

Abstract
Background
Genome-wide association studies have identified novel type 2 diabetes loci, each of which has a modest impact on risk.
Objective
To examine the joint effects of several type 2 diabetes risk variants and their combination with conventional risk factors on type 2 diabetes risk in 2 prospective cohorts.
Design
Nested case–control study.
Setting
United States.
Participants
2809 patients with type 2 diabetes and 3501 healthy control participants of European ancestry from the Health Professionals Follow-up Study and Nurses’ Health Study.
Measurements
A genetic risk score (GRS) was calculated on the basis of 10 polymorphisms in 9 loci.
Results
After adjustment for age and body mass index (BMI), the odds ratio for type 2 diabetes with each point of GRS, corresponding to 1 risk allele, was 1.19 (95% CI, 1.14 to 1.24) and 1.16 (CI, 1.12 to 1.20) for men and women, respectively. Persons with a BMI of 30 kg/m2 or greater and a GRS in the highest quintile had an odds ratio of 14.06 (CI, 8.90 to 22.18) compared with persons with a BMI less than 25 kg/m2 and a GRS in the lowest quintile after adjustment for age and sex. Persons with a positive family history of diabetes and a GRS in the highest quintile had an odds ratio of 9.20 (CI, 5.50 to 15.40) compared with persons without a family history of diabetes and with a GRS in the lowest quintile. The addition of the GRS to a model of conventional risk factors improved discrimination by 1% (P < 0.001).
Limitation
The study focused only on persons of European ancestry; whether GRS is associated with type 2 diabetes in other ethnic groups remains unknown.
Conclusion
Although its discriminatory value is currently limited, a GRS that combines information from multiple genetic variants might be useful for identifying subgroups with a particularly high risk for type 2 diabetes.
Type 2 diabetes is a rapidly growing public health issue with a major impact on morbidity and premature mortality worldwide (1). The recent increase in the prevalence of this disease is largely attributable to environmental factors; however, convincing evidence shows that genetic factors also play an important role in causing type 2 diabetes (2, 3). Initial efforts to identify type 2 diabetes susceptibly genes favored genome-wide linkage and candidate gene association studies. These approaches identified common single nucleotide polymorphisms (SNPs) in PPARG, KCNJ11, and TCF7L2, which have been widely replicated in populations of various ethnicity (4 – 6). The advent of genome-wide association studies promises more efficient identification of susceptibility genes. Recent genome-wide association studies have discovered several new potential loci, including HHEX, CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8, and WFS1 (7–14). Variants in FTO and MC4R were also associated with type 2 diabetes, but the associations were entirely mediated by body mass index (BMI) (15, 16).
Given our growing knowledge of the genetic factors that predispose to type 2 diabetes and the decreasing costs of genotyping, genetic screening for persons at high risk for type 2 diabetes has received considerable attention. The risk attributable to an individual variant is modest and unlikely to have important clinical utility. However, a combination of the major genetic factors may contribute substantially to the disease risk and will be useful in characterizing high-risk populations.
Although the joint effects of type 2 diabetes loci identified from genome-wide association studies have been investigated previously (17–22), few studies have comprehensively investigated the impact of conventional risk factors, such as BMI, lifestyle, and family history, on these genetic effects. We sought to confirm associations reported by genome-wide association studies and to examine the joint genetic effects of established type 2 diabetes risk variants and their combination with conventional risk factors on type 2 diabetes risk in 2 prospective cohorts: the Health Professionals Follow-up Study (HPFS) and the Nurses’ Health Study (NHS).
METHODS
Study Design and Participants
The NHS was established in 1976 when 121 700 female registered nurses age 30 to 55 years and residing in 11 large U.S. states completed a mailed questionnaire on their medical history and lifestyle characteristics (23). Beginning in 1980, on a 2- to 4-year cycle, dietary information has been updated by using validated semiquantitative food-frequency questionnaires. Every 2 years, follow-up questionnaires have been sent to update information on potential risk factors and to identify newly diagnosed cases of type 2 diabetes and other diseases (24). The HPFS began in 1986 when 51 529 male U.S. health professionals age 40 to 75 answered a detailed questionnaire that included a comprehensive diet survey and items on lifestyle practice and medical history (25). The cohort is followed through biennial mailed questionnaire. Dietary information is updated every 4 years (26). Blood was collected from 32 826 NHS members between 1989 and 1990 and from 18 159 HPFS members between 1993 and 1999.
Participants for our study were selected from those who provided blood samples (27, 28). Demographic characteristics and health status of participants who provided blood samples were generally similar to those who did not. To minimize potential bias due to population stratification, we restricted our analyses to white persons of European ancestry. We defined diabetes cases as initially self-reported diabetes subsequently confirmed by a validated supplementary questionnaire (29, 30). Cases before 1998 were diagnosed by using criteria consistent with those proposed by the National Diabetes Data Group (31), which included 1 of the following: 1 or more classic symptoms (excessive thirst, polyuria, weight loss, hunger, pruritus, or coma) plus fasting plasma glucose level of 7.8 mmol/L (140 mg/dL) or greater, random plasma glucose level of 11.1 mmol/L (200 mg/dL) or greater, or plasma glucose level 2 hours after an oral glucose tolerance test of 11.1 mmol/L (200 mg/dL) or greater; at least 2 elevated plasma glucose levels (as described previously) on different occasions in the absence of symptoms; or treatment with hypoglycemic medication (insulin or oral hypoglycemic agent). We used the American Diabetes Association’s diagnostic criteria for the diagnosis of diabetes cases during the 1998 and 2002 cycles (32). These criteria were the same as those proposed by the National Diabetes Data Group except for the elevated fasting plasma glucose criterion, for which the cut-point was changed from 7.8 mmol/L (140 mg/dL) to 7.0 mmol/L (126 mg/dL). This study included 1297 male case patients and 1612 female case patients followed through 2002. We matched the case patients to 1338 and 2163 nondiabetic control male and female participants, respectively, on age, month and year of blood draw, and fasting status. All participants provided written informed consent, and the Human Research Committee at the Brigham and Women’s Hospital, Boston, approved the study.
Genotyping
A QIAmp blood kit (Qiagen, Chatsworth, California) was used to extract DNA from the buffy coat fraction of centrifuged blood. Samples of DNA were genotyped by using the OpenArray SNP Genotyping System (BioTrove, Woburn, Massachusetts) according to the manufacturer’s instructions; primers and probes are available on request. We selected 17 SNPs from 13 loci that had a nominal to strong association with type 2 diabetes in recently published genome-wide association studies: HHEX (rs7923837, rs1111875), CDKAL1 (rs7756992, rs7754840), IGF2BP2 (rs4402960), SLC30A8 (rs13266634), WFS1 (rs6446482, rs10010131), CDKN2A/B (rs564398, rs10811661), EXT2 (rs11037909), PKN2 (rs6698181), FLJ39370 (rs17044137), LOC387761 (rs7480010), TCF7L2 (rs12255372), PPARG (rs1801282), and KCNJ11 (rs5219). Genotyping success rates exceeded 90% for both case patients and control participants from each cohort. Duplicate samples (n = 484) were included and geno-typed with greater than 95% concordance. To improve the success rate, we chose duplicates with the most complete genotyping (<4 missing per sample) for study inclusion. With the exception of rs17044137 (FLJ39370; P < 0.001), we detected no significant departures from Hardy–Weinberg equilibrium among control participants after adjustment for multiple testing (P < 0.003: α = 0.05/17 SNPs).
