

Abstract
The aim of this study was to synthesize and evaluate novel biodegradable polyesters namely; poly(ethylene glycol)-Poly(glycerol adipate-co-ω-pentadecalactone), PEG-PGA-co-PDL-PEG, and poly(ethylene glycol methyl ether)-Poly(glycerol adipate-co-ω-pentadecalactone), PGA-co-PDL-PEGme as an alternative sustained release carrier for lung delivery compared with non-PEG containing polymer PGA-co-PDL. The co-polymers were synthesized through lipase catalysis ring opening polymerization reaction and characterized using GPC, FT-IR, 1H-NMR and surface contact angle. Furthermore, microparticles containing a model hydrophilic drug, sodium diclofenac, were prepared via spray drying from a modified single emulsion and characterized for their encapsulation efficiency, geometrical particle size, zeta potential, tapped density, primary aerodynamic diameter, amorphous nature, morphology, in vitro release and the aerosolization performance. Microparticles fabricated from mPEG-co-polymer can be targeted to the lung periphery with an optimum in vitro deposition. Furthermore, a significantly higher in vitro release (p > 0.05, ANOVA/Dunnett’s) was observed with the PEG and mPEG-co-polymers compared to PGA-co-PDL. In addition, these co-polymers have a good safety profile upon testing on human bronchial epithelial, 16HBE14o- cell lines.
Keywords: PGA-co-PDL, PEG-co-polymers, Microparticles, Lung delivery, Sodium diclofenac
1. Introduction
Polymeric microparticles produced using poly(ether anhydride) and poly(lactide-co-glycolide), PLGA, have been investigated to achieve sustained or controlled release of intended therapeutic agents to the lungs (Kawashima et al., 1999; Edwards et al., 1997; Fu et al., 2002; Learoyd et al., 2010; Fiegel et al., 2004). However, drug incorporation with these carriers is quite low which makes it difficult to encapsulate sufficient amounts of drugs for efficient therapy (Govender et al., 1999; Leo et al., 2004). Also PLGA based particles are typically associated with a burst release which is undesirable when used as controlled release formulations (Kim and Martin, 2006). Moreover, the triphasic release pattern is not desirable for lung delivery and the acidic environment evolved during the degradation of PLGA and PLA carriers affects the stability of loaded macromolecules (Van de Weert et al., 2000; Batycky et al., 1997). This is partly due to the fact that PLGA and PLA were not specifically designed for use in the lungs. Thus a new polymer which overcomes these problems is imperative in the formulation of carriers for pulmonary delivery.
Newly functionalized biodegradable co-polyesters, PGA-co-PDL, have proven to be a promising carrier for hydrophilic (Puri et al., 2008), hydrophobic (Thompson et al., 2007) drugs as well as macromolecules (Tawfeek et al., 2010). More recently, they were shown to be a useful carrier for lung delivery (Tawfeek et al., 2011). However, the higher hydrophobicity of this carrier can greatly affect their lung deposition through microparticles aggregation, hence limiting their deep deposition into the alveoli region as well as its in vitro release pattern. Additionally, the hydrophobic nature of the produced PGA-co-PDL microparticles, and their size make them undergo rapid phagocytosis by antigen-presenting cells (APC) like macrophages and dendritic cells (Ahsan et al., 2002; Foged et al., 2005; Thiele et al., 2001) which limits their residence in the lung airways. Additionally, the lower encapsulation efficiency which was recorded previously in the preparation of sodium fluorescein (a model hydrophilic drug) loaded PGA-co-PDL microparticles was an issue in drug delivery (Tawfeek et al., 2011). One of the most important approaches to obtain microparticles, becoming repellent to the adsorption of asponic proteins and resistant to unspecific phagocytosis, relies on polymer PEGylation (Donald et al., 2006). The purpose of these PEG chains is to create a barrier layer to block the adhesion of asponins present in blood serum, so that the particles can remain camouflaged or invisible to phagocyotic cells. PEG is an uncharged, hydrophilic polymer, which is soluble in water as well as in many organic solvents. Due to its low toxicity and immunogenicity, PEG is highly suitable for biomedical applications (Harris and Zalipsky, 2004; Bentley et al., 2005; Harris and Chess, 2003). PEGylation can also promote pulmonary delivery to achieve long-lasting peptide effects (Onoue et al., 2008; Youn et al., 2008). BioAir™ (Biosante Pharma, USA) comprises calcium phosphate nanoparticulates (CaP) of insulin, and CaP–PEG particles significantly reduced the elimination of insulin in rats following pulmonary administration. Consequently, the amount of bioavailable insulin from this pulmonary formulation was equivalent to or higher than that of insulin injected subcutaneously (Garcia-Contreras et al., 2003).
PEG was incorporated into the polymer backbone to reduce the interparticle adhesion forces and decrease the density of polymer aerosols, as well as to render the particles less susceptible to phagocytosis (Gref et al., 1994). Recently, PEGylated poly(lactic-co-glycolic acid) (PEG-PLGA) has been reported as carriers for both hydrophobic and hydrophilic drugs (Dorati et al., 2005; Cheng et al., 2007). Furthermore, PEG can increase the polymer hydrophilicity as well as its degradation profile which is suitable for lung delivery (Fu et al., 2002). Fiegel et al. (2004) studied the incorporation of different concentrations of PEG to poly(sebacic acid) to form poly(ether-anhydride) for dry powder aerosol inhalation. The authors found that addition of 10% PEG to the polymer backbone significantly enhanced the deposition in the lower stages of an in vitro lung model following aerosolization from the DPI. Moreover, it has the ability to avoid the phagocytic clearance and provide a controlled release of bovine serum albumin for prolonged pulmonary delivery. Nowadays, using functionalized PEG enables researchers to change the polymer properties by exploiting the functional groups for attaching, cross linking and conjugation with various chemical moieties. The terminal hydroxyl group of PEG can react either by chemical catalysis (Dong and Feng, 2004; Zhou et al., 2004), enzyme/lipase catalysis (Kumar et al., 2002; He et al., 2003) or through catalyst free synthesis to form a part of a polyester backbone (Lin et al., 2005).
The aim of this work was to study the modification of PGA-co-PDL backbone through incorporation of either poly(ethylene glycol), PEG, or poly(ethylene glycol methyl ether), mPEG, into the PGA-co-PDL backbone. The polymerization process was proceeded utilizing the terminal hydroxyl groups of PEG or mPEG with monomers namely, glycerol, divinyl adipate and pentadecalactone through lipase catalysis. Then, microparticles were prepared using either PEG-PGA-co-PDL-PEG or PGA-co-PDL-PEGme via spray drying the modified single emulsion (o/w) incorporating sodium diclofenac (SD) as a model hydrophilic drug. The prepared microparticles were evaluated for their spray drying yield, encapsulation efficiency, particle size, zeta potential, tapped density, theoretical primary aerodynamic diameter (dae), morphology and in vitro release in phosphate buffered saline of pH 7.4. Moreover, Percentage fine particle fraction, %FPF, fine particle dose, FPD, and mean median aerodynamic diameter, MMAD, for the dry powder inhalation microparticles, were calculated from impaction study using the Next Generation Impactor, NGI (flow rate 60 L/min, HandiHaler®). Cell viability study was also performed using human bronchial epithelium cell lines “16HBE14o” for PEG-PGA-co-PDL-PEG, PGA-co-PDL-PEGme in comparison with microparticles fabricated from PGA-co-PDL.
