paraganglioma, the stroma of the MTC usually contains amyloid material which can be stained by Congo red. MTC originates in the parafollicular C cells of the thyroid gland. Therefore, MTC is characterized by positive staining with calcitonin. The positivity of epithelial markers and lack of S100 positive sustentacular cells also facilitate the differential diagnosis. Occasionally, rare cases of MTC may be negative for calcitonin [13], and paraganglioma may be positive for calcitonin or cytokeration [7], which might make the distinguishing problematic. Indeed, without the preoperative presentation of elevated calcitonin and CEA levels, the case termed as paraganglioma-like MTC by Bockhorn et al. [14] is almost indistinguishable from a paragangolioma. In such instances, the biochemical markers play an important role in the final diagnosis. A recent study based on complementary DNA arrays suggested combination of calcitonin gene-related protein/calcitonin with three proteins expressed in paraganglioma, namely nicotinamide adenine dinucleotide dehydrogenase 1 alpha subcomplex, 4-like 2, cytochrome c oxidase subunit IV isoform 2 and vesicular monoamine transporter 2, are considered to be useful in the differential diagnosis [15]. However, more cases are needed to reach a final conclusion.
Another thyroid neoplasm that may cause confusion with paraganlioma is hyalinizing trabecular tumor (HTT), also known as paraganglioma-like adenoma. HTT is a thyroid tumor of follicular cell origin with a trabecular pattern consisting of cells arranged around delicate vessels. There is marked intratrabecular hyalinization in the tumor. By immunohistochemistry, HTT is positive for thyroglobulin and TTF1 with unique membraneous expression of Ki-67. In addition, parathyroid adenoma can lie within the thyroid parenchyma, in which neoplastic cells are arranged in solid sheets, nodular, trabecular or follicular patterns, and the stroma is generally sparse but richly vascularized, which sometimes mimics paraganglioma. Moreover, both neoplasms show positive immunostaining for chromogranin A, making the differential diagnosis more difficult. Fortunately, immunostaining with PTH is of great help in differentiating parathyroid adenoma from paraganglioma, since the former one shows positive staining for PTH. Other lesions which might be less confused with paraganglioma include atypical follicular adenoma, Hürthle cell neoplasm, metastatic carcinoid tumors and rarely metastatic alveolar soft part sarcoma. The microscopic findings in combination with a panel of immnostains will allow distinction in most cases [8, 16].
Although Van Duinen et al. [31] demonstrated that up to 29 % of head and neck paraganglioma had detectable active catecholamine synthesis, the vast majority of paraganglioma in the head and neck region are non-secretory [32]. Cases of functional thyroid paraganglioma associated with multiple endocrine neoplasia had never been reported. In spite of this, postoperatively biochemical evaluation of catecholamines and metanephrines secretion are needed in patients with thyroid paragangliomas so as to exclude another possible primary functional tumor, as well as imaging evaluation for multicentric and metastatic disease. As the diagnosis of the current 3 cases was made post-operatively, we were not able to measure 24-h urinary catecholamines, vanilmandelic acid (VMA), and metanephrine before surgery. However, these hormone excretions were measured after surgery and all were within the normal range. CT scan of the whole-body did not demonstrate any evidence of multicentric tumor, local recurrence, distant metastasis or concurrent neoplasia.
Approximately 10 % of patients with paragangliomas are familial, which are a consequence of germline mutations in succinate dehydrogenase B (SDHB), C (SDHC), D(SDHD), or SDH assembly factor 2 (SDHAF2) [11]. In the current study, the absence of family history or other chromaffin tumors (e.g. pheochromocytoma) excluded the possibility of inherited disease. However, Fishbein et al. [32] demonstrated that all patients with head and neck paraganglioma should have genetic testing, since over half of these patients have an identified germline mutation in one of the known susceptibility genes. They also indicated that germline mutation in SDHB is the only currently reliable predictor of malignancy in pheochromocytomas and paragangliomas [32]. In addition, mutations in succinate dehydrogenase may activate an angiogenic pathway that ultimately leads to tumorigenesis [11]. Given these important implications, it is suggested that all patients with these rare tumors undergo possibly genetic testing. However, germline mutation testing in primary thyroid paraganglioma is seldom performed, with only one report by Zantour et al. [1] discovering a new germline mutation in SDHB gene. We also failed to perform genetic testing of germline mutations in these genes. Genetic testing and the signification of germline mutation of the succinate dehydrogenase genes in thyroid paraganglioma are worthy to be investigated further in larger series.