Covariate Assessment
Information about medical history, anthropometric data, lifestyle factors, and family history of diabetes in first-degree relatives was derived from the baseline questionnaires (25, 33). Body mass index was calculated as weight (in kilograms) divided by the square of height (in meters). For men, physical activity was expressed as metabolic equivalent task hours of moderate to vigorous exercise per week and was calculated by using the reported time spent on various activities, weighting each activity by its intensity level. For women, physical activity was expressed as hours per week because metabolic equivalent task hours were not measured at baseline in the NHS. Self-administered questionnaires about body weight and physical activity have been validated as described elsewhere (34–36). Self-reported and measured weights were highly correlated (r = 0.97) (34), and the correlation between physical activity as reported in 1-week recalls and on questionnaires was 0.79 (35). Energy-adjusted intakes of dietary cereal fiber (grams per day), trans fatty acids (grams per day), and ratio of polyunsaturated–saturated fatty acids were calculated on the basis of food-frequency questionnaires administered in 1986 for men and 1980 for women. The reproducibility and validity of the food-frequency questionnaires has been published elsewhere (24).
Genetic Risk Score Computation
The Genetic Risk Score (GRS) was calculated on the basis of SNPs tagging reproducibly associated loci reaching genome-wide levels of significance (10, 37). These loci included HHEX (rs1111875), CDKAL1 (rs7756992), IGF2BP2 (rs4402960), SLC30A8 (rs13266634), WFS1 (rs10010131), CDKN2A/B (rs564398, rs10811661), TCF7L2 (rs12255372), PPARG (rs1801282), and KCNJ11 (rs5219). Among SNPs in linkage disequilibrium, only the SNP with the most significant main effect in our study was included in the score. A GRS that included the alternate SNP of those in linkage disequilibrium was evaluated, and similar results were observed (data not shown). Two methods were used to create the GRS: a simple count method (count GRS) and a weighted method (weighted GRS). Both methods assumed each SNP to be independently associated with risk. We assumed an additive genetic model for each SNP, applying a linear weighting of 0, 1, and 2 to genotypes containing 0, 1, or 2 risk alleles, respectively. This model performs well, even when the true genetic model is unknown or wrongly specified (38). The count method assumes that each SNP in the panel contributes equally to the risk for type 2 diabetes and was calculated by summing the values for each of the SNPs, producing a score out of 20 (the total number of risk alleles). For the weighted GRS, each SNP was weighted by β-coefficients obtained from a recent meta-analysis by Frayling (37): 0.1044 (WFS1, rs10010131), 0.1398 (HHEX, rs1111875), 0.1310 (CDKAL1, rs7756992), 0.1310 (IGF2BP2, rs4402960), 0.1398 (SLC30A8, rs13266634), 0.1823 (CDKN2A/B, rs10811661), 0.3148 (TCF7L2, rs12255372), 0.1310 (PPARG, rs1801282), and 0.1310 (KCNJ11, rs5219). For the second SNP (rs564398) in CDKN2A/B, which was not in linkage disequilibrium with rs10811661, the β-coefficient was obtained from a genome-wide association studies (10). The weighted GRS was calculated by multiplying each β-coefficient by the number of corresponding risk alleles (0, 1, or 2) and then summing the products. Because this produced a score out of 3.04 (twice the sum of the β-coefficients), all values were divided by 3.04 and multiplied by 20 to simplify interpretation and facilitate comparison with the count GRS. To simulate the likelihood of incomplete genotyping data in the clinic, only persons for whom data were missing on 5 or more of the 10 SNPs were excluded (41 case patients and 54 control participants), and scores for the remaining persons with missing genotypes (573 case patients and 684 control participants) were standardized to those for persons with complete data (count GRS = [total number risk alleles/number of non-missing genotypes × 2] × 20). In sensitivity analyses, we excluded all participants with missing genotypes or assigned missing genotypes the average genotype at that locus for case patients and control participants, separately. We observed similar results for both analyses.
Statistical Analysis
Statistical analyses were performed by using the SAS statistical package (version 9.1 for UNIX; SAS Institute, Cary, North Carolina), unless indicated otherwise. Chi-square tests and t tests were used for comparisons of means and proportions between case patients and control participants. To determine the effect of each variant on risk for type 2 diabetes, odds ratios and 95% CIs were estimated by conditional and unconditional logistic regression, after adjustment for age (unconditional only) and BMI (<23, 23 to 24.9, 25 to 29.9, 30 to 34.9, or ≥35 kg/m2). Codominant and additive genetic models were evaluated with reference to the “nonrisk” allele as defined elsewhere (7–13). Because conditional and unconditional analysis yielded similar results, we present results only from the latter to maximize the number of participants included. In multivariate analysis, we further adjusted for family history of diabetes (yes or no), smoking (never, past, or current), alcohol intake (0, 0.1 to 4.9, 5.0 to 9.9, 10.0 to 14.9, or ≥15 g/d), menopausal status (pre- or postmenopausal [hormone use: never, past, or current]; women only), and quintiles of physical activity—all established risk factors for diabetes. We also considered dietary factors, including ratio of polyunsaturated–saturated fatty acids and intake of cereal fiber and trans fat, because they have been previously associated with type 2 diabetes (39). Similar analyses were repeated after pooling individual-level data from the 2 cohorts and further adjusting for sex.
Power calculations were performed by using Quanto 1.2.3 (University of Southern California, Los Angeles, California; accessed at http://hydra.usc.edu/gxe). The combined data set provided 80% power to detect SNPs with risk ratios greater than 1.18, 1.12, and 1.11, given an α value of 0.05 and allele frequencies of 0.1, 0.3, and 0.5, respectively. Two-way gene–gene interactions were tested in each cohort by using the likelihood ratio test. We considered 2-sided P values less than 0.050 to be statistically significant. Adjustments for multiple comparison tests were not performed because SNPs were selected on the basis of an a priori hypothesis.
The GRS was modeled as a continuous variable or categorized into quintiles. Multiplicative interactions between conventional risk factors and the GRS for pooled data of men and women were tested by using the likelihood ratio test comparing a “main effects only” model with a model with the product interaction terms for GRS and conventional risk factors. We estimated 80% power to detect gene–environment interaction odds ratios of 1.20 or greater. Risk factors considered for interactions included BMI, family history of diabetes, smoking, alcohol intake, physical activity, ratio of polyunsaturated–saturated fatty acids, and cereal fiber and trans fat intake (39).
To measure the discriminative improvement attributable to the GRS, we plotted receiver-operating characteristic curves and calculated corresponding areas under the curve (AUCs) for a logistic regression model including conventional risk factors and a model including conventional risk factors and the GRS (40). The conventional model included age (quintiles), sex, family history of diabetes (yes or no), smoking (never, past, or current), alcohol intake (0, 0.1 to 4.9, 5.0 to 9.9, 10.0 to 14.9, or ≥15 g/d), BMI (quintiles), and physical activity (quintiles).