2. Materials and methods
2.1. Materials
Glycerol, ω-pentadecalactone, Poly(ethylene glycol) (PEG, Mw 4500Da), Poly(ethylene glycol methyl ether) (mPEG, Mw 2000Da), Novozyme 435 (a lipase from Candida antarctica immobilized on a microporous acrylic resin), poly(vinyl alcohol) (PVA, MW 9–10 K, 80%), l-leucine, RPMI-1640 medium with l-glutamine and sodium hydrogen carbonate (NaHCO3), (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), sodium diclofenac were all obtained from Sigma–Aldrich, UK. Dichloromethane (DCM) and Methanol were purchased from BDH laboratory supplies, UK. Tetrahydrofuran (THF), 75 cm2/tissue culture flask with vented cap (IWAKI brand), 24 tissue culture test plates, 96 well flat bottom plates, Antibiotic/Antimycotic Solution (100X) were purchased from Fisher Scientific, UK. Phosphate buffered saline tablets (PBS), pH 7.4, were obtained from Oxoid, UK. Divinyl adipate was obtained from Fluorochem, UK. Fetal Calf Serum heat inactivated was purchased from Biosera, UK. 16HBE14o- cells were produced by Dr. Dieter Gruenert from the California Pacific Medical Center, University of California San Francisco, USA.
2.2. Polymer synthesis
The PEG and mPEG co-polymers, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, were synthesized using the enzyme catalyzed polycondensation and ring opening co-polymerization reactions as described previously (Thompson et al., 2006) with slight modification. Briefly, PEG (4500Da) or mPEG (2000Da) was added to the reaction medium (Tetrahydrofuran,THF) incorporating monomers (glycerol, divinyl adipate and ɷ-pentadecalactone) prior to addition of novozyme 435 (1 g). The theoretical molar ratio of PEG or mPEG: glycerol: divinyl adipate: ɷ-pentadecalactone was 0.005:1:1:1. Approximately 0.0006 M, 2.6 g of PEG and 0.00025 M, 0.5 g of mPEG were added to 0.05 M of each monomer. The synthesized co-polymer was characterized by gel permeation chromatography, GPC (Viscotek TDA Model 300 using OmniSEC3 operating software), calibrated with polystyrene standards (polystyrene standards kit, Supelco, USA). FT-IR spectrum was obtained using a Perkin Elmer Spectrum BX spectrometer fitted with a PIKE technologies MIRacle sampling accessory and using Spectrum v5.0.1 for data processing, and 1H-NMR spectroscopy (Bruker AVANCE 300, Inverse probe with B-ACS 60, Auto sampler with gradient chemming) as described by Thompson et al. (2006). Additionally the degree of hydrophilicity was determined through measuring the surface contact angle (θ). A very small drop of water (6–10 μL) was dropped on the polymer film sample via pre-calibrated syringe needle. Photographs of dropping water droplet were taken until the droplet stabilized on the surface via a built-in digital camera at 16 ms frame interval for 60 ms using Attension (Theta Lite, CAM-101) by KSV instruments. Then, the surface contact angle (θ) was measured using the circular model of the drop profile fitting method applied to these images.
2.3. Microparticle preparation
Microparticles were prepared by spray drying directly from a modified single emulsion (o/w). Briefly, 50 mg SD was dissolved in the organic phase, composed of 2.5 ml methanol and 7.5 ml DCM (1:3 ratio), containing 450 mg polymer followed by addition to an aqueous phase, 150 ml distilled water containing 1% w/v PVA as an emulsifier and l-leucine (1.5% w/w of polymer weight) as a dispersing agent, under moderate stirring conditions (Silverson L5RT mixer, 2000 rpm at room temperature, 25 °C) to form the o/w single emulsion. The produced single emulsion was then spray dried using a mini-spray dryer (Büchi, B-290 Flawil, Switzerland). Spray drying conditions were performed as previously reported (Tawfeek et al., 2011). The spray drying parameters were set to preserve the outlet temperature in the range of 44–48 °C, as DSC analysis indicated a low melting point for PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme.
2.4. Microparticles characterization
2.4.1. Yield, drug loading and encapsulation efficiency
The yields of spray dried PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme microparticles were quantified as the percentage mass of anticipated total powder yields. The drug loading (DL) and percentage encapsulation efficiency (EE%) were calculated using Eqs. (1) and (2), respectively:
| (1) |
| (2) |
Briefly, 10 mg of the microparticles was weighed and solubilized in DCM/water mixture (2:1, 8:4 ml of DCM: water) to dissolve the polymer and extract the drug. The amount of water added has the ability to dissolve SD either encapsulated or present on the surface of microparticles as the solubility of SD in water reported to be 14.18 mg/ml (Maja et al., 2004). The two phases were then separated by centrifugation (5 min at 16200g, accuSpin Micro 17) and the aqueous layer analyzed for SD content spectrophotometrically at 275.5 nm (n = 3).
2.4.2. Particle size, zeta potential, powder density and primary aerodynamic diameter (dae)
Spray dried microparticles were sized using a Zetaplus, Brookhaven Instruments, UK. Microparticles suspension (100 μl) was diluted to 4 ml using double distilled water and the measurements recorded at 25 °C (n = 3). The zeta potential was determined using the same instrument with 50 μl of the suspension added to 2 ml of distilled water and the measurement was performed using a gold-plated zeta dip probe at 25 °C (n = 3). The poured density of spray-dried microparticle powders was determined by adding approximately 0.5 g of powder to a 10 ml graduated cylinder and recording the volume. The tapped density was determined by tapped density measurements on the same samples in a 10 ml graduated measuring cylinder until constant volume was obtained (Grenha et al., 2005) (n = 3). dae was calculated using data acquired from geometric particle size (d) and tapped density (p) according to (Eq. (3)) (Bosquillon et al., 2004).
| (3) |
2.4.3. Amorphous nature and water content
The degree of amorphous material from the spray dried formulations was performed using differential scanning calorimetry (DSC, Perkin Elmer Pyris 1). Briefly, 3–5 mg of sample was placed into a hermetically sealed and crimped pan. The samples were subjected to two scanning programs in the DSC using a heating rate of 20 °C/min purged with nitrogen at 20 ml/min as described previously by Thompson et al. (2007). The weight loss of the powders as a function of temperature was determined using a thermogravimetric analyzer (TGA 2050-Thermogravimetric analyzer, UK). Approximately 6–8 mg of each sample was weighed in a platinum pan and heated at the temperature range of 25–260 °C using a scanning rate of 10 °C/min purged under nitrogen at 20 ml/min (n = 3).
2.4.4. Particle morphology
Microparticles were visualized by scanning electron microscopy (FEI – Inspect S Low VAC Scanning Electron Microscope). Briefly, particles were mounted on aluminum stubs (pin stubs, 13 mm) layered with a sticky conductive carbon tab and air dried. An atomic layer of gold (10–15 nm) was deposited onto the particle containing stubs using an EmiTech K 550X Gold Sputter Coater, 25 mA for 3 min.
