

Abstract
Study Objectives:
The basal forebrain (BF) has been implicated as an important brain region that regulates the sleep-wake cycle of animals. Gamma-aminobutyric acidergic (GABAergic) neurons are the most predominant neuronal population within this region. However, due to the lack of specific molecular tools, the roles of the BF GABAergic neurons have not been fully elucidated. Previously, we have found high expression levels of the Kv2.2 voltage-gated potassium channel on approximately 60% of GABAergic neurons in the magnocellular preoptic area and horizontal limb of the diagonal band of Broca of the BF and therefore proposed it as a potential molecular target to study this neuronal population. In this study, we sought to determine the functional roles of the Kv2.2-expressing neurons in the regulation of the sleep-wake cycle.
Design:
Sleep analysis between two genotypes and within each genotype before and after sleep deprivation.
Setting:
Animal sleep research laboratory.
Participants:
Adult mice. Wild-type and Kv2.2 knockout mice with C57/BL6 background.
Interventions:
EEG/EMG recordings from the basal state and after sleep-deprivation which was induced by mild aggitation for 6 h.
Results:
Immunostaining of a marker of neuronal activity indicates that these Kv2.2-expressing neurons appear to be preferentially active during the wake state. Therefore, we tested whether Kv2.2-expressing neurons in the BF are involved in arousal using Kv2.2-deficient mice. BF GABAergic neurons exhibited augmented expression of c-Fos. These knockout mice exhibited longer consolidated wake bouts than wild-type littermates, and that phenotype was further exacerbated by sleep deprivation. Moreover, in-depth analyses of their cortical electroencephalogram revealed a significant decrease in the delta-frequency activity during the nonrapid eye movement sleep state.
Conclusions:
These results revealed the significance of Kv2.2-expressing neurons in the regulation of the sleep-wake cycle.
Citation:
Hermanstyne TO; Subedi K; Le WW; Hoffman GE; Meredith AL; Mong JA; Misonou H. Kv2.2: a novel molecular target to study the role of basal forebrain GABAergic neurons in the sleep-wake cycle. SLEEP 2013;36(12):1839-1848.
Keywords: Channel, basal forebrain, GABA, cortex
INTRODUCTION
The maintenance of the sleep-wake cycle requires multiple brain regions and neuronal populations.1 It has been proposed that the sleep-wake cycle is regulated by a balance between a sleep and an arousal circuit. The basal forebrain (BF) is one of the brain regions implicated in the regulation of sleep-wake dynamics.2,3 Lesions of the BF significantly affect the sleep-wake cycle and electroencephalogram.4,5 Cholinergic and gamma-aminobutyric acidergic (GABAergic) neurons are the major neuronal populations in the BF that provide robust projections to the cerebral cortex. Specific lesions of the BF cholinergic neurons decrease the percentage of wakefulness as characterized by somnographic recordings,6 indicating that BF cholinergic neurons are involved in promoting wakefulness. These results led to the idea that the BF is involved in the arousal circuit. However, the GABAergic neurons are the predominant population in the BF and outnumber the cholinergic neurons.7 Therefore, without elucidating the role of these GABAergic neurons, it would be difficult to make a conclusion regarding the roles of the BF in the regulation of the sleep-wake cycle. Immunohistochemical and electrophysiological analyses have revealed that GABAergic neurons in the BF are heterogeneous in molecular identities8,9 as well as in their firing behaviors,10 unlike cholinergic neurons that are thought to be homogeneous.11 Therefore, specific markers and molecular tools are required to distinguish and study multiple subpopulations of GABAergic neurons within the BF.