Role of the Funding Source
The National Institutes of Health funded this study. The funding source had no role in the design and conduct of the study; the collection, analysis, or interpretation of the data; or the decision to submit the manuscript for publication.
RESULTS
Case patients with type 2 diabetes had a significantly higher BMI, engaged in less physical activity, and were more likely to smoke and have a family history of diabetes than control participants, among both men and women (Table 1). Among men, case patients consumed a diet with a lower ratio of polyunsaturated–saturated fatty acids and lower fiber content than control participants. Among women, case patients consumed less alcohol and were more likely to be older and postmenopausal than control participants.
Table 1.
Risk Factor Characteristics of Case Patients and Control Participants at Baseline*
| Characteristic | Men† |
P Value‡ | Women† |
P Value‡ | ||
|---|---|---|---|---|---|---|
| Case Patients |
Control Participants |
Case Patients |
Control Participants |
|||
| Age, y | 55.7 (8.5) | 55.4 (8.6) | 0.36 | 44.1 (6.8) | 43.6 (6.8) | 0.030 |
| BMI, kg/m2 | 27.8 (4.1) | 25.1 (2.8) | <0.001 | 27.7 (5.3) | 23.9 (4.2) | <0.001 |
| Family history of diabetes, % | 36.0 | 15.3 | <0.001 | 52.3 | 22.6 | <0.001 |
| Current smoking, % | 11.7 | 7.2 | <0.001 | 29.5 | 21.7 | <0.001 |
| Alcohol intake, g/d | 11.2 (16.6) | 12.2 (15.5) | 0.110 | 4.1 (8.4) | 6.4 (9.7) | <0.001 |
| Physical activity§ | 14.6 (18.8) | 21.3 (27.4) | <0.001 | 3.6 (2.8) | 4.1 (2.9) | <0.001 |
| Current PMH users, % | – | – | – | 29.5 | 27.1 | 0.32 |
| Postmenopausal, % | – | – | – | 38.9 | 33.8 | 0.001 |
| Ratio of polyunsaturated–saturated fatty acids | 0.55 (0.20) | 0.58 (0.21) | 0.012 | 0.35 (0.12) | 0.35 (0.14) | 0.140 |
| Trans fat intake, g/d | 2.9 (1.1) | 2.8 (1.2) | 0.40 | 4.1 (1.3) | 4.0 (1.3) | 0.31 |
| Cereal fiber intake, g/d | 5.8 (3.7) | 6.3 (4.5) | 0.002 | 2.5 (1.5) | 2.6 (1.6) | 0.130 |
BMI = body mass index; PMH = postmenopausal hormone.
Values are means (SDs) unless otherwise indicated. Nutrient values represent the mean energy-adjusted intake.
Men: 1197 case patients and 1338 control participants; women: 1612 case patients and 2163 control participants.
Test of differences between case patients and control participants: chi-square for categorical variables and t tests for continuous variables.
Metabolic equivalent task hours per week for men and hours per week for women.
Table 2 shows allele and genotype frequencies of the 17 SNPs and results from the logistic regression analysis, assuming codominant or additive effect models. Consistent with previous reports on white persons of European ancestry, strong pairwise linkage disequilibrium was observed for SNPs in HHEX, CDKAL1, and WFS1 (r2 = 0.78 to 0.97), but not CDKN2A/B (r2 = 0.067 for men and 0.033 for women). After pooling data from the 2 cohorts, we confirmed significant associations between type 2 diabetes and SNPs for HHEX (rs1111875), CDKAL1 (rs7756992, rs7754840), IGF2BP2 (rs4402960), SLC30A8 (rs13266634), CDKN2A/B (rs564398, rs10811661), TCF7L2 (rs12255372), PPARG (rs1801282), and KCNJ11 (rs5219) loci (Figure 1). Further adjustment for conventional risk factors, including family history of diabetes, smoking, alcohol intake, menopausal status (women only), physical activity, or diet, did not change the results substantially (data not shown). No significant and consistent (that is, observed in both cohorts) 2-way gene–gene interactions were observed (data not shown).
Table 2.
Association of Candidate SNP Loci and Risk for Type 2 Diabetes Among Women and Men
| SNP | Loci | Major Allele (A) |
Minor Allele (a) |
Case Patients |
Control Participants |
Odds Ratio (95% CI)* |
P Value* | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | AA/Aa/aa | MAF | AA/Aa/aa | AA | Aa | aa | Additive | |||||
| Women | ||||||||||||
| rs7923837 | HHEX | G† | A | 0.37 | 588/716/205 | 0.37 | 779/1000/263 | 1.00 | 0.90 (0.72–1.12) | 0.96 (0.76–1.21) | 1.01 (0.90–1.12) | 0.85 |
| rs1111875 | HHEX | G† | A | 0.40 | 571/782/237 | 0.40 | 724/1040/324 | 1.00 | 1.05 (0.85–1.29) | 1.11 (0.89–1.38) | 1.06 (0.95–1.17) | 0.31 |
| rs7756992 | CDKAL1 | A | G† | 0.31 | 739/716/124 | 0.28 | 1074/842/149 | 1.00 | 1.19 (1.03–1.39) | 1.31 (0.99–1.73) | 1.17 (1.04–1.31) | 0.007 |
| rs7754840 | CDKAL1 | G | C† | 0.32 | 718/705/157 | 0.30 | 1004/869/193 | 1.00 | 1.09 (0.94–1.26) | 1.19 (0.93–1.53) | 1.09 (0.98–1.22) | 0.10 |
| rs4402960 | IGF2BP2 | G | T† | 0.36 | 590/711/168 | 0.31 | 933/919/175 | 1.00 | 1.24 (1.06–1.45) | 1.60 (1.24–2.07) | 1.26 (1.12–1.40) | <0.001 |