2.5. In-vitro release
Approximately, 10 mg of spray dried microparticles was added to 1.5 ml microtubes, containing 1 ml phosphate buffer saline pH 7.4 under sink conditions (Maja et al., 2004), and incubated at 37 °C on an orbital shaker (IKA KS 130) at 250 rpm (n = 3). The supernatants were collected to quantify the release of SD over 24 h by centrifugation (5 min at 13500 rpm, accuSpin Micro 17) and analyzed spectrophotometrically as above.
2.6. In-vitro aerosolization performance
Aerodynamic particle size distribution was determined using NGI. Microparticle samples (20 ± 0.4 mg) were manually loaded into hydroxypropyl methylcellulose capsules (HPMC size 3) and placed in a HandiHaler®. A pump (Copley Scientific, Nottingham, UK) was operated at a flow rate of 60 L/min for 4 s and the NGI plates were coated with 1% w/w glycerol/methanol solution. Following inhalation all parts of NGI were washed with DCM/water (2:1), and analyzed as above. The %FPF (defined as the mass of drug deposited (dae < 4.6 μm), expressed as a percentage of the emitted dose), MMAD (Feddah et al., 2000), and the FPD (expressed as the mass of drug deposited in the NGI (dae < 4.6 μm), were determined (n = 3).
2.7. Cell viability study
The toxicity profiles of the prepared formulations compared with non-PEGylated PGA-co-PDL microparticles (0–5 mg/ml) were evaluated over 24 h in the normal human bronchial epithelial (16HBE14o-) cell line (passage no. 22) as previously reported (Tawfeek et al., 2011). The relative cell viability (%) was calculated as presented in Eq (4):
| (4) |
where A is the absorbance of the test substance concentrations, S is the absorbance obtained for the (isopropanol) and CM is the absorbance obtained for untreated cells incubated with medium (control).
2.8. Statistical analysis
The formulations were compared with each other by means of a one-way ANOVA with Tukey’s and Dunnett multiple comparison tests assuming an equal variance or with Dunnett’s C test assuming non-equal variance. The statistical significance level was set at p ⩽ 0.05.
3. Results
3.1. Polymer synthesis and characterization
The chemical structure of PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme co-polymers is presented in (Fig. 1a and b, respectively).
Figure 1.

Chemical structure of (a) PEG-PGA-co-PDL-PEG & (b) PGA-co-PDL-PEGme.
The random nature of the co-polymers was confirmed from the integration pattern of peaks obtained from 1H-NMR spectra. PEG-PGA-co-PDL-PEG; 1H-NMR (δH CDCl3, 300 MHz): 1.26 (s, 21.7H, H-g), 1.59 (m)-1.68 (m) (8.2H, H-e, e’, h), 2.26 (m)-2.38 (m) (6H, H-d, d’, i), 3.64 (s, 3.9H, H-j, k, l), 4.03 (m)-4.17 (m) (6H, H-a, b, c, f), 5.1 (s, H, H-m). PGA-co-PDL-PEGme; 1H-NMR (δH CDCl3, 300 MHz): 1.26 (s, 21.0H, H-g), 1.57 (m)-1.67 (m) (8.0H, H-e, e’, h), 2.26 (m)-2.39 (m) (6H, H-d, d’, i), 3,38 (s, 4H, H-l), 3.68 (s, 2.5H, H-j, k, l), 4.03 (m)-4.34 (m) (5H, H-a, b, c, f), 5.1 (s, H, H-m). In addition, the co-polymers had the following characteristic FT-IR bands at υ max: typical broad shallow –OH band appeared at 3447.0 cm−1, –CH2 groups of DVA, PDL, PEG and mPEG appeared at 2915.7 cm−1, –CH group of glycerol appeared at 2848.4 cm−1, –CH3 group of methyl ether symmetric bending vibration appeared at 1365.7 cm−1, the carbonyl group of DVA and lactone monomers appeared at 1730.7 cm−1, C–O group of lactone, glycerol and PEG appeared at 1417.0 and 1164.8 cm−1, respectively. Furthermore, data from GPC confirmed unimodal mass distribution with no peaks related to free PEG or mPEG. The calculated co-polymers’ Mws were found to be 21.0, 13.6 and 7.6 KDa corresponding to PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively as shown in Fig. 2.
Figure 2.

Gel permeation chromotrgrams for PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme.
The actual co-polymer ratios were found to be (1:1:1:0.01 and 1:1:1:0.017) for PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively with PEG and mPEG content 9.05% and 11.64%, calculated from the ratios of PEG, mPEG Mwt and the total co-polymer Mwt. In addition, the surface contact angle (θ) was found to be 70.02 ± 0.1, 48.17 ± 0.7 and 49.63 ± 1.3 for PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively.
3.2. Microparticle preparation and characterization
Spray drying from a modified single emulsion produced microparticles with a high yield of 70.23 ± 2.5% and 75.14 ± 3.6%, SD loading of 67.64 ± 3.6 and 66.84 ± 2.7 μg/mg particle and encapsulation efficiency 60.88 ± 3.3% and 60.16 ± 2.5% for PEG-PGA-co-PDL and PGA-co-PDL-PEGme, respectively (Table 1). In addition, all formulations had a geometrical particle size less than 4.0 μm suitable for targeting the respiratory bronchioles (Table 1). Moreover, the prepared microparticles had a higher negative surface charge indicating a greater degree of colloidal stability within the dispersion medium (Table 1). The tapped densities for all formulations ranged from 0.24 ± 0.2–0.26 ± 0.2 g/cm3, Table 1 and were used together with the geometrical particle size to calculate the theoretical aerodynamic diameter (dae). As shown in (Table 1), the calculated dae was between (1.17 ± 0.31 and 1.25 ± 0.42 μm). The water contents for all formulations were within the range of moisture contents of spray dried powders as reported previously (Ståhl et al., 2002; Chew et al., 2005a).
Table 1.
Physical characteristics of spray dried microparticles. The results are the mean ± S.D.
| Formulation | Yield (%) | Drug loading (μg/mg polymer) | EE (%) | Particle size (μm) | Zeta potential (mv) | Tapped density (g/cm3) | dae(μm) | Water content (%) |
|---|---|---|---|---|---|---|---|---|
| PGA-co-PDL | 68.88 ± 5.5 | 76.52 ± 2.6 | 68.87 ± 9.0 | 2.30 ± 0.18 | −32.28 ± 2.6 | 0.26 ± 0.2 | 1.17 ± 0.31 | 2.56 ± 0.2 |
| PEG-PGA-co-PDL | 70.23 ± 2.5 | 67.64 ± 3.6 | 60.88 ± 3.3 | 3.92 ± 0.12 | −29.98 ± 1.4 | 0.25 ± 0.3 | 1.96 ± 0.15 | 1.48 ± 0.4 |
| PGA-co-PDL-PEGme | 75.14 ± 3.6 | 66.84 ± 2.7 | 60.16 ± 2.5 | 2.57 ± 0.23 | −31.26 ± 1.8 | 0.24 ± 0.2 | 1.25 ± 0.42 | 3.56 ± 0.7 |
Yield, drug loading, encapsulation efficiency (n = 6). Particles size, zeta potential, tapped density, primary aerodynamic diameter and water content (n = 3).