In summary, thyroid paraganglioma is a rare neuroendocrine tumor which may be confused with other common types of thyroid neoplasms. Preoperative examinations are usually not diagnostic. Immunohistochemistry plays an important role in the differential diagnosis. Although atypical features may be observed in some cases of thyroid paraganglioma, they seem to have no association with clinical behavior. Nevertheless, long-term follow-up should be scheduled and postoperative surveillance, including cervical ultrasonography, whole-body CT, and 24-h urine catecholamine levels, is recommended. Increasing understanding of the underlying somatic genetic and genomic changes is also needed to provide insights into the biology of this tumor, as well as new targets for novel therapeutics.
Acknowledgments
The authors are very grateful to Dr. Bai-Zhou Li (Department of Pathology, the 2nd Affiliated Hospital of Zhejiang University, Medical College, Hangzhou, China) for kindly providing the ultrasound image and CT scan figure of case 1.
Conflict of interest
None.
Contributor Information
Bao-Hua Yu, Email: yubh@shca.org.cn.
Wei-Qi Sheng, Email: shengweiqi2006@yahoo.com.cn.
Jian Wang, Phone: +86-21-64175590, FAX: 86-21-64170067, Email: jwang@shca.org.cn.
References
- 1.Zantour B, Guilhaume B, Tissier F, et al. A thyroid nodule revealing a paraganglioma in a patient with a new germline mutation in the succinate dehydrogenase B gene. Eur J Endocrinol. 2004;151:433–438. doi: 10.1530/eje.0.1510433. [DOI] [PubMed] [Google Scholar]
- 2.Ferri E, Manconi R, Armato E, Ianniello F. Primary paraganglioma of thyroid gland: a clinicopathologic and immunohistochemical study with review of the literature. Acta Otorhinolaryngol Ital. 2009;29:97–102. [PMC free article] [PubMed] [Google Scholar]
- 3.Haegert DG, Wang NS, Farrer PA, et al. Non-chromaffin paragangliomatosis manifesting as a cold thyroid nodule. Am J Clin Pathol. 1974;61:561–570. doi: 10.1093/ajcp/61.4.561. [DOI] [PubMed] [Google Scholar]
- 4.Schmit GD, Gorman B, van Heerden JA, Gharib H. Inferior laryngeal paraganglioma mimicking a primary thyroid tumor. Endocr Pract. 2006;12:432–435. doi: 10.4158/EP.12.4.432. [DOI] [PubMed] [Google Scholar]
- 5.Erem C, Kocak M, Nuhoglu İ, et al. Primary thyroid paraganglioma presenting with double thyroid nodule: a case report. Endocrine. 2009;36:368–371. doi: 10.1007/s12020-009-9238-3. [DOI] [PubMed] [Google Scholar]
- 6.Cayot F, Bastien H, Justrabo E, et al. Multiple paragangliomas of the neck localized in the thyroid region. Papillary thyroid cancer associated with parathyroid adenoma. Sem Hop. 1982;58:2004–2007. [PubMed] [Google Scholar]
- 7.Kronz JD, Argani P, Udelsman R, et al. Paraganglioma of the thyroid: two cases that clarify and expand the clinical spectrum. Head Neck. 2000;22:621–625. doi: 10.1002/1097-0347(200009)22:6<621::AID-HED12>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.Yano Y, Nagahama M, Sugino K, et al. Paraganglioma of the thyroid: report of a male case with ultrasonographic imagings, cytologic, histologic, and immunohistochemical features. Thyroid. 2007;17:575–578. doi: 10.1089/thy.2006.0284. [DOI] [PubMed] [Google Scholar]
- 9.Tiong HY, White SA, Roop L, et al. Paraganglioma-an unusual solitary nodule of the thyroid. Eur J Surg Oncol. 2000;26:720–721. doi: 10.1053/ejso.2000.0990. [DOI] [PubMed] [Google Scholar]
- 10.Foppiani L, Marugo A, Del Monte P, et al. Thyroid paraganglioma manifesting as hot toxic nodule. J Endocrinol Invest. 2005;28:479–480. doi: 10.1007/BF03347231. [DOI] [PubMed] [Google Scholar]
- 11.Phitayakorn R, Faquin W, Wei N, et al. Thyroid-associated paragangliomas. Thyroid. 2011;21:725–733. doi: 10.1089/thy.2010.0362. [DOI] [PubMed] [Google Scholar]
- 12.Evankovich J, Dedhia RC, Mastaki JM, et al. Primary sclerosing paraganglioma of the thyroid gland: a case report. Ann Otol Rhinol Laryngol. 2012;121:510–515. doi: 10.1177/000348941212100803. [DOI] [PubMed] [Google Scholar]