| rs13266634 | SLC30A8 | C† | T | 0.27 | 838/633/115 | 0.30 | 1001/905/184 | 1.00 | 1.15 (0.87–1.51) | 1.38 (1.05–1.82) | 1.19 (1.06–1.33) | 0.003 |
| rs6446482 | WFS1 | G† | C | 0.38 | 601/738/237 | 0.40 | 716/1041/308 | 1.00 | 0.92 (0.75–1.14) | 1.09 (0.87–1.36) | 1.07 (0.97–1.19) | 0.190 |
| rs10010131 | WFS1 | G† | A | 0.37 | 614/731/222 | 0.40 | 728/1053/299 | 1.00 | 0.96 (0.76–1.18) | 1.14 (0.91–1.42) | 1.10 (0.99–1.22) | 0.090 |
| rs564398 | CDKN2B | A† | G | 0.40 | 573/733/255 | 0.42 | 694/1025/359 | 1.00 | 1.05 (0.86–1.29) | 1.22 (0.99–1.51) | 1.12 (1.01–1.24) | 0.040 |
| rs10811661 | CDKN2B | T† | C | 0.15 | 1137/416/34 | 0.18 | 1408/600/70 | 1.00 | 1.54 (0.97–2.45) | 1.66 (1.06–2.60) | 1.14 (1.00–1.31) | 0.050 |
| rs11037909 | EXT2 | T† | C | 0.27 | 835/639/104 | 0.27 | 1097/815/138 | 1.00 | 1.10 (0.82–1.49) | 1.00 (0.74–1.34) | 0.95 (0.85–1.07) | 0.39 |
| rs6698181 | PKN2 | C | T† | 0.39 | 598/748/243 | 0.37 | 837/939/314 | 1.00 | 1.09 (0.93–1.27) | 1.11 (0.89–1.37) | 1.06 (0.96–1.17) | 0.27 |
| rs17044137 | FLJ39370 | T | A† | 0.24 | 881/592/85 | 0.24 | 1191/769/117 | 1.00 | 1.02 (0.88–1.18) | 0.97 (0.70–1.34) | 1.00 (0.89–1.13) | 0.92 |
| rs7480010 | LOC387761 | A | G† | 0.28 | 813/636/124 | 0.28 | 1088/811/187 | 1.00 | 1.03 (0.89–1.20) | 0.86 (0.66–1.12) | 0.97 (0.87–1.08) | 0.53 |
| rs12255372 | TCF7L2 | G | T† | 0.33 | 715/666/182 | 0.27 | 1117/827/151 | 1.00 | 1.29 (1.11–1.50) | 2.00 (1.55–2.59) | 1.36 (1.22–1.52) | <0.001 |
| rs1801282 | PPARG | C† | G | 0.11 | 1252/319/18 | 0.13 | 1617/464/43 | 1.00 | 1.75 (0.96–3.21) | 1.98 (1.10–3.57) | 1.19 (1.02–1.39) | 0.030 |
| rs5219 | KCNJ11 | G | A† | 0.38 | 584/721/227 | 0.35 | 858/934/237 | 1.00 | 1.16 (0.99–1.35) | 1.36 (1.09–1.71) | 1.17 (1.05–1.30) | 0.007 |
| Men | ||||||||||||
| rs7923837 | HHEX | G† | A | 0.36 | 472/499/155 | 0.38 | 493/606/190 | 1.00 | 1.06 (0.81–1.37) | 1.31 (1.01–1.71) | 1.17 (1.03–1.32) | 0.010 |
| rs1111875 | HHEX | G† | A | 0.38 | 452/542/175 | 0.40 | 467/631/217 | 1.00 | 1.15 (0.90–1.47) | 1.41 (1.09–1.82) | 1.20 (1.06–1.35) | 0.003 |
| rs7756992 | CDKAL1 | A | G† | 0.35 | 452/522/131 | 0.28 | 657/520/94 | 1.00 | 1.48 (1.23–1.77) | 2.01 (1.48–2.74) | 1.44 (1.26–1.65) | <0.001 |
| rs7754840 | CDKAL1 | G | C† | 0.35 | 489/514/143 | 0.30 | 626/565/108 | 1.00 | 1.11 (0.92–1.32) | 1.62 (1.21–2.18) | 1.21 (1.06–1.38) | 0.004 |
| rs4402960 | IGF2BP2 | G | T† | 0.36 | 451/529/137 | 0.31 | 602/542/127 | 1.00 | 1.38 (1.15–1.65) | 1.47 (1.10–1.96) | 1.26 (1.11–1.44) | <0.001 |
| rs13266634 | SLC30A8 | C† | T | 0.26 | 629/449/76 | 0.31 | 613/571/123 | 1.00 | 1.54 (1.10–2.15) | 2.06 (1.48–2.88) | 1.39 (1.22–1.60) | <0.001 |
| rs6446482 | WFS1 | G† | C | 0.38 | 448/525/179 | 0.40 | 479/621/209 | 1.00 | 0.96 (0.75–1.23) | 1.04 (0.81–1.34) | 1.03 (0.91–1.16) | 0.75 |
| rs10010131 | WFS1 | G† | A | 0.37 | 449/535/160 | 0.40 | 471/616/201 | 1.00 | 1.07 (0.83–1.38) | 1.13 (0.87–1.47) | 1.06 (0.94–1.20) | 0.43 |
| rs564398 | CDKN2B | A† | G | 0.40 | 423/546/185 | 0.41 | 455/634/211 | 1.00 | 0.98 (0.77–1.25) | 1.11 (0.86–1.43) | 1.07 (0.95–1.21) | 0.30 |
| rs10811661 | CDKN2B | T† | C | 0.15 | 826/304/24 | 0.17 | 882/393/29 | 1.00 | 0.87 (0.48–1.58) | 1.11 (0.62–1.99) | 1.21 (1.03–1.43) | 0.030 |
| rs11037909 | EXT2 | T† | C | 0.24 | 664/413/72 | 0.27 | 700/515/95 | 1.00 | 0.95 (0.67–1.36) | 1.12 (0.79–1.58) | 1.11 (0.97–1.27) | 0.140 |
| rs6698181 | PKN2 | C | T† | 0.36 | 478/505/160 | 0.36 | 539/594/179 | 1.00 | 0.97 (0.81–1.16) | 0.96 (0.74–1.24) | 0.97 (0.86–1.10) | 0.75 |
| rs17044137 | FLJ39370 | T | A† | 0.29 | 515/556/51 | 0.27 | 651/574/67 | 1.00 | 1.29 (1.08–1.53) | 0.97 (0.64–1.43) | 1.15 (1.00–1.33) | 0.140 |
| rs7480010 | LOC387761 | A | G† | 0.29 | 583/469/106 | 0.30 | 639/549/117 | 1.00 | 0.92 (0.77–1.09) | 0.96 (0.71–1.30) | 0.95 (0.84–1.09) | 0.46 |
| rs12255372 | TCF7L2 | G | T† | 0.34 | 493/542/128 | 0.28 | 671/533/92 | 1.00 | 1.52 (1.27–1.82) | 2.08 (1.52–2.84) | 1.48 (1.29–1.69) | <0.001 |
| rs1801282 | PPARG | C† | G | 0.10 | 930/206/11 | 0.11 | 1039/260/19 | 1.00 | 2.28 (0.97–5.32) | 2.41 (1.05–5.55) | 1.15 (0.95–1.40) | 0.160 |
| rs5219 | KCNJ11 | G | A† | 0.36 | 471/554/152 | 0.37 | 524/602/189 | 1.00 | 1.06 (0.88–1.27) | 0.92 (0.71–1.20) | 0.99 (0.87–1.11) | 0.84 |
MAF = minor allele frequency; SNP = single nucleotide polymorphism.
Models are adjusted for age and body mass index (5 categories); P values are for the additive model.
Risk allele.
Figure 1. Association of reported loci and risk for type 2 diabetes in pooled analysis of men and women.
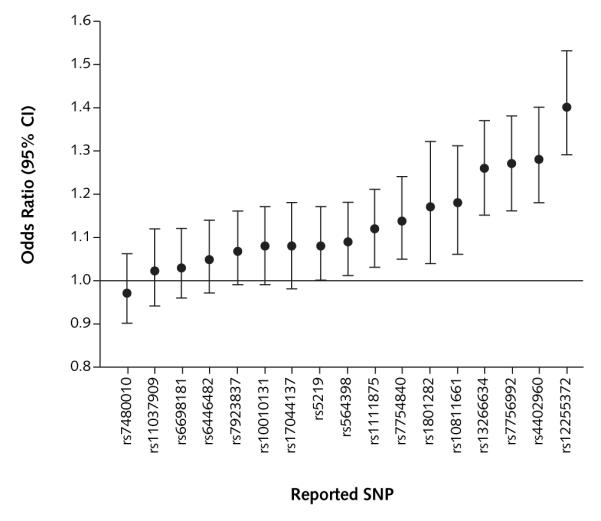
Odds ratios (95% CIs) are adjusted for age (quintiles), sex, and body mass index (quintiles). SNP = single nucleotide polymorphism.