Fig. 3 represents the DSC thermograms of PEG-PGA-co-PDL-PEG, PGA-co-PDL-PEGme co-polymers (control) and PEG-PGA-co-PDL-PEG, PGA-co-PDL-PEGme spray dried microparticles loaded with SD. The outlet temperature on the spray dryer was adjusted to be between 45 and 48 °C due to the low melting of the co-polymer as noted in the DSC thermograms (Fig. 3). It is worth noting that the spray drying process changed the thermal behaviors of the blank polymers, resulting in a lower onset of melting, 58.32 °C (PEG-PGA-co-PDL-PEG) and 56.45 °C (PGA-co-PDL-PEGme) compared to 60.53 °C and 60.12 °C for the co-polymers alone (control). In addition, the endothermic peaks became broader in shape with spray dried formulations coupled with a decrease in area under the endothermic curve and the heat of fusion (ΔH) 2.54 J/g and 1.88 J/g for PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme formulations, respectively (Fig. 3).
Figure 3.

DSC thermograms for (A) PEG-PGA-co-PDL-PEG, (B) PGA-co-PDL-PEGme, (C) PEG-PGA-co-PDL-PEG spray dried microparticles with 1.5% w/w l-leucine and (D) PGA-co-PDL-PEGme spray dried microparticles with 1.5% w/w l-leucine.
Scanning electron microscopy, SEM, showed spherical particles with wrinkled morphology for all the investigated spray dried microparticles (Fig. 4, a, b and c, PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively). Furthermore, the prepared microparticles appeared to be deformed with some aggregation and there is no observed difference between the produced spray dried microparticles using PGA-co-PDL or the PEG, mPEG co-polymers.
Figure 4.

Scan electron micrographs comparing (a) PGA-co-PDL (b) PEG-PGA-co-PDL-PEG and (c) PGA-co-PDL-PEGme spray dried microparticles. The scale bar represents 5 μm.
3.3. In vitro release
The release of SD from spray dried microparticles prepared from different co-polymers is presented in Fig. 5. Basically the release profile was characterized with a higher initial burst release phase, especially with PEG and mPEG co-polymers, followed by a sustained released phase till 24 h. A non-significant (p > 0.05, ANOVA/Tukey’s) higher release was found with PGA-co-PDL-PEGme compared to PEG-PGA-co-PDL-PEG 94.64 ± 1.2% and 87.83 ± 4.4%, respectively after 24 h release in PBS buffered saline at 37 °C. Furthermore, microparticles prepared using PGA-co-PDL showed a significant (p < 0.05, ANOVA/Dunnett’s C) lower burst and continuous release 29.80 ± 6.1% and 74.65 ± 6.3%, respectively compared to PEG-PGA-co-PDL-PEG 42.55 ± 2.7%, 87.83 ± 4.4% and PGA-co-PDL-PEGme 50.47 ± 1.7%, 94.64 ± 1.2%. Moreover, by analyzing the release data of SD, from different microparticle formulations, in different release kinetic models namely; Zero, First and Higuchi diffusion models, it was found that the best model fit was according to the Higuchi diffusion model with the highest R2 value of 0.931, 0.859 and 0.929 and the release rate constant, Kh = 7.302, 7.231 and 6.890 mg/cm2 min1/2 for PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively.
Figure 5.
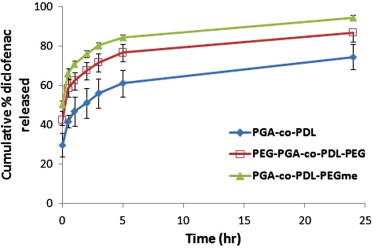
Cumulative in vitro release of sodium diclofenac from spray dried microparticles in PBS buffer at 37 °C. Data represents mean ± S.D. (n = 3).
3.4. In-vitro aerosolization performance
SD deposition data, obtained from spray dried formulations, indicated there was a difference in aerosolization performance between the different used co-polymers as shown in (Figs. 6 and 7). For example, PGA-co-PDL and PEG-PGA-co-PDL-PEG showed a significantly higher powder deposit in the capsule compared with PGA-co-PDL-PEGme (p < 0.05, ANOVA/Tukey’s, Fig. 6). At the same time, there were no significant differences between different carriers (co-polymers) in the amount of powder deposited in the inhaler, mouth piece and throat (p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 6).
Figure 6.

Comparison of sodium diclofenac deposition in capsule, inhaler, mouth piece and throat via different formulations. Data represents mean ± S.D. (n = 3). ∗p < 0.05 (capsule) PGA-co-PDL and PEG-PGA-co-PDL-PEG compared to PGA-co-PDL-PEGme (ANOVA/Tukey’s).
Figure 7.

Comparison of sodium diclofenac deposition in different stages of NGI, (Cut-off diameter, 7.8–0.13 μm) for different formulations. Data represents mean ± S.D. (n = 3). ∗p < 0.05 (stages 1, 2 and 4) PGA-co-PDL-PEGme compared to PGA-co-PDL and PEG-PGA-co-PDL-PEG (ANOVA/Tukey’s). ∗p < 0.05 (stages 6 and 7) PGA-co-PDL-PEGme comapred to PGA-co-PDL (ANOVA/Tukey’s, Dunnett C). ∗p < 0.05 (stage 8) PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme compared to PGA-co-PDL (ANOVA/Tukey’s).
Regarding the SD deposition in different stages of NGI, significantly higher amounts of PGA-co-PDL-PEGme microparticles were deposited in stages 1 and 2, (Cut-off diameter, 7.8 and 4.6 μm), compared to PGA-co-PDL and PEG-PGA-co-PDL-PEG fabricated microparticles (p < 0.05, ANOVA/Tukey’s, Fig. 7). At the same time, there was no significant difference between PGA-co-PDL and PEG-PGA-co-PDL-PEG formulations (p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 7). Furthermore, there were no significant differences between all investigated carriers on the amount of microparticles deposited on stage 3 and stage 5 (Cut-off diameter, 2.7, 0.96 μm, p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 7). However, on stage 4, (Cut-off diameter, 1.6 μm), significantly higher amounts of PGA-co-PDL-PEGme microparticles were deposited compared to PGA-co-PDL and PEG-PGA-co-PDL-PEG microparticles (p < 0.05, ANOVA/Tukey’s, Fig. 7).
In lower stages 6 and 7 (Cut-off diameter, 0.57 and 0.33 μm), it was found that, PGA-co-PDL-PEGme showed a significantly higher deposition compared to PGA-co-PDL microparticles (p < 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 7) with no significant difference between PGA-co-PDL-PEGme and PEG-PGA-co-PDL-PEG. Furthermore, there was no significant difference between PGA-co-PDL and PEG-PGA-co-PDL-PEG microparticles deposited on those stages (p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 7). PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme showed significantly higher amounts of microparticles deposited on MOC, stage 8 (Cut-off diameter, 0.13 μm), compared to PGA-co-PDL microparticles (p < 0.05, ANOVA/Tukey’s, Fig. 7). Moreover, there is no significant difference between PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme fabricated microparticles (p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 7).