- 13.Corrado S, Montanini V, De Gaetani C, et al. Primary paraganglioma of the thyroid gland. J Endocrinol Invest. 2004;27:788–792. doi: 10.1007/BF03347525. [DOI] [PubMed] [Google Scholar]
- 14.Bockhorn M, Sheu SY, Frilling A, et al. Paraganglioma-like medullary thyroid carcinoma: a rare entity. Thyroid. 2005;15:1363–1367. doi: 10.1089/thy.2005.15.1363. [DOI] [PubMed] [Google Scholar]
- 15.Castelblanco E, Gallel P, Ros S, et al. Thyroid paraganglioma. Report of 3 cases and description of an immunohistochemical profile useful in the differential diagnosis with medullary thyroid carcinoma, based on complementary DNA array results. Hum Pathol. 2012;43:1103–1112. doi: 10.1016/j.humpath.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 16.Vodovnik A. Fine needle aspiration cytology of primary thyroid paraganglioma. Report of a case with cytologic, histologic and immunohistochemical features and differential diagnostic considerations. Acta Cytol. 2002;46:1133–1137. doi: 10.1159/000327120. [DOI] [PubMed] [Google Scholar]
- 17.Mitsudo SM, Grajower MD, Balbi H, Silver C. Malignant paraganglioma of the thyroid gland. Arch Pathol Lab Med. 1987;111:378–380. [PubMed] [Google Scholar]
- 18.de Vries EJ, Watson CG. Paraganglioma of the thyroid. Head Neck. 1989;11:462–465. doi: 10.1002/hed.2880110514. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong MJ, Chiosea SI, Carty SE, et al. Thyroid paragangliomas are locally aggressive. Thyroid. 2012;22:88–93. doi: 10.1089/thy.2011.0110. [DOI] [PubMed] [Google Scholar]
- 20.Brownlee RE, Shockley WW. Thyroid paraganglioma. Ann Otol Rhinol Laryngol. 1992;101:293–299. doi: 10.1177/000348949210100402. [DOI] [PubMed] [Google Scholar]
- 21.Banner B, Morecki R, Eviatar A. Chemodectoma in the mid-thyroid region. J Otolaryngol. 1979;8:271–273. [PubMed] [Google Scholar]
- 22.Buss DH, Marshall RB, Baird FG, Myers RT. Paraganglioma of the thyroid gland. Am J Surg Pathol. 1980;4:589–593. doi: 10.1097/00000478-198012000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Hughes JH, El-Mofty S, Sessions D, Liapis H. Primary intrathyroidal paraganglioma with metachronous carotid body tumor: report of a case and review of the literature. Pathol Res Pract. 1997; 193: 791–6; discussion 797–9. [DOI] [PubMed]
- 24.Napolitano L, Francomano F, Angelucci D, Napolitano AM. Thyroid paraganglioma: report of a case and review of the literature. Ann Ital Chir. 2000; 71:511–3; discussion 513–4. [PubMed]
- 25.Skiadas PK, Kakavoulis TN, Gikonti IJ. Normalisation of blood pressure and heart rate after excision of a thyroid paraganglioma. Eur J Surg. 2001;167:392–394. doi: 10.1080/110241501750215320. [DOI] [PubMed] [Google Scholar]
- 26.González Poggioli N, López Amado M, Pimentel MT. Paraganglioma of the thyroid gland: a rare entity. Endocr Pathol. 2009;20:62–65. doi: 10.1007/s12022-009-9066-2. [DOI] [PubMed] [Google Scholar]
- 27.Mun KS, Pailoor J, Chan KS, Pillay B. Extra-adrenal paraganglioma: presentation in three uncommon locations. Malays J Pathol. 2009;31:57–61. [PubMed] [Google Scholar]
- 28.Basu S, Viswanathan S. Primary paraganglioma of thyroid presenting as solitary thyroid mass. J Cancer Res Ther. 2011;7:385–387. doi: 10.4103/0973-1482.87028. [DOI] [PubMed] [Google Scholar]
- 29.LaGuette J, Matias-Guiu X, Rosai J. Thyroid paraganglioma: a clinicopathologic and immunohistochemical study of three cases. Am J Surg Pathol. 1997;21:748–753. doi: 10.1097/00000478-199707000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Massaioli N, Balbo G, Fausone G, Negro D. Endothyroid (non-chromaffin) branchiomeric paraganglioma. Description of a clinical case. Minerva Chir. 1979;34:867–874. [PubMed] [Google Scholar]
- 31.Van Duinen N, Steenvoorden D, Kema IP, Jansen JC, Vriends AHJT, Bayley JP, Smit JWA, Romijn JA, Corssmitt EPM. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. J Clin Endocrinol Metab. 2010;95:209–214. doi: 10.1210/jc.2009-1632. [DOI] [PubMed] [Google Scholar]
- 32.Fishbein L, Nathanson KL. Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet. 2012;205:1–11. doi: 10.1016/j.cancergen.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]