

Abstract
Our previous studies demonstrated that mutations in type IX and type XI collagens in mice caused osteoarthritis (OA)-like changes in knee and temporomandibular (TM) joints. We also found that the overexpression of matrix metalloproteinase 13 (Mmp-13) was probably due to the up-regulation of a collagen receptor, discoidin domain receptor 2 (Ddr2), which was responsible for knee cartilage degeneration in mutant mice. The objective of our study was to determine whether the expression of Mmp-3, Mmp-13 and Ddr2 was increased in OA-like TM joints in mutant mice using immunohistochemistry. We found that the staining for Ddr2, Mmp-13 and Mmp-derived type II collagen fragments in tissue sections from 6 month-old mice was increased in TM joints of the mutant mice. In contrast, we found no difference in the staining for Mmp-3 amongst the two mutant mice and their wild-type littermates. We conclude that, similar to previous observations in knee joints, the overexpression of Ddr2 and Mmp-13 may be responsible for the OA-like change in TM joints in mutant mice.
Keywords: temporomandibular joint, cartilage, Mmp-13, Ddr2, type II collagen
Introduction
Although the precise etiology of temporomandibular (TM) joint disorders is unknown, the osteoarthritic change in the articular cartilage of the condyle in TM joints is one of the common causes of the joint disorder (1). Thus, an understanding of the cellular and molecular basis of osteoarthritis (OA) in TM joints will provide novel insights into the pathogenesis of the TM joint disorder.
OA is characterized as a group of overlapping but distinct diseases (2). Epidemiological studies have indicated a number of risk factors for the initiation of OA, including local biomechanical, as well as systemic factors. Interestingly, regardless of the nature of the factor(s) that initiate the disease, the pathological change in OA is very distinct. That change is characterized by proteoglycan degradation at the early stage of the disease followed by type II collagen degradation as the disease progresses (3). The pathological similarity of the disease indicates that there may be a common chain of events in OA progression. It has been reported that the expression of MMP-3 and MMP-13 is increased in TM joints of OA patients (4,5). We have been investigating the pathogenesis of OA using two murine mutant models: type IX collagen-deficient (Col9a1-/-) and type XI collagen-haploinsufficient (heterozygous chondrodysplasia, cho/+) mice (6-8). Both mutant mouse strains developed normally, but exhibited age-dependent OA-like changes in knee and TM joints starting at 3 months of age. More importantly, we found that the protein expression of matrix metalloproteinase-13 (Mmp-13) and the amount of Mmp-derived type II collagen fragments were increased in knee joints of mutant mice at 6 months of age. A high level of expression of Mmp-13 was most likely due to the increased expression of a cell membrane tyrosine kinase receptor, discoidin domain receptor 2 (Ddr2), which bound preferentially to type II collagen (7,9). Based on our previous studies and those from others, we hypothesize that the exposure of chondrocytes to type II collagen fibrils is increased as a result of the proteoglycan degradation in OA articular cartilage. This causes the enhanced interaction of chondrocytes with type II collagen fibrils. The increased interaction can induce signaling through Ddr2, resulting in the increased expression of Mmp-13 and the irreversible destruction of the articular cartilage. To further support our hypothesis, we examined the expression of Ddr2, Mmp-3, Mmp-13 and the level of Mmp-derived type II collagen fragments in the articular cartilage of TM joints in Col9a1-/- and cho/+ mice.
Materials & Methods
Genotyping of Col9a1-/- and cho/+ mice
Genotyping procedures of Col9a1-/- and cho/+ mice have been described previously (8,10). Col9a1-/-, cho/+ and wild-type mice were separated and maintained under a 12-hour lighting schedule (12 hours with light and 12 hours without light).
Immunohistochemistry
TM joints were obtained from Col9a1-/-, cho/+ and wild-type mice (n=8 in each group) at the ages of 3 and 6 months. Each mouse head was cut in half along the mid-sagittal plane and the right half of the head was collected and embedded in paraffin (the left side was stored in a storage medium for a possible future use). Serial sections were cut at a thickness of 6 μm. Sections were taken immediately after the condyle was seen to be continuous with the ramus. This enabled us to obtain sections from the middle of the condyle. Approximately 100 sections represented an entire TM joint. Every twentieth section was used for immunohistochemistry. Five sections from each joint were mounted on one slide. There were 8 slides from each group at the age of 3 or 6 months. Thus, twenty-four slides from each age were used for each antibody (MMP-3, MMP-13, DDR2, and the polyclonal antibody against degraded type II collagen).