Higher FPD and FPF (%) were obtained using PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme compared to PGA-co-PDL microparticles as shown in (Table 2) being only significant with PGA-co-PDL-PEGme compared to the other carriers (p < 0.05, ANOVA/Tukey’s). The MMAD obtained from cascade impaction studies ranged from (4.58 ± 0.7 to 2.92 ± 0.1 μm, Table 2) indicating particle aggregation (duplicate) compared to geometric particle size (Table 1). Furthermore, PGA-co-PDL-PEGme microparticles showed a significantly lower MMAD value compared to PGA-co-PDL microparticles (Table 2, p < 0.05, ANOVA/Tukey’s).
Table 2.
Percentage fine particle fraction (FPF%), fine particle dose (FPD, μg) and mean median aerodynamic diameter (MMAD, μm) for sodium diclofenac loaded microparticles formulations. The results are the mean ± S.D.
| Formulation | FPF% | FPD (μg) | MMAD (μm) |
|---|---|---|---|
| PGA-co-PDL | 30.73 ± 3.8 | 20.89 ± 3.2 | 4.58 ± 0.77 |
| PEG-PGA-co-PDL | 34.07 ± 5.4 | 25.81 ± 7.0 | 4.36 ± 0.75 |
| PGA-co-PDL-PEGme | ⁎44.06 ± 1.8 | ⁎⁎33.93 ± 0.58 | ⁎⁎⁎2.92 ± 0.17 |
p > 0.05 (FPF%) PGA-co-PDL-PEGme compared to PGA-co-PDL, PEG-PGA-co-PDL-PEG (ANOVA/Tukey’s).
p > 0.05 (FPD) PGA-co-PDL-PEGme compared to PGA-co-PDL, PEG-PGA-co-PDL-PEG (ANOVA/Tukey’s).
p > 0.05 (MMAD) PGA-co-PDL-PEGme compared to PGA-co-PDL (ANOVA/Tukey’s).
3.5. Cell viability study
The investigated co-polymer microparticles appear to be well tolerated by normal lung bronchial epithelial cells in vitro. Higher percentage cell viability was observed (88.14 ± 14.0%, 96.07 ± 5.2% and 89.27 ± 8.5% for PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme, respectively) even at higher microparticle concentrations of 5 mg/ml. Moreover, there was no significant difference between PGA-co-PDL and PEG or mPEG containing PGA-co-PDL polymer in the percentage of cell viability (p > 0.05, ANOVA/Tukey’s, Dunnett’s C, Fig. 8).
Figure 8.

Cell viability of human bronchial epithelium cell line (16HBE14o) measured by MTT cytotoxicity assay following 24 h exposure to different concentrations of PGA-co-PDL, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme microparticles suspension. Data represent mean ± S.D., (n = 6).
4. Discussion
Recently, Poly(glycerol adipate-co-ω-pentadecalactone), PGA-co-PDL, a biodegradable polyester, was studied as a promising new sustained release carrier for lung delivery (Tawfeek et al., 2011). However, its higher hydrophobicity is still an issue which limits its residence in the lung, release profile and deposition to alveoli region. In this study we are trying to solve these problems through modification of the PGA-co-PDL backbone via introduction of poly(ethylene glycol), PEG 4500Da, or poly(ethylene glycol methyl ether) mPEG 2000Da. This modification can be performed via lipase catalyzed ring opening polymerization and polycondensation reaction as the method described previously (Thompson et al., 2006). Lipase from Candida antarctica efficiently produced PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme co-polymers within high yield reaching 12–15 g.
The lower Mwt of PGA-co-PDL-PEGme, 7.5 KDa, could be attributed to the mode of attachment of mPEG into the PGA-co-PDL. mPEG is attached to the PGA-co-PDL on its side chains and after certain polymer growth, mPEG blocks the terminal positions and there is no possibility for further attachment of monomers hence, the chain stopped and the polymerization reaction was terminated. However, in case of PEG-PGA-co-PDL-PEG, PEG molecules can attach to PGA-co-PDL backbone in different positions. They can attach to DVA or PDL subunits, hence being incorporated inside the polymer chain and this cannot impede the chain progress which leads to a higher co-polymer Mwt (13.6 KDa) compared to PGA-co-PDL-PEGme (7.5 KDa). Furthermore, PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme had 9.05% and 11.5% PEG and mPEG contents which are suitable for drug delivery applications for PEG-containing polymers. In addition the higher mPEG content compared to PEG could be attributed to the higher feed ratio (1:1:1:0.017, 1:1:1:0.01 for PGA-co-PDL-PEGme and PEG-PGA-co-PDL-PEG, respectively). The incorporation of either PEG or mPEG leads to a significant reduction on the surface contact angle of both PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme compared to PGA-co-PDL and thus being reversed on the polymer hydrophilicity as well as its in vitro deposition.
The method of w/o/w multiple emulsion solvent evaporation is considered as the most widely used method for encapsulation of hydrophilic compounds. However, the diffusion and partitioning of such compounds to the external aqueous phase limit its encapsulation and loading (Uchida et al., 1997). In this study, we have tried to enhance the encapsulation of SD, a hydrophilic molecule, by making a co-solvent mixture that aids in solubilization of SD without the need to use an aqueous phase. Higher encapsulation efficiency was attained compared with what we had obtained with encapsulation of sodium fluorescein (18.94 ± 0.01–25.70 ± 0.09%) with PGA-co-PDL using spray drying from w/o/w multiple emulsion (Tawfeek et al., 2011). However, 100% encapsulation efficiency cannot be achieved due to some partitioning still occurring which could be attributed to the miscibility of methanol with water.
The negative surface charge demonstrated the anionic nature of the produced microparticles, which may be associated with incomplete removal of PVA present in the external aqueous phase of the single emulsion. It is accepted that spray drying products are mainly characterized by their amorphous nature or the disordered crystalline phase due to rapid drying of droplets (Corrigan, 1995). This behavior was demonstrated in our study by the broadening of the melting endothermic peaks for spray-dried formulations. Furthermore, the accumulation of l-leucine at the air–liquid interface and the surface of microparticles may contribute to enhance broadening of the endothermic melting peak compared with untreated polymers (control). Similar results were also found in our previous work using PGA-co-PDL and different amino-acids as dispersibility enhancers (Tawfeek et al., 2011). In addition, the shift of endothermic peak to a lower temperature and intensity (peak height) indicated distribution of the drug inside the PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme microparticles.