To evaluate the joint effects of reproducibly associated variants, we calculated a GRS by using either a simple count (count GRS) or a weighted (weighted GRS) approach. The median count GRS for both control cohorts was 11.0, whereas the median weighted GRS was 10.4 for men and 10.2 for women. Demographic and risk factor characteristics did not significantly differ across quintiles of count or weighted GRS (data not shown). Based on the count GRS, the odds ratio for type 2 diabetes associated with each point scored, corresponding to 1 risk allele, was 1.19 (95% CI, 1.14 to 1.24) and 1.16 (CI, 1.12 to 1.20) for men and women, respectively, after adjustment for age and BMI (Table 3). Odds ratios increased across quintiles of count GRS for both men and women (P < 0.001 for trend). Compared with persons in the lowest quintile of GRS, men in the highest quintile had an odds ratio for type 2 diabetes of 2.76 (CI, 2.06 to 3.68) and women in this quintile had an odds ratio of 2.17 (CI, 1.76 to 2.69). Risk estimates were slightly increased in both cohorts when analyses were performed by using the weighted GRS (Table 3). In multivariate analysis, odds ratios remained significant. Further adjustment for dietary factors, including ratio of polyunsaturated–saturated fatty acids and intake of cereal fiber and trans fat, did not substantially change the results (data not shown). Data from both cohorts were pooled to further investigate the relationship between the count GRS and type 2 diabetes. Compared with individuals with a GRS of 11 (median GRS of the population), those with a GRS of 7 or less (n = 254) had an odds ratio for type 2 diabetes of 0.43 (CI, 0.31 to 0.60), whereas those with a GRS of 15 or more (n = 301) had an odds ratio of 2.05 (CI, 1.55 to 2.70) (Figure 2). Multivariate adjustment had a negligible effect on these risk estimates (odds ratio, 0.49 [CI, 0.35 to 0.68] and 2.05 [CI, 1.53 to 2.74] for GRS of ≤7 and ≥15, respectively).
Table 3.
Association Between GRS and Risk for Type 2 Diabetes
| Characteristic | Continuous GRS | Quintile of GRS |
||||
|---|---|---|---|---|---|---|
| First | Second | Third | Fourth | Fifth | ||
| Count GRS* | ||||||
| Men | ||||||
| Total, n | – | 511 | 417 | 389 | 790 | 391 |
| Median GRS (range) | – | 8.75 (5.0–9.0) | 10.0 (9.1–10.0) | 11.0 (10.1–11.0) | 12.0 (11.1–13.0) | 14.0 (13.1–18.0) |
| OR (95% CI) | ||||||
| Age- and BMI-adjusted | 1.19 (1.14–1.24) | 1.00 | 1.29 (0.97–1.72) | 1.61 (1.20–2.14) | 2.07 (1.62–2.65) | 2.76 (2.06–3.68) |
| Multivariate-adjusted† | 1.19 (1.14–1.24) | 1.00 | 1.28 (0.95–1.72) | 1.59 (1.18–2.15) | 2.09 (1.62–2.70) | 2.72 (2.01–3.68) |
| Women | ||||||
| Total, n | – | 851 | 616 | 600 | 788 | 862 |
| Median GRS (range) | – | 8.75 (4.0–9.0) | 10.0 (9.1–10.0) | 11.0 (10.1–11.0) | 12.0 (11.1–12.5) | 13.3 (12.6–18.0) |
| OR (95% CI) | ||||||
| Age- and BMI-adjusted | 1.16 (1.12–1.20) | 1.00 | 1.05 (0.83–1.34) | 1.61 (1.27–2.03) | 2.08 (1.67–2.58) | 2.17 (1.76–2.69) |
| Multivariate-adjusted† | 1.15 (1.11–1.19) | 1.00 | 1.01 (0.79–1.29) | 1.50 (1.17–1.92) | 1.95 (1.55–2.45) | 2.02 (1.62–2.53) |
| Weighted GRS * | ||||||
| Men | ||||||
| Total, n | – | 411 | 453 | 486 | 536 | 612 |
| Median GRS (range) | – | 7.7 (4.1–8.7) | 9.2 (8.9–9.9) | 10.4 (10.1–11.0) | 11.6 (11.1–12.3) | 13.5 (12.4–17.9) |
| OR (95% CI) | ||||||
| Age- and BMI-adjusted | 1.20 (1.15–1.25) | 1.00 | 1.37 (1.02–1.85) | 1.66 (1.24–2.22) | 2.07 (1.56–2.75) | 2.95 (2.23–3.90) |
| Multivariate-adjusted† | 1.19 (1.14–1.24) | 1.00 | 1.29 (0.94–1.75) | 1.58 (1.17–2.15) | 1.98 (1.47–2.66) | 2.79 (2.07–3.73) |
| Women | ||||||
| Total, n | – | 619 | 678 | 729 | 805 | 806 |
| Median GRS (range) | – | 7.5 (3.8–8.3) | 9.1 (8.4–9.8) | 10.2 (9.9–10.8) | 11.4 (10.9–12.2) | 13.3 (12.3–18.4) |
| OR (95% CI) | ||||||
| Age- and BMI-adjusted | 1.16 (1.12–1.20) | 1.00 | 1.25 (0.97–1.60) | 1.60 (1.25–2.04) | 1.94 (1.53–2.46) | 2.46 (1.95–3.10) |
| Multivariate-adjusted† | 1.15 (1.11–1.19) | 1.00 | 1.20 (0.92–1.56) | 1.52 (1.17–1.96) | 1.78 (1.39–2.29) | 2.26 (1.77–2.90) |
BMI = body mass index; GRS = genetic risk score; OR = odds ratio.
See the Methods section for count GRS and weighted GRS computations.
Adjusted for age, BMI (5 categories), family history of diabetes (yes or no), smoking (never, past, or current), menopausal status (pre- or postmenopausal [hormone use: never, past, or current]; women only), alcohol (5 categories), and quintiles of physical activity (metabolic equivalent task hours per week for men, hours per week for women).
Figure 2. Genetic risk score and risk for type 2 diabetes.
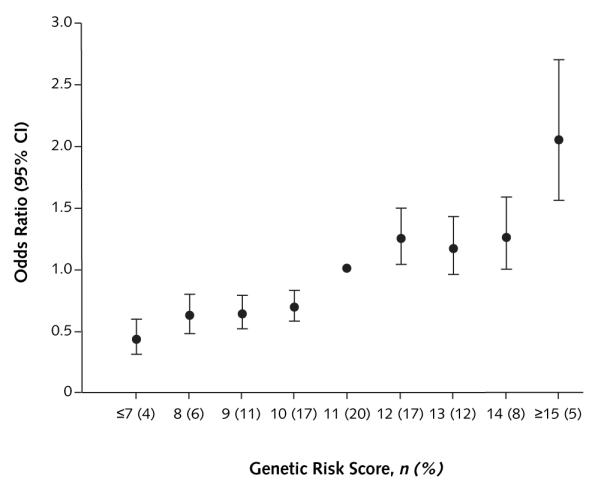
Results are based on the count genetic risk score for pooled data from men and women. Adjusted for age (quintiles), sex, and body mass index (quintiles).