Paraffin sections were deparaffinized, quenched for endogenous peroxidase activity using a 1% hydrogen peroxide solution, and blocked for 60 minutes using a 1.5% goat serum for Ddr2 and type II collagen fragments and donkey serum for Mmp-3 and Mmp-13. Sections were incubated with primary antibodies at 4°C overnight. The first set of sections was incubated with a polyclonal antibody (1:300 dilution) against Ddr2 (Cat. No. sc-7554, Santa Cruz Biotechnology, CA). The second set of sections (next to the section for Ddr2) was incubated with a polyclonal antibody (1:200 dilution) against Mmp-3 (Cat. No. sc-6839, Santa Cruz Biotechnology, CA). The third set of sections (next to the section for Mmp-3) was incubated with a polyclonal antibody (1:200 dilution) against Mmp-13 (Cat. No. AB-8120, Chemicon, Temecula, CA). For immunostaining of Mmp-derived type II collagen fragments, a set of sections from those samples that showed positive Mmp-13 staining was collected and incubated with a polyclonal antibody (1:200 dilution) against the carboxyl terminus of the three-quarter fragment produced by Mmp (including Mmp-13)-induced cleavage of type II collagen (IBEX Tech., Inc, Montreal, Quebec, Canada). After the incubation, sections were washed with PBS three times followed by the treatment with a biotinylated secondary antibody (goat anti-rabbit IgG-B for Ddr2 and degraded type II collagen, Cat.No. sc-2040 and donkey anti-goat IgG-B for Mmp-3 and Mmp-13, Cat.No. sc-2042, Santa Cruz, Biotechnology, CA). Color was developed with a peroxidase substrate (VECTOR NovaRED Substrate, Cat. No. SK-4800, Laboratories, Burlingame, CA) after sections were treated with a mixture of avidin and biotinylated horseradish peroxidase (VECTASTAIN ABC Kit, Cat.No. PK-4000, Vector Laboratories, Burlingame, CA). Sections were counterstained with 0.2% Fast Green solution, except for sections of Mmp-derived type II collagen fragments that were stained with hematoxylin. Staining without a primary antibody was performed as a negative control.
Results
Immunohistochemistry
To determine whether the expression of Mmp-3 was increased in mutant mice, we examined the level of Mmp-3 in the articular cartilage of TM joints of Col9a1-/-, cho/+ and wild-type mice at the ages of 3 and 6 months. Results indicated positive staining for Mmp-3. There was no difference in immunostaining intensity of Mmp-3 in the articular cartilage of all mice at both ages, (see Figure 1).
Figure 1.

Immunostaining of Mmp-3 in TM joint articular cartilages of Col9a1-/- and cho/+ and wild-type mice at the ages of 3 and 6 months. The immunostaining for Mmp-3 (brown color staining) was seen in Col9a1-/-, cho/+ and wild-type mice at the both ages. Individual positive staining cells were seen in the enlarged inserts (2× enlargement). No differences in the immunostaining intensity between mutant and wild-type mice were observed. Bar = 50 μm.
The expression of Mmp-13 was increased in the articular cartilage of TM joints of both mutant mice at the age of 6 months, compared to that of wild-type mice (see Figure 2). Increased staining in the articular cartilage was local and variable among individual samples. No immunostaining for Mmp-13 was detected in the articular cartilage of TM joints of Col9a1-/-, cho/+ and wild-type mice at the age of 3 months.
Figure 2.

Immunostaining of Mmp-13 in TM joint articular cartilages of Col9a1-/-, cho/+ and wild-type mice at the ages of 3 and 6 months. The regional increase in immunostaining intensity of Mmp-13 (brown color staining) was seen in both mutant mice at the age of 6 months. Individual positive staining cells were seen in the enlarged inserts (2× enlargement). There was no detectable immunostaining of Mmp-13 in the TM articular cartilage of the mutant and wild-type mice at the age of 3 months. Bar = 50 μm.