The geometric particle size, particle shape and morphology are known to affect the aerodynamic properties and pulmonary deposition (Bean et al., 1967). The dae calculated from tapped density indicated the spray-dried particles generated are suitable for targeting the alveolar region. However, in vitro aerosolization results suggested that the formulations did not aerosolize as individual particles, but rather as particle aggregates, as indicated when comparing geometric particle size with MMAD. This most likely occurred due to an incomplete powder de-aggregation as van der Waals forces between particles were not completely overcome upon inhalation. The wrinkled morphology of the produced microparticles has been observed by many researchers (Seville et al., 2007). The reason for that behavior is possibly due to the excessive built up of vapor pressure during solvent evaporation in the spray drying process especially with hydrophobic amino acids, such as l-leucine (Alder et al., 2000). It was worth noting that, leucine not only acts as a dispersibility enhancer but it also affects the in vitro release of SD from the prepared microparticles. Once again, this could be attributed to the surface activity of l-leucine coating the microparticles during the spray drying process. Hence reduced surface adsorption of the drug, decreased the continuous release especially marked with hydrophobic PGA-co-PDL. Similar results have been reported for l-leucine and sodium fluorescein loaded PGA-co-PDL microparticles (Tawfeek et al., 2011). Also this behavior was found with other surfactants, such as polysorbate 20 and sodium dodecyl sulfate, which reduced the surface accumulation of certain proteins in a concentration-dependant manner (Alder et al., 2000; Maa et al., 1998). However, the higher burst release of SD associated with PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme microparticles may be due to the migration of some drug crystals toward the microparticle surface by residual solvent during the spray drying process. Furthermore, the higher amounts of SD released from PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme compared to PGA-co-PDL could be attributed to the presence of either PEG or mPEG within the polymer backbone which affected the polymer matrix and hydrophobicity. Additionally, it can also influence its plasticity and porosity (Jiang et al., 2002; Chew et al., 2005b), resulting in more rapid entry of water into the microparticles, eventually accelerating the release of the drug (Castellanos et al., 2005). Also, we cannot neglect the effect of polymer Mwt, hence PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme have a lower Mwt (13.6, 7.6 KDa, respectively, compared to PGA-co-PDL, 21.0 KDa).
In this current investigation, the release of SD from different formulations followed Higuchi’s model, and mediated through the diffusion process with very little contribution to the degradation of the polymer. Whether drug release is governed primarily via diffusion or degradation depends on whether drug dissolution/diffusion or polymer degradation is faster (Hillery et al., 2001). Hence, to achieve zero-order release, it is recommended that the rate of polymer degradation controls the rate of release (Göpferich, 1996; Edlund and Albertsson 2002). Degradable microparticles do not give zero order release because a constant surface area for drug release cannot be maintained due to the degradation of the rate controlling membrane. It should not be possible to get zero-order release from degradable microparticles unless they have surface-only erosion because, in order for the drug path length to stay constant, zero order release requires the microparticles to degrade from the outside (Tice and Cowsar, 1984). This means that, in order to be released, the drug at the center of the microparticles should have the same distance to travel as the drug near the surface, assuming that the drug is homogeneously dispersed within the matrix (Leach, 1999). Additionally the incomplete release of SD from the prepared microparticles may be associated with the slow hydrolyzation of the ester linkages in the polymer backbone (Thompson et al., 2008) especially with PGA-co-PDL microparticles having a very stable ester linkage between monomers. So, further investigations are required to optimize the release profile by making a di-or tri-block PEGylated co-polymer or by attaching the PEG molecules to the hydroxyl group of glycerol subunit.
Recently, the addition of various dispersing agents such as, l-arginine and l-leucine amino acids, as potential dispersibility enhancers to improve the aerosol performance was investigated (Li et al., 1996, 2005; Seville et al., 2007; Tawfeek et al., 2011). Also l-leucine was found to be superior to any other investigated amino acids in enhancing the aerosolization performance of PGA-co-PDL loaded sodium fluorescein microparticles (Tawfeek et al., 2011). Thus all formulations were prepared utilizing l-leucine (1.5% w/w of polymer weight). Moreover, higher amounts of powder deposited in the throat, this possibly attributed to the aerosolization conditions (flow rate 60 L/min, HandiHaler®) that cannot overcome the Van der Waal forces between particles. Also, some aggregation of microparticles, especially those prepared using PGA-co-PDL and PEG-PGA-co-PDL-PEG, was noticed and demonstrated through the lower values of FPD and FPF% (Table 2). The higher throat deposition could also be attributed to the higher strength of Van der Waals forces. These forces are directly proportional to the contact surface area of a particle; hence, an increase in Van der Waals forces strength is observed with small particle size (Table 2) due to their larger surface area. Furthermore, all the investigated carriers show a higher deposition on different stages of NGI as seen in Fig. 7 due to the coating effect of l-leucine together with lower tapped density and water content values. In addition to the enhanced physical stability of microparticles presented in relatively higher zeta potential values (Table 1), all of these factors can improve the aerosolization performance. Moreover, PGA-co-PDL-PEGme spray dried microparticles would be expected to perform well during inhalation and to deliver a large proportion of the dose to the central and even peripheral regions of the lungs due to the higher amounts deposited on lower stages of NGI, 6–8 (Cut off diameter, 0.57–0.13 μm). The lower value of MMAD (2.92 ± 0.17 μm), higher FPD (33.39 ± 0.58) and FPF% (44.06 ± 0.17) might be responsible for this optimum deposition. Furthermore, the enhanced hydrophilic characters of PEG and mPEG containing polymers compared to the hydrophobic PGA-co-PDL generated particles with a higher deposition in lower stages of NGI, hence, they can be used as successful carriers for lung periphery. However, the safety of the carrier used for pulmonary drug delivery is an important issue. Normal bronchial epithelial cells (16HBE14o-) were chosen in accordance with the aerosolization and particle size distribution (MMAD) results for the particles generated (Canal-Raffin et al., 2007). The higher% cell viability and good cells tolerability observed from different carriers even at higher microparticle concentration (5 mg/ml) provide an indication about the feasibility of using PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme as alternative safe sustained release carriers for pulmonary drug delivery. This higher microparticle concentration (5 mg/ml) cannot be found in the lung in our study. However, it was prepared and used to show that even higher concentrations of microparticles, generated using a different technique or more advanced inhaler, reached into the lung still safe and tolerated.
5. Conclusion
The present study suggests that PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme could be considered as alternative novel biodegradable sustained release carriers for lung delivery. They showed an enhanced encapsulation efficiency and lower hydrophobicity. Furthermore, they have the ability to enhance the release of SD from the prepared microparticles compared to PGA-co-PDL microparticles. Utilizing lipase enzyme from Candida antarctica successfully produced the PEG and mPEG containing polymers within higher yields and lower surface contact angle (θ) compared to PGA-co-PDL hence, an optimum deposition on lung periphery was obtained from NGI data. In addition, incorporation of l-leucine was found to enhance the aerosolization performance and decrease the continuous release of SD from different used carriers. PGA-co-PDL-PEGme has the ability to deliver higher amounts of SD to the central and peripheral regions of the lungs. Furthermore, cell viability study revealed the safety of PEG-PGA-co-PDL-PEG and PGA-co-PDL-PEGme spray dried microparticles. Future studies will be conducted to determine if the polymers elicit an immune response and the drug transport through different lung cell lines e.g., calu-3 cell lines. In addition, we will investigate the possibility of making nanoparticles to enhance the aerosolization performance and interaction with the lung cells.