We next determined whether the importance of genetic susceptibility varies across subgroups of the population by testing interactions between the GRS and conventional risk factors. The interaction between the count GRS and BMI (P = 0.023) was significant, indicating a stronger genetic effect among obese participants than normal-weight participants. After adjustment for age and sex, persons with a BMI of 30 kg/m2 or greater and a GRS in the highest quintile had an odds ratio of 14.06 (CI, 8.90 to 22.18) compared with persons with a BMI less than 25 kg/m2 and a GRS in the lowest quintile (Figure 3, top). Persons with a positive family history of diabetes and a GRS in the highest quintile had an odds ratio of 9.20 (CI, 5.50 to 15.40) compared with those without a family history of diabetes and with a GRS in the lowest quintile (Figure 3, bottom) (P for interaction = 0.63). Smoking, physical activity, alcohol use, or dietary intake did not significantly interact with the GRS (data not shown).
Figure 3. Joint effects of conventional risk factors and genetic risk score on risk for type 2 diabetes.

Values on bars indicate sample size. Top. Joint effects of body mass index and count genetic risk score (adjusted for age and sex) for pooled data from men and women. Bottom. Joint effects of family history of diabetes and count genetic risk score (adjusted for age, sex, and body mass index) for pooled data from men and women.
Figure 4 shows the receiver-operating characteristic curves for the logistic regression model incorporating conventional risk factors with and without inclusion of the GRS. The AUC for the conventional model was 0.78 (CI, 0.77 to 0.79) and significantly increased to 0.79 (CI, 0.78 to 0.80) when the GRS was added (P < 0.001). Results were the same when dietary risk factors (ratio of polyunsaturated–saturated fatty acids and intake of cereal fiber and trans fat) were added to the conventional model (data not shown).
Figure 4. Receiver-operating characteristic curves for type 2 diabetes.
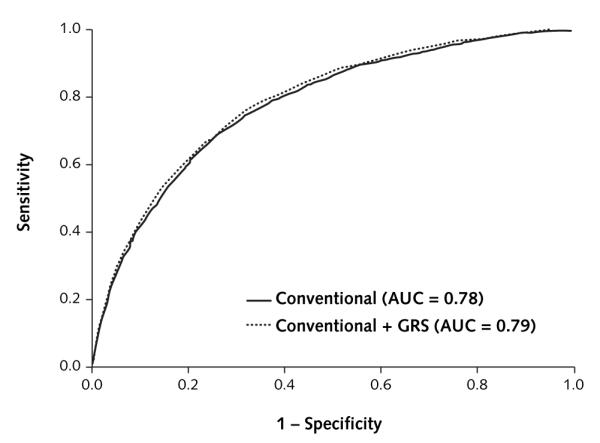
The curves are based on logistic regression models incorporating conventional risk factors (age, sex, body mass index, family history of diabetes, smoking, alcohol intake, and physical activity) with and without the count GRS. AUC = area under the curve; GRS = genetic risk score.
DISCUSSION
In these 2 large, well-established cohort studies, we extended support for many of the candidate loci identified from genome-wide association studies and demonstrated that a GRS that aggregates information from multiple genetic variants might be useful for assessing genetic predisposition to type 2 diabetes. Despite its limited discriminatory value, the GRS in combination with conventional risk factors, such as obesity and family history of diabetes, identifies subgroups of a population with a particularly high risk for type 2 diabetes.
Among the 13 type 2 diabetes susceptibility loci that we examined, we found significant evidence for association with type 2 diabetes for 7: HHEX, CDKAL1, IGF2BP2, SLC30A8, CDKN2A/B, TCF7L2, and PPARG. The direction and magnitude of these associations were consistent with previous reports (37). Although significantly associated with type 2 diabetes, the modest risk that each locus confers limits the clinical utility of each when considered independently. Taken collectively, however, they provide a global measure of an individual’s genetic predisposition to type 2 diabetes. We took a conservative approach to creating a GRS by including only loci that reached genome-wide significance in meta-analysis (10, 37). Each risk allele (1 point) was associated with an approximately 16% to 19% increased risk for type 2 diabetes. Persons with 13 or more risk alleles (the highest quintile) had a greater than 2-fold increased risk for type 2 diabetes compared with those with 9 or less (the lowest quintile). Because the former group constitutes approximately 20% of the study population, the GRS may be a useful tool for identifying a substantial proportion of people with a high genetic risk for type 2 diabetes. To account for the different magnitudes of effect attributable to each SNP, we computed a weighted GRS by using β-coefficients reported in previous genome-wide meta-analyses (10, 37). These β-coefficients should represent the best estimates of risk available and account for different genotype and environment backgrounds of the populations studied. The results of the weighted GRS were very similar to those of the count GRS score, possibly because the range of risk effects attributable to each of the loci was narrow.
In addition to its large sample size and prospective setting, a major strength of our study is the availability of comprehensive, validated environmental exposures in both cohorts, which allowed us to investigate the interplay between the GRS and traditional risk factors for type 2 diabetes. Our results indicate that the GRS may be useful in conjunction with certain conventional risk factors, such as obesity, to uncover subsets of the population with a markedly increased risk for type 2 diabetes that merit more aggressive intervention and monitoring. These data need to be confirmed in further studies.
A family history of diabetes is a commonly used surrogate for genetic susceptibility to type 2 diabetes and remains one of the strongest risk factors for the disease. This risk factor, however, encompasses both genetic and shared environmental components. For this reason, explicit assessment of an individual’s genetic constitution should provide a better measurement of genetic risk than family history. In our study, the strong association between family history of diabetes and risk for type 2 diabetes persisted when adjusting for GRS. These findings suggest that other risk loci remain to be discovered or that family history has a much larger shared environmental component than previously thought.
Although knowledge gained from genome-wide association studies may further our understanding of type 2 diabetes pathogenesis and development of prevention and treatment interventions, its incremental usefulness over conventional risk factors for predicting type 2 diabetes is uncertain. Given the design of our study, we could not precisely estimate the predictive power of the GRS and were limited to discriminatory analysis. The GRS significantly improved case–control discrimination beyond that afforded by conventional risk factors, but the magnitude of this improvement was marginal: Addition of the GRS increased the AUC by only 1%. Minimal improvements in the AUC with the addition of genetic information derived from genome-wide association studies have also been reported by other studies (18–21). Clinical risk models that incorporate fasting glucose level or other measures of insulin sensitivity have shown the least discriminative improvement with the addition of genetic information (19, 20). Collinearity might account for this effect, because the GRS might be increasing risk through these intermediate traits. Alternatively, the AUC of these models is already high, and the addition of any variable is unlikely to increase it. Consistent with this notion are concerns that change in the AUC may be an insensitive measure of the improvement in risk prediction when a novel risk factor is considered (41). Net reclassification improvement may be a more sensitive measure (41) but has not been sufficiently applied to the study of a type 2 diabetes GRS. Although our risk model did not include a measure of glucose metabolism, it is larger than the other study and considered a broader range of behavioral factors, including smoking, alcohol, and physical activity (19). The risk factors we included are established risk factors for type 2 diabetes (42) and are relatively easy to measure in the clinic.