To determine whether the activity of the Mmp-13 was increased, we examined the amount of type II collagen fragments cleaved by Mmps (including Mmp-13). We observed that more intensive staining for degraded type II collagen appeared in the superficial layer of the articular cartilage in TM joints of Col9a1-/- and cho/+ mice at the age of 6 months, compared to that of wild-type mice (see Figure 3). We did not observe immunostaining for type II collagen fragments in the articular cartilage of TM joints of Col9a1-/-, cho/+ and wild-type mice at the age of 3 months (data not shown).
Figure 3.
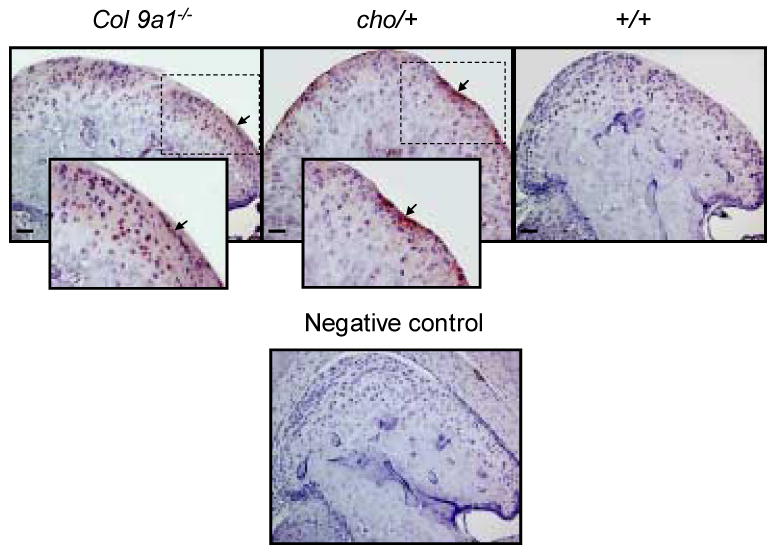
Immunostaining of Mmp-derived type II collagen fragments. More type II collagen fragments (brown color staining) were observed in the superficial layer region of the articular cartilage in mutant mice compared with wild-type mice at the age of 6 months. The positive staining for type II collagen fragments was shown in the enlarged inserts (2× enlargement). Bar = 50 μm.
Our previous studies indicated that the increased expression of Mmp-13 was associated with the elevated expression of Ddr2 in knee articular cartilage of Col9a1-/- and cho/+ mice at the age of 6 months. To determine whether the increased expression of Mmp-13 was also associated with a high-level expression of Ddr2 in TM joints of Col9a1-/- and cho/+ mice, we examined the expression of Ddr2. Results showed that immunostaining intensity for Ddr2 was also increased in the articular cartilage of TM joints in Col9a1-/- and cho/+ mice at the age of 6 months (see Figure 4). There was no positive staining observed in the articular cartilage of TM joints of the mutant and wild-type mice at the age of 3 months.
Figure 4.

Immunostaining of Ddr2 in TM joint articular cartilages of Col9a1-/-, cho/+ and wild-type mice at the ages of 3 and 6 months. The increase in immunostaining intensity of Ddr2 (brown color staining) was seen in both mutant mice at the age of 6 months. Individual positive staining cells were seen in the enlarged inserts (2× enlargement). There was no detectable immunostaining of Ddr2 in the TM articular cartilage of the mutant and wild-type mice at the age of 3 months. Bar = 50 μm.
Discussion
In previous studies, we found the increased staining for proteoglycans in TM joints of Col9a1-/- and cho/+ mice at 3 months of age, indicating that degeneration of articular cartilage started in the mutant mice. It is likely that the enhanced staining for proteoglycans resulted from increased expression levels of proteoglycans. It is believed that the over-production of the proteoglycan is one of the earliest signs of articular cartilage degeneration.