We reported previously that a large subpopulation of neurons within the magnocellular preoptic area (MCPO) and the horizontal limb of the diagonal band of Broca (HDB) of the BF express the Kv2.2 voltage-gated potassium channel.12 Kv2 channels (Kv2.1 and Kv2.2) are the major constituents of the somatic delayed rectifier potassium current,13,14 with Kv2.1 as the dominant Kv2-family protein in virtually all areas of the brain including the cerebral cortex, hippocampus, and striatum.12 These somatic potassium channels are thought to be important in determining the overall excitability of neurons.15,16 In contrast to the widespread expression of Kv2.1, Kv2.2 exhibits preferential expression in a few brain nuclei including the BF and the medial nucleus of the trapezoid body.12,17 Particularly, we found that Kv2.2 is expressed in approximately 60% of GABAergic neurons in the MCPO and HDB of the BF at very high levels, defining them as a novel and major subpopulation in BF sleep/wake-related areas. Interestingly, in these neurons, Kv2.2 is the predominant Kv2-family protein, because the expression of Kv2.1 is negligible.12 We proposed that Kv2.2 is a viable molecular target to study the functional role of these GABAergic neurons in sleep behavior. In the current study, using Kv2.2 knockout mice as a model system, we sought to test the hypothesis that Kv2.2-expressing neurons regulate the sleep-wake cycle, particularly arousal, of the mouse.
MATERIALS AND METHODS
Animals
All animal use procedures were in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, and approved by the institutional animal use committee. Male wild-type (WT) and Kv2.2 knockout (KO) mice were used in the study.18 These mice were obtained from Kv2.2 heterozygous breeding pairs on a C57BL/6 background generated by Texas A&M Institute for Genomic Medicine. In these mice, the second exon of the Kv2.2 gene was replaced with a targeting vector containing β-geo (LacZ/neomycin) cassette. Forty-two animals were used in this study. At the time of the experiments, all animals were between the ages of 4-5 mo old and were housed individually in a 12:12 h light-dark (LD) cycle. To determine the genotype of the mice generated from the heterozygous breeding pairs, DNA extracts from tail biopsies were used in a qualitative polymerase chain reaction (PCR) experiment (Figure 1B). Briefly, small (0.5 cm) tail snips were digested with 300 μg of Proteinase K overnight at 55°C. Genomic DNA was extracted and purified for PCR using a standard phenol-chloroform extraction method. PCR was performed with the following primers:
Mutant Primer Pairs: Neo3a and Primer #17; detects a 472-bp product
Wild-type Primer Pairs: Primer #18 and #19; detects a 312-bp product
Primer Neo3a: GCAGCGCATCGCCTTCTATC
Primer #17: GTGTCTCAGAAATGGCGTGTC
Primer #18: GGATACTGAAACTCGCCAGAC
Primer #19: GTATAGGAAGGGCAATAACCAG
Figure 1.
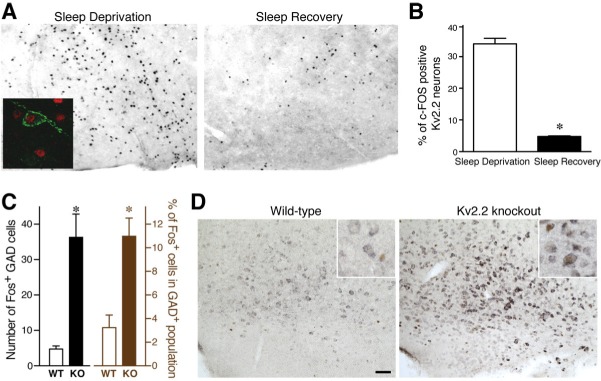
Activity of Kv2.2-gamma-aminobutyric acidergic (GABAergic) neurons in the basal forebrain (BF). (A) The differential expression patterns of c-Fos in coronal sections that include the magnocellular preoptic area/horizontal limb of the diagonal band of Broca (MCPO/HDB) of the basal forebrain from sleep deprived and sleep recovered wild type (WT) mice. Inset: Double immunolabeling with anti-Kv2.2 (green) and anti–c-Fos (red) antibodies. (B) Quantitative analysis of three consecutive coronal brain sections (spanning 120 μm) containing MCPO/HDB from sleep deprived and sleep recovered WT mice. The percentages of c-Fos positive Kv2.2 neurons are shown (unpaired Student t-test; P = 0.0001; n = 4). (C) Augmented expression of c-Fos in GABAergic neurons in the BF of Kv2.2 knockout (KO) mice. The number (left) and percentage (right) of cells positive for both c-Fos protein and glutamic acid decarboxylase 67 (GAD67) messenger RNA (mRNA) in the MCPO are shown. *P < 0.01. (D) Double labeling of GAD67 mRNA (purple) and c-Fos protein (brown) in the MCPO. Scale bar, 100 μm. Inset: high-magnification images of neurons in the MCPO. Note that there are neurons double-positive for GAD67 mRNA and c-Fos protein in the KO section but not in the WT section.