Our study has several limitations. Some of the control participants may have undiagnosed type 2 diabetes that would bias the results toward the null. However, in a previous validation study (43), the prevalence of undiagnosed type 2 diabetes in these health professionals (approximately 2%) was substantially lower than that in the general population (approximately 30%) (44). As type 2 diabetes is a relatively late-onset disease, some of the control participants in our study may develop the disease later in life. To further explore the effect this may have had on our results, we conducted analyses using subsets of our control group defined by different minimum age thresholds but observed similar results. Population stratification may also affect the observed associations. Because participants were selected from well-characterized cohorts with a defined study base and because the analysis was restricted to white persons of European ancestry, biases due to population stratification are probably minimized. Nonetheless, because of population differences in allele frequencies, linkage disequilibrium patterns, and risk factor prevalence, whether our GRS is significantly associated with type 2 diabetes in other ethnic groups merits further investigation. The GRS may be especially relevant to groups in which established risk factors have not yet developed, such as children or young adolescents. Although this warrants further study, targeting young patients for genetic screening must be viewed with caution and should not undermine efforts to promote a proper diet and exercise for type 2 diabetes prevention.
In conclusion, data obtained from 2 large cohort studies showed that loci with modest individual effects could have a significant impact on risk for type 2 diabetes when their contributions are combined. Information provided by a GRS based on multiple variants might be combined with knowledge on environmental risk factors to identify subsets of the population at high risk for type 2 diabetes who are likely to benefit most from more aggressive preventive interventions. Significant risks attributable to a GRS must be tempered with its clinical utility, which has much to gain with further research. Since the advent of genome-wide association studies, the rate of type 2 diabetes loci discovery has increased considerably. As additional novel loci are added to the GRS, our ability to characterize genetically susceptible individuals will continue to improve.
Context
Individual genetic polymorphisms are weakly associated with type 2 diabetes. It is unclear whether genetic information adds usefully to the assessment of diabetes risk using only conventional risk factors.
Contribution
After developing a genetic risk score for type 2 diabetes that combined data on 10 polymorphisms, the investigators examined the score’s contribution to the prediction of type 2 diabetes in 2 cohorts. The genetic risk score improved risk prediction modestly when considered in addition to conventional risk factors.
Caution
The study included only persons of European ancestry. Whether the genetic risk score is associated with type 2 diabetes in other ethnic groups remains unknown. The study did not examine the contribution of glucose levels in identifying diabetes risk.
—The Editors
Acknowledgment
The authors thank Patrice Soule and Dr. Hardeep Ranu of the Dana Farber/Harvard Cancer Center Genotyping Core for sample preparation and genotyping and the participants in the NHS and HPFS for their dedication and commitment.
Grant Support: By the National Institutes of Health (grants DK58845 and CA87969). Dr. Cornelis is a recipient of a Canadian Institutes of Health Research Fellowship. Dr. Qi is a recipient of the American Heart Association Scientist Development Award. Dr. Zhang is supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Primary Funding Source: National Institutes of Health.
Footnotes
Potential Financial Conflicts of Interest: None disclosed.
Reproducible Research Statement: Study protocol and data set: Not available. Statistical code: Available from Dr. Cornelis (mcorneli@hsph.harvard.edu).
Current author addresses and author contributions are available at www.annals.org.
Current Author Addresses: Drs. Cornelis, Qi, Kraft, Cai, Hunter, and Hu: Harvard School of Public Health, Building II, 665 Huntington Avenue, Boston, MA 02115.
Dr. Zhang: Division of Epidemiology, Statistics, and Prevention Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892.
Dr. Manson: 900 Commonwealth Avenue East, Boston, MA 02215.
Author Contributions: Conception and design: L. Qi, F.B. Hu.
Analysis and interpretation of the data: M.C. Cornelis, L. Qi, C. Zhang, P. Kraft, D.J. Hunter, F.B. Hu.
Drafting of the article: M.C. Cornelis, L. Qi.
Critical revision of the article for important intellectual content: M.C. Cornelis, L. Qi, C. Zhang, P. Kraft, J. Manson, D.J. Hunter, F.B. Hu.
Final approval of the article: L. Qi, C. Zhang, P. Kraft, J. Manson, D.J. Hunter, F.B. Hu.
Provision of study materials or patients: F.B. Hu.
Statistical expertise: M.C. Cornelis, L. Qi, P. Kraft, D.J. Hunter.
Obtaining of funding: L. Qi, F.B. Hu.
Administrative, technical, or logistic support: J. Manson, F.B. Hu.
Collection and assembly of data: L. Qi, J. Manson, F.B. Hu.
References
- 1.Hogan P, Dall T, Nikolov P, American Diabetes Association Economic costs of diabetes in the US in 2002. Diabetes Care. 2003;26:917–32. doi: 10.2337/diacare.26.3.917. [PMID: 12610059] [DOI] [PubMed] [Google Scholar]
- 2.Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35:1060–7. doi: 10.1007/BF02221682. [PMID: 1473616] [DOI] [PubMed] [Google Scholar]
- 3.Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet. 1990;46:222–8. [PMID: 2301392] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dam RM, Hoebee B, Seidell JC, Schaap MM, de Bruin TW, Feskens EJ. Common variants in the ATP-sensitive K+ channel genes KCNJ11 (Kir6.2) and ABCC8 (SUR1) in relation to glucose intolerance: population-based studies and meta-analyses. Diabet Med. 2005;22:590–8. doi: 10.1111/j.1464-5491.2005.01465.x. [PMID: 15842514] [DOI] [PubMed] [Google Scholar]
- 5.Ludovico O, Pellegrini F, Di Paola R, Minenna A, Mastroianno S, Cardellini M, et al. Heterogeneous effect of peroxisome proliferator-activated receptor gamma2 Ala12 variant on type 2 diabetes risk. Obesity (Silver Spring) 2007;15:1076–81. doi: 10.1038/oby.2007.617. [PMID: 17495182] [DOI] [PubMed] [Google Scholar]
- 6.Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, et al. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med. 2007;85:777–82. doi: 10.1007/s00109-007-0203-4. [PMID: 17476472] [DOI] [PubMed] [Google Scholar]
- 7.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–5. doi: 10.1038/nature05616. [PMID: 17293876] [DOI] [PubMed] [Google Scholar]
- 8.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–5. doi: 10.1038/ng2043. [PMID: 17460697] [DOI] [PubMed] [Google Scholar]