Numerous studies have reported that Mmp-3 and Mmp-13 may play an important role in cartilage degeneration at the early stages of OA (11-14). However, our results suggest that Mmp-3 and Mmp-13 may not be responsible for the cartilage degeneration of TM joints in both mutant mice at 3 months of age. Based on our findings, there is no difference in the protein expression of Mmp-3 in the articular cartilage of TM joints between mutant and wild-type mice. Furthermore, the protein expression of Mmp-13 is not detectable in the cartilage of mutant mice at this stage. The protein expression of Mmp-3 within cartilage of TM joints in normal mice at 3 months of age may reflect a function of Mmp-3 in the extracellular matrix remodeling in the cartilage during development, rather than a role in the cartilage degeneration in OA progression in TM joints. In addition, no increased expression of Mmp-3 in the cartilage of TM joints in both mutant mice is contrary to reports that the expression of MMP-3 is increased in human OA TM joints. This may be due to cartilage samples taken from patients with the advanced disease. The expression patterns of Mmp-3 and Mmp-13 in the articular cartilage of TM joints in both mutant mice at 3 months of age suggests that other molecules, such as aggrecanases or other Mmps, may play a significant role in the cartilage degeneration at this stage. Interestingly, an in vitro study shows that aggrecanase inhibitors can block aggrecan degradation in OA cartilage, whereas MMP inhibitors do not (15).
We also observed that the decreased staining for proteoglycans and increased staining for type II collagen fragments became evident in TM joints of both mutant mice at 6 months of age. However, we did not find increased expression of Mmp-3 in the mutant mice, suggesting that Mmp-3 may not be a critical factor involved in the proteoglycan degradation in OA progression, at least in the case of TM joints. A high level of the type II collagen fragments in TM joints of mutant mice likely resulted from increased expression and activity of Mmps, including Mmp-13. Interestingly, increased expression of Ddr2 was also found in TM joints of mutant mice at this stage. These findings are consistent with our previous observations in knee joints of both mutant mice. These results indicate that lack of type IX collagen or reduction of type XI collagen may affect multiple joints, including weight-bearing knee joints, as well as non-weight-bearing TM joints.
Although we do not know whether initial events of TM joint degeneration in mutant mice are similar, the increased expression of Ddr2 was observed in both mutant mice. This suggests that Ddr2 may be one of the common factors involved in OA progression, at least in those mutant mice. Based on results from this and previous studies, we speculate that the collagen network in the extracellular matrix of articular cartilage of a joint is crucial to maintain joint integrity. For example, if the collagen network in the matrix is disrupted, in mice lacking type IX collagen, or having the reduced level of type XI collagen, normal mechanical loads can activate chondrocytes. We observed signs of increased chondrocyte activities, such as increased levels of proteoglycans, in TM joints in both mutant mice at 3 months of age. Over time, the chondrocytes synthesize and release matrix-degrading enzymes that degrade proteoglycans. We also observed the focal disappearance of proteoglycans in the articular cartilage of mutant mice at 6 months of age. One of the consequences of proteoglycan degradation is to enhance the exposure of chondrocytes to type II collagen fibrils. Normally, little or no type II collagen is located close to chondrocyte surfaces (pericellularly) (16). The enhanced exposure of chondrocytes to type II collagen fibrils may result in increased signaling through Ddr2. The activation of Ddr2 induces the expression of Mmp-13 (which then cleaves type II collagen) as well as expression of DDR2 itself. A number of clinical studies of OA have shown that there is no type II collagen breakdown until most of proteoglycans are depleted from extracellular matrix (17,18). The resulting type II collagen fragments may in turn bind to integrins such as α2βı to activate signals that further increase the synthesis of Mmps (19). The result is a feedback amplification that enhances the damage to articular cartilage, eventually leading to its irreversible destruction.