Acknowledgments
The author is highly grateful to Dr. Gillian Hutcheon and Dr. Imran Saleem, School of Pharmacy and Bimolecular Science, Liverpool John Moore University, Liverpool, UK., for their guidance and expertise support. The author also acknowledges Dr. Andy Evans for his support in cell culture work.
Footnotes
Peer review under responsibility of King Saud University.

References
- Ahsan F., Rivas I.P., Khan M.A., Torres Suárez A.I. Targeting to macrophages: role of physicochemical properties of particulate carriers-liposomes and microspheres-on the phagocytosis by macrophages. J. Controlled Release. 2002;79:29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- Alder M., Unger M., Lee G. Surface composition of spray dried particles of bovine serum albumin/trehalose/surfactant. Pharm. Res. 2000;17:863–870. doi: 10.1023/a:1007568511399. [DOI] [PubMed] [Google Scholar]
- Batycky R.P., Hanes J., Langer R., Edwards D.A. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J. Pharm. Sci. 1997;86:1464–1477. doi: 10.1021/js9604117. [DOI] [PubMed] [Google Scholar]
- Bean H.S., Beckett A.H., Carless J.E. vol. 2. Academic Press; London: 1967. (Advances in Pharmaceutical Sciences). pp. 181–221. [Google Scholar]
- Bentley M.D., Bossard M.J., Burton K.W., Viegas T.X. Poly(ethylene) glycol conjugates of biopharmaceuticals in drug delivery. Mod. Biopharm. 2005;4:1393–1418. [Google Scholar]
- Bosquillon C., Préat V., Vanbever R. Pulmonary delivery of growth hormone using dry powders and visualization of its local fate in rats. J. Controlled Release. 2004;96:233–244. doi: 10.1016/j.jconrel.2004.01.027. [DOI] [PubMed] [Google Scholar]
- Canal-Raffin M., L’Azou B., Martinez B., Sellier E., Fawaz F., Robinson P. Physicochemical characteristics and bronchial epithelial cell cytotoxicity of Folpan 80 WG(R) and Myco 500(R), two commercial forms of folpet. Part Fibre Toxicol. 2007;8(4):1743–89778. doi: 10.1186/1743-8977-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos I.J., Flores G., Griebenow K. Effect of the molecular weight of poly(ethylene glycol) used as emulsifier on alpha-chymotrypsin stability upon encapsulation in PLGA microspheres. J. Pharm. Pharmacol. 2005;57:1261–1269. doi: 10.1211/jpp.57.10.0004. [DOI] [PubMed] [Google Scholar]
- Cheng J., Teply B.A., Sherifi I., Sung J., Luther G., Gu F.X., Levy-Nissenbaum E., Radovic-Moreno A.F., Langer R., Farokhzad O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew N.Y.K., Shekunov B.Y., Tong H.H.Y., Chow A.H.L., Savage C., Wu J., Chan H.K. Effect of amino acids on the dispersion of disodium cromoglycate powders. J. Pharm. Sci. 2005;94:2289–2300. doi: 10.1002/jps.20426. [DOI] [PubMed] [Google Scholar]
- Chew N.Y.K., Tang P., Chan H.K., Raper J.A. How much particle surface corrugation is sufficient to improve aerosol performance of powders? Pharm. Res. 2005;22:148–152. doi: 10.1007/s11095-004-9020-4. [DOI] [PubMed] [Google Scholar]
- Corrigan O.I. Thermal-analysis of spray-dried products. Thermochim. Acta. 1995;24:245–258. [Google Scholar]
- Donald E., Owens I.I.I., Nicholas A. Peppas Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Dong Y.C., Feng S.S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEGPLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25:2843–2849. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- Dorati R., Genta I., Montanari L., Cilurzo F., Buttafava A., Faucitano A., Conti B. The effect of [gamma]-irradiation on PLGA/PEG microspheres containing ovalbumin. J. Controlled. Release. 2005;107:78–90. doi: 10.1016/j.jconrel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Edlund U., Albertsson A.C. Degradable polymer microspheres for controlled drug delivery. Adv. Polym. Sci. 2002;157:67–112. [Google Scholar]
- Edwards D.A., Hanes J., Caponetti G., Hrkach J., BenJebria A., Eskew M.L. Large porous particles for pulmonary drug delivery. Science. 1997;276:1868–1871. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- Feddah M.R., Brown K.F., Gipps E.M., Davies N.M. In-vitro characterisation of metered dose inhaler versus dry powder inhaler glucocorticoid products: influence of inspiratory flow rates. J. Pharm. Pharm. Sci. 2000;3:317–324. [PubMed] [Google Scholar]
- Fiegel J., Fu H., Hanes J. Poly(ether-anhydride) dry powder aerosols for sustained drug delivery in the lungs. J. Controlled Release. 2004;96:411–423. doi: 10.1016/j.jconrel.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Foged C., Brodin B., Frokjaer S., Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Fu J., Fiegel J., Krauland E., Hanes J. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials. 2002;23:4425–4433. doi: 10.1016/s0142-9612(02)00182-5. [DOI] [PubMed] [Google Scholar]
- Garcia-Contreras L., Morcol T., Bell S.J.D., Hickey A.J. Evaluation of novel particles as pulmonary delivery systems for insulin in rats. AAPS Pharm. Sci. 2003;5:1–11. doi: 10.1208/ps050209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpferich A. Mechanisms of polymer degradation and erosion. Biomaterials. 1996;17:103–114. doi: 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- Govender T., Stolnik S., Garnett M.C., Illum L., Davis S.S. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J. Controlled Release. 1999;57:171–185. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Gref R., Minamitake Y., Peracchia M.T., Trubetskoy V., Torchilin V., Langer R. Biodegradable long-circulating polymeric nanosphers. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- Grenha A., Seijo B., Remuñán-López C. Microencapsulated chitosan nanoparticles for lung protein delivery. Eur. J. Pharm. Sci. 2005;25:427–437. doi: 10.1016/j.ejps.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Harris J.M., Chess R.B. Effect of PEGylation on pharmaceuticals. Nat. Rev. Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- Harris J.M., Zalipsky S. Polyethylene glycol: Chemistry and biological applications. ACS Symp. Ser. 2004;680:489. [Google Scholar]
- He F., Li S.M., Vert M., Zhuo R.X. Enzyme-catalyzed polymerization and degradationof copolymers prepared from epsilon-caprolactone and poly(ethylene glycol) Polymer. 2003;44:5145–5151. [Google Scholar]
- Hillery A.M., Lloyd A.W., Swarbrick J. Tylor & Francis; London: 2001. Drug Delivery and Targeting for Pharmaceutical and Pharmaceutical Scientists. [Google Scholar]