Analysis of Sleep Behavior
Electrodes for electroencephalography (EEG) were implanted in the skull of Kv2.2 KO mice and WT littermates under continuous anesthesia with isoflurane. Two stainless-steel electrodes (Plastics One, Roanoke, VA) were placed to make contact with the dura from the motor cortex and the somatosensory cortex. Electrode coordinates are as follows: The first screw is placed 1.0 mm anterior from bregma and 1.0 mm lateral from the central suture. The second screw is placed on the contralateral side 3.0 mm posterior from bregma and 3.0 mm lateral from the central suture. The third electrode is placed in the nuchal muscles of the neck to measure electromyographic (EMG) recordings. These electrodes were connected to a telemetry transmitter (DSI, Saint Paul, MN) located subcutaneously on the lateral side of the abdomen for the continuous recording of EEG and EMG in freely moving animals. Following surgery, the animals were allowed to recover for 1 w.
Signals were acquired by using the Neuroscore program (DSI Analysis Software, Saint Paul, MN) at a sampling rate of 500 Hz. Sleep (nonrapid eye movement [NREM]) and rapid eye movement [REM]) and wake states were determined by visual inspection of EEG and EMG waveforms of 10-sec epochs for 9 h to determine scoring parameters as previously described.19,20 Wake was distinguished by low-amplitude, high-frequency EEG and high-amplitude EMG, non-REM sleep was distinguished by high-amplitude, low-frequency EEG and low-amplitude EMG, and REM sleep was distinguished by low-amplitude, high-frequency EEG and muscle atonia. The vigilant states for the rest of the recording (150 h) were determined by the Neuroscore program based on the scoring parameters.
We recorded the baseline EEG/EMG for 2 days (days 1 and 2). On the third day of recording (day 3), animals were sleep deprived for 6 h (starting at lights on, 07:00) by gentle agitation (introduction of novel objects, tapping on the cage, and gently touching the animal with a paint brush). The recording continued for the additional 2.5 days after sleep deprivation. The hypnograms in Figure 4A were generated from the recording of day 2 during the dark phase using the Neuroscore program.
Figure 4.