Acknowledgments
This study was supported by funds from the National Institutes of Health, R01-AR051989 (to L. X.) and R01-AR36819 (to B. R. O.) and R01-AR051989 and P01-AR050245 (to Y. L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greene CS. The etiology of temporomandibular disorders: implications for treatment. J Orofacial Pain. 2001;15:93–105. [PubMed] [Google Scholar]
- 2.Felson DT, Lawrence RC, Dieppe PA, Hirsch R, Helmick CG, Jordan JM, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133:635–646. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 3.Hamerman D. The biology of osteoarthritis. N Engl J Med. 1989;320:1322–1330. doi: 10.1056/NEJM198905183202006. [DOI] [PubMed] [Google Scholar]
- 4.Tiilikainen P, Pirttiniemi P, Kainulainen T, Pernu H, Raustia A. MMP-3 and -8 expression is found in the condylar surface of temporomandibular joints with internal derangement. J Oral Pathol Med. 2005;34:39–45. doi: 10.1111/j.1600-0714.2004.00262.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee YJ, Lee EB, Kwon YE, Lee JJ, Cho WS, Kim HA, et al. Effect of estrogen on the expression of matrix metalloproteinase (MMP)-1, MMP-3, and MMP-13 and tissue inhibitor of metalloproternase-1 in osteoarthritis chondrocytes. Rheumatol Int. 2003;23:282–288. doi: 10.1007/s00296-003-0312-5. [DOI] [PubMed] [Google Scholar]
- 6.Xu L, Flahiff CM, Waldman BA, Wu D, Olsen BR, Setton LA, et al. Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of mice with the chondrodysplasia gene (cho) Arthritis Rheum. 2003;48:2509–2518. doi: 10.1002/art.11233. [DOI] [PubMed] [Google Scholar]
- 7.Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, et al. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem. 2005;280:548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- 8.Hu K, Xu L, Cao L, Flahiff CM, Setton LA, Youn I, et al. Pathogenesis of Osteoarthritis-like Changes in Joints of Type IX Collagen-Deficient Mice. Arthritis Rheum. 2006;54:2891–2900. doi: 10.1002/art.22040. [DOI] [PubMed] [Google Scholar]
- 9.Leitinger B, Steplewski A, Fertala A. The D2 period of collagen II contains a specific binding site for the human discoidin domain receptor, DDR2. J Mol Biol. 2004;344:993–1003. doi: 10.1016/j.jmb.2004.09.089. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Lacerda DL, Warman ML, Beier DR, Oxford JT, Morris NP, et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- 11.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–543. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- 12.Stove J, Gerlach C, Huch K, Gunther KP, Brenner R, Puhl W, et al. Gene expression of stromelysin and aggrecan in osteoarthritic cartilage. Pathobiology. 2001;69:333–338. doi: 10.1159/000064641. [DOI] [PubMed] [Google Scholar]
- 13.Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997;40:2065–2074. doi: 10.1002/art.1780401120. [DOI] [PubMed] [Google Scholar]
- 14.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J Clin Invest. 1996;97:2011–2019. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malfait AM, Liu RQ, Ijiri K, Komiya S, Tortorella MD. Inhibition of ADAM-TS4 and ADAM-TS5 prevents aggrecan degradation in osteoarthritic cartilage. J Biol Chem. 2002;277:22201–22208. doi: 10.1074/jbc.M200431200. [DOI] [PubMed] [Google Scholar]
- 16.Buckwalter JA, Hunziker EB. Articular cartilage morphology and biology. In: Archer C, Caterson B, Benjamin M, Ralphs J, editors. Biology of synovial joints. UK: CRC press; 1999. pp. 75–100. [Google Scholar]
- 17.Dijkgraaf LC, de Bont LG, Boering G, Liem RS. The structure, biochemistry, and metabolism of osteoarthritic cartilage: a review of the literature. J Oral Maxillofac Surg. 1995;53:1182–92. doi: 10.1016/0278-2391(95)90632-0. [DOI] [PubMed] [Google Scholar]
- 18.Mankin HJ, Brandt KD. Biochemistry and metabolism of articular cartilage in osteoarthritis. In: Moskowitx RW, Howell DS, Goldberg VM, et al., editors. Osteoarthritis: Diagnosis and Medical/Surgical Management. Philadelphia: Saunders; 1992. pp. 109–154. [Google Scholar]
- 19.Loeser RF, Sadiev S, Tan L, Goldring MB. Integrin expression by primary and immortalized human chondrocytes: evidence of a differential role for alpha1beta1 and alpha2beta1 integrins in mediating chondrocyte adhesion to types II and VI collagen. Osteoarthritis Cartilage. 2000;8:96–105. doi: 10.1053/joca.1999.0277. [DOI] [PubMed] [Google Scholar]