- 9.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–5. doi: 10.1126/science.1142382. [PMID: 17463248] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Wellcome Trust Case Control Consortium (WTCCC) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–41. doi: 10.1126/science.1142364. [PMID: 17463249] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6. doi: 10.1126/science.1142358. [PMID: 17463246] [DOI] [PubMed] [Google Scholar]
- 12.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [PMID: 17554300] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–3. doi: 10.1038/ng2067. [PMID: 17603484] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Wellcome Trust Case Control Consortium Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45. doi: 10.1038/ng.120. [PMID: 18372903] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi L, Kang K, Zhang C, van Dam RM, Kraft P, Hunter D, et al. Fat mass-and obesity-associated (FTO) gene variant is associated with obesity: longitudinal analyses in two cohort studies and functional test. Diabetes. 2008;57:3145–51. doi: 10.2337/db08-0006. [PMID: 18647953] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, et al. Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75. doi: 10.1038/ng.140. [PMID: 18454148] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cauchi S, Meyre D, Durand E, Proença C, Marre M, Hadjadj S, et al. Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS ONE. 2008;3:e2031. doi: 10.1371/journal.pone.0002031. [PMID: 18461161] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, Oostra BA, et al. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–8. doi: 10.2337/db08-0425. [PMID: 18694974] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–19. doi: 10.1056/NEJMoa0804742. [PMID: 19020323] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–32. doi: 10.1056/NEJMoa0801869. [PMID: 19020324] [DOI] [PubMed] [Google Scholar]
- 21.Lango H, Palmer CN, Morris AD, Zeggini E, Hattersley AT, McCarthy MI, et al. UK Type 2 Diabetes Genetics Consortium Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57:3129–35. doi: 10.2337/db08-0504. [PMID: 18591388] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weedon MN, McCarthy MI, Hitman G, Walker M, Groves CJ, Zeggini E, et al. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med. 2006;3:e374. doi: 10.1371/journal.pmed.0030374. [PMID: 17020404] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colditz GA, Hankinson SE. The Nurses’ Health Study: lifestyle and health among women. Nat Rev Cancer. 2005;5:388–96. doi: 10.1038/nrc1608. [PMID: 15864280] [DOI] [PubMed] [Google Scholar]
- 24.Willett WC. Nutritional Epidemiology. Oxford University Pr; New York: 1998. [Google Scholar]
- 25.Rimm EB, Giovannucci EL, Willett WC, Colditz GA, Ascherio A, Rosner B, et al. Prospective study of alcohol consumption and risk of coronary disease in men. Lancet. 1991;338:464–8. doi: 10.1016/0140-6736(91)90542-w. [PMID: 1678444] [DOI] [PubMed] [Google Scholar]
- 26.Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135:1114–26. doi: 10.1093/oxfordjournals.aje.a116211. discussion 1127-36. [PMID: 1632423] [DOI] [PubMed] [Google Scholar]
- 27.Hu FB, Doria A, Li T, Meigs JB, Liu S, Memisoglu A, et al. Genetic variation at the adiponectin locus and risk of type 2 diabetes in women. Diabetes. 2004;53:209–13. doi: 10.2337/diabetes.53.1.209. [PMID: 14693717] [DOI] [PubMed] [Google Scholar]
- 28.Qi L, van Dam RM, Meigs JB, Manson JE, Hunter D, Hu FB. Genetic variation in IL6 gene and type 2 diabetes: tagging-SNP haplotype analysis in large-scale case-control study and meta-analysis. Hum Mol Genet. 2006;15:1914–20. doi: 10.1093/hmg/ddl113. [PMID: 16644865] [DOI] [PubMed] [Google Scholar]
- 29.Hu FB, Leitzmann MF, Stampfer MJ, Colditz GA, Willett WC, Rimm EB. Physical activity and television watching in relation to risk for type 2 diabetes mellitus in men. Arch Intern Med. 2001;161:1542–8. doi: 10.1001/archinte.161.12.1542. [PMID: 11427103] [DOI] [PubMed] [Google Scholar]
- 30.Manson JE, Rimm EB, Stampfer MJ, Colditz GA, Willett WC, Krolewski AS, et al. Physical activity and incidence of non-insulin-dependent diabetes mellitus in women. Lancet. 1991;338:774–8. doi: 10.1016/0140-6736(91)90664-b. [PMID: 1681160] [DOI] [PubMed] [Google Scholar]
- 31.National Diabetes Data Group Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes. 1979;28:1039–57. doi: 10.2337/diab.28.12.1039. [PMID: 510803] [DOI] [PubMed] [Google Scholar]
- 32.Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183–97. doi: 10.2337/diacare.20.7.1183. [PMID: 9203460] [DOI] [PubMed] [Google Scholar]
- 33.Colditz GA, Manson JE, Hankinson SE. The Nurses’ Health Study: 20-year contribution to the understanding of health among women. J Womens Health. 1997;6:49–62. doi: 10.1089/jwh.1997.6.49. [PMID: 9065374] [DOI] [PubMed] [Google Scholar]
- 34.Willett W, Stampfer MJ, Bain C, Lipnick R, Speizer FE, Rosner B, et al. Cigarette smoking, relative weight, and menopause. Am J Epidemiol. 1983;117:651–8. doi: 10.1093/oxfordjournals.aje.a113598. [PMID: 6859020] [DOI] [PubMed] [Google Scholar]
- 35.Wolf AM, Hunter DJ, Colditz GA, Manson JE, Stampfer MJ, Corsano KA, et al. Reproducibility and validity of a self-administered physical activity questionnaire. Int J Epidemiol. 1994;23:991–9. doi: 10.1093/ije/23.5.991. [PMID: 7860180] [DOI] [PubMed] [Google Scholar]
- 36.Rimm EB, Stampfer MJ, Colditz GA, Chute CG, Litin LB, Willett WC. Validity of self-reported waist and hip circumferences in men and women. Epidemiology. 1990;1:466–73. doi: 10.1097/00001648-199011000-00009. [PMID: 2090285] [DOI] [PubMed] [Google Scholar]
- 37.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–62. doi: 10.1038/nrg2178. [PMID: 17703236] [DOI] [PubMed] [Google Scholar]
- 38.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781–91. doi: 10.1038/nrg1916. [PMID: 16983374] [DOI] [PubMed] [Google Scholar]
- 39.Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, et al. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001;345:790–7. doi: 10.1056/NEJMoa010492. [PMID: 11556298] [DOI] [PubMed] [Google Scholar]
- 40.Pencina MJ, D’Agostino RB. Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat Med. 2004;23:2109–23. doi: 10.1002/sim.1802. [PMID: 15211606] [DOI] [PubMed] [Google Scholar]
- 41.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–72. doi: 10.1002/sim.2929. discussion 207-12. [PMID: 17569110] [DOI] [PubMed] [Google Scholar]
- 42.Schulze MB, Hu FB. Primary prevention of diabetes: what can be done and how much can be prevented? Annu Rev Public Health. 2005;26:445–67. doi: 10.1146/annurev.publhealth.26.021304.144532. [PMID: 15760297] [DOI] [PubMed] [Google Scholar]
- 43.Field AE, Coakley EH, Must A, Spadano JL, Laird N, Dietz WH, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med. 2001;161:1581–6. doi: 10.1001/archinte.161.13.1581. [PMID: 11434789] [DOI] [PubMed] [Google Scholar]
- 44.Gregg EW, Cadwell BL, Cheng YJ, Cowie CC, Williams DE, Geiss L, et al. Trends in the prevalence and ratio of diagnosed to undiagnosed diabetes according to obesity levels in the U.S. Diabetes Care. 2004;27:2806–12. doi: 10.2337/diacare.27.12.2806. [PMID: 15562189] [DOI] [PubMed] [Google Scholar]