- Jiang G., Woo B.H., Kang F.R., Singh J., DeLuca P.P. Assessment of protein release kinetics, stability and protein polymer interaction of lysozyme encapsulated poly (d, l-lactide-co-glycolide) microspheres. J. Controlled Release. 2002;79:137–145. doi: 10.1016/s0168-3659(01)00533-8. [DOI] [PubMed] [Google Scholar]
- Kawashima Y., Yamamoto H., Takeuchi H., Fujioka S., Hino T. Pulmonary delivery of insulin with nebulized DL-lactide/glycolide copolymer (PLGA) nanospheres to prolong hypoglycemic effect. J. Controlled Release. 1999;62:279–287. doi: 10.1016/s0168-3659(99)00048-6. [DOI] [PubMed] [Google Scholar]
- Kim D.H., Martin D.C. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials. 2006;27:3031–3037. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Kumar R., Shakil N.A., Chen M.H., Parmar V.S., Saumelson L.A., Kumar J., Watterson A.C. Chemo-enzymatic synthesis and characterization of novel Functionalized amphiphilic polymers. J. Macromol. Sci.-Pure Appl. Chem. 2002;A39:1137–1149. [Google Scholar]
- Leach K.J. Cancer, drug delivery to treat-local and systemic. In: Mathiowitz E., editor. Encyclopaedia of Contolled Drug Delivery. John Wiley & Sons; New York: 1999. pp. 123–191. [Google Scholar]
- Learoyd T.P., Burrows J.L., French E., Seville P.C. Sustained delivery of salbutamol and beclometasone from spray-dried double emulsions. J. Microencapsulation. 2010;27:162–170. doi: 10.1080/02652040903052044. [DOI] [PubMed] [Google Scholar]
- Leo E., Brina B., Forni F., Angela M. In vitro evaluation of PLA nanoparticles containing a lipophilic drug in water-soluble or insoluble form. Int. J. Pharm. 2004;278:133–141. doi: 10.1016/j.ijpharm.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Li W.I., Perzl M., Heyder J., Langer R., Brain J.D., Englmeier K.H. Aerodynamics and aerosol particle deaggregation phenomena in model oral-pharyngeal cavities. J. Aerosol Sci. 1996;27:1269–1286. [Google Scholar]
- Li H.Y., Seville P.C., Williamson I.J., Birchall J.C. The use of amino acids to enhance the aerosolisation of spary dried powders for pulmonary gene therapy. J. Gene Med. 2005;7:1035–1043. doi: 10.1002/jgm.654. [DOI] [PubMed] [Google Scholar]
- Lin W.J., Wang C.L., Chen Y.C. Comparison of two pegylated copolymeric micelles and their potential as drug carriers. Drug Delivery. 2005;12:223–227. doi: 10.1080/10717540590952672. [DOI] [PubMed] [Google Scholar]
- Maa Y.F., Nguyen P.A.T., Hsu S.W. Spray-drying of air-liquid interface sensitive recombinant human growth hormone. J. Pharm. Sci. 1998;87:152–159. doi: 10.1021/js970308x. [DOI] [PubMed] [Google Scholar]
- Maja K., Marija M., Marjan V., Franc V. Study of the physicochemical parameters affecting the release of diclofenac sodium from lipohilic matrix tablets. Acta Chim. Slov. 2004;51:409–425. [Google Scholar]
- Onoue S., Hashimoto N., Yamada S. Dry powder inhalation systems for pulmonary delivery of therapeutic peptides and proteins. Expert Opin. Ther. Patents. 2008;18:429–442. [Google Scholar]
- Puri S., Kallinteri P., Higgins S., Hutcheon G.A., Garnett M.C. Drug incorporation and release of water soluble drugs from noval functionalized poly(glycerol adipate) nanoparticles. J. Controlled Release. 2008;125:59–67. doi: 10.1016/j.jconrel.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Seville P.C., Learoyd T.P., Li H.Y., Williamson I.J., Birchall J.C. Amino acid-modified spray-dried powders with enhanced aerosolisation properties for pulmonary drug delivery. Powder Tech. 2007;178:40–50. [Google Scholar]
- Ståhl K., Claesson M., Lilliehorn P., Lindén H., Bäckström K. The effect of process variables on the degradation and physical properties of spray dried insulin intended for inhalation. Int. J. Pharm. 2002;233:227–237. doi: 10.1016/s0378-5173(01)00945-0. [DOI] [PubMed] [Google Scholar]
- Tawfeek H.M., Khidr S.H., Samy E.M., Ahmed S.M., Shabir A., Mohammed A.R., Hutcheon G.A., Saleem I. Pulmonary delivery of alpha-chymotrypsin via novel polyester microparticles. J. Pharm. Pharmacol. 2010;62:1294–1295. [Google Scholar]
- Tawfeek H., Khidr S., Samy E., Ahmed S., Murphy M., Mohammed A., Shabir A., Hutcheon G., Saleem I. Poly(glycerol adipate-co-ω-pentadecalactone) spary-dried microparticles as sustained release carriers for pulmonary delivery. Pharm. Res. 2011;28:2086–2097. doi: 10.1007/s11095-011-0433-6. [DOI] [PubMed] [Google Scholar]
- Thiele L., Rothen-Rutishauser B., Jilek S., Wunderli-Allenspach H., Merkle H.P., Walter E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J. Controlled Release. 2001;76:59–71. doi: 10.1016/s0168-3659(01)00412-6. [DOI] [PubMed] [Google Scholar]
- Thompson C.J., Hansford D., Higgins S., Hutcheon G.A., Rostron C., Munday D.L. Enzymatic synthesis and evaluation of new novel omega-pentadecalactone polymers for the production of biodegradable microspheres. J. Microencapsulation. 2006;23:213–226. doi: 10.1080/02652040500444123. [DOI] [PubMed] [Google Scholar]
- Thompson C.J., Hansford D., Higgins S., Rostron C., Hutcheon G., Munday D.L. Evaluation of ibuprofen-loaded microspheres prepared from novel copolyesters. Int. J. Pharm. 2007;329:53–61. doi: 10.1016/j.ijpharm.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Thompson C.J., Hansford D., Munday D.L., Higgins S., Rostron C., Hutcheon G.A. Synthesis and evaluation of novel polyesteribuprofen conjugates for modified drug release. Drug Dev. Ind. Pharm. 2008;34:877–884. doi: 10.1080/03639040801929075. [DOI] [PubMed] [Google Scholar]
- Tice T.R., Cowsar D.R. Biodegradable contolled-release parenteral systems. Pharm. Tech. 1984;8:26–34. [Google Scholar]
- Uchida T., Yoshida K., Nakada Y., Nagareyan N., Konishi Y., Nakai A. Preparation and characterization of polylactic acid microspheres containing water-soluble anesthetics with small molecular weight. Chem. Pharm. Bull. Tokyo. 1997;45:513–517. doi: 10.1248/cpb.45.1539. [DOI] [PubMed] [Google Scholar]
- Van de Weert M., Hennink W.E., Jiskoot W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm. Res. 2000;17:1159–1167. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- Youn Y.S., Kwon M.J., Na D.H., Chae S.Y., Lee S., Lee K.C. Improved intrapulmonary delivery of site-specific PEGylated salmon calcitonin: optimization by PEG size selection. J. Controlled Release. 2008;125:68–75. doi: 10.1016/j.jconrel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhuo R.X., Liu Z.L. Synthesis and properties of novel biodegradable triblock copolymers of poly(5-methyl-5-methoxycarbonyl-1,3-dioxan-2-one) and poly(ethylene glycol) Polymer. 2004;45:5459–5463. [Google Scholar]