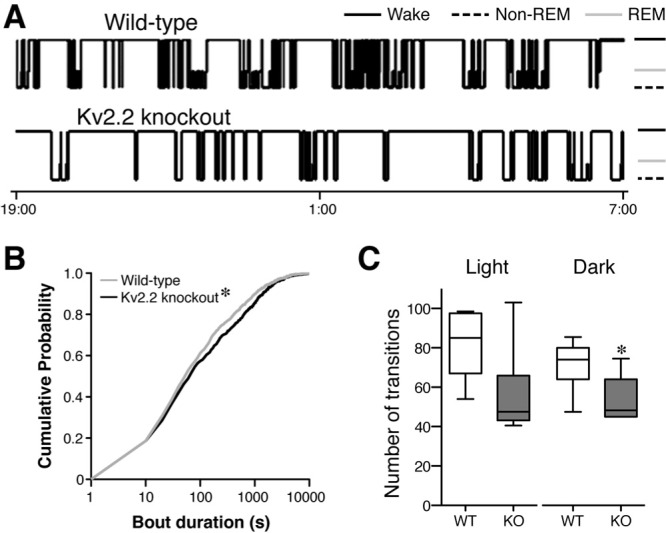
Quantitative analysis of the hypnograms. (A) Representative 12-h hypnograms (19:00 to 07:00) from the baseline recording revealing the architectural parameters of the sleep-wake cycle. The height of the horizontal line depicts the vigilant state the mouse is in at the time. Solid line, wake state; gray line, rapid eye movement (REM) sleep; dashed line, nonrapid eye movement (non-REM) sleep. (B) Cumulative probability distribution plot of the duration of wake bout episodes during the dark periods. Kv2.2 knockout (KO) mice exhibited a rightward shift indicating longer wake bouts (Kolmogorov-Smirnov test; P < 0.05; n = 7). (C) The average number of transitions from the wake state to any of the sleep states (non-REM and REM) during the light and dark periods reveals that the Kv2.2 KO mice transition less frequently from the wake state in the dark state (unpaired Student t-test; P = 0.02; n = 7). WT, wild-type.
EEG Signal Analysis
Wake EEG and sleep EEG were obtained from the original EEG recordings of 3 h using a script running on Scilab (Paris, France). The script can be provided from H.M. upon request. Basically, wake EEG and sleep EEG were extracted by thresholding of the signal. Because the signal amplitude varied among animals, the threshold was set for each recording as follows. First, the total power of EEG and EMG signals was computed in 4-sec epochs for the frequency range between 0.1 to 100 Hz. Then, the difference (Δ) of the highest total power over the lowest total power (L) in the 3-h recording was calculated for EEG and EMG. To obtain wake EEG, we extracted EEG signals, where EMG power was above a threshold (L+Δ/5). To obtain sleep EEG, we extracted EEG signals, where EEG power was above L+Δ/2.5. The examples of extractions are shown in Figure 7A. A discrete fast Fourier-transform (FFT) algorithm was applied to 30 min of sleep and wake EEG to obtain frequency and amplitude values of the overall power spectrum. Then the integral of the power spectrum was calculated by using the trapezoidal rule, which provides the percentage of each distinct frequency band within that power spectrum. The distinct frequency bands are as follows: slow band (0.1-1 Hz), delta band (1-4 Hz), theta band (4-7 Hz), alpha band (8-12 Hz), beta band (12-30 Hz), and gamma band (30-90 Hz).
Figure 7.
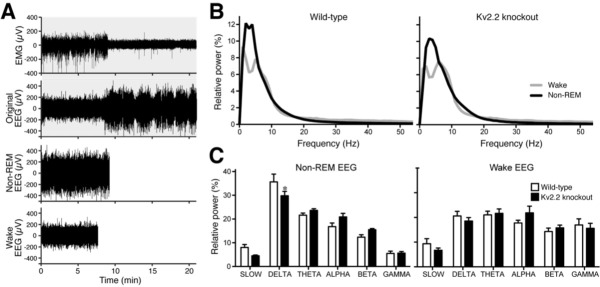
Altered delta oscillations observed in Kv2.2 knockout mice. (A) An example of the extraction of nonrapid eye movement (non-REM) electroencephalographic (EEG) and wake EEG from the original baseline EEG recording of a wild-type animal. The actual analyses in B and C were done with at least 30 min long non-REM EEG and wake EEG. Only 20 min of the original EEG and EMG were used for this example for better visualization of the signals. (B) The averaged power spectra for both wake (gray curve) and non-REM (black) EEGs after sleep deprivation were plotted. The overall difference between the wake and non-REM curves for the Kv2.2 knockout mice was much smaller than wild-type littermates. (C) Kv2.2 knockout mice showed a significant reduction in the delta frequency band during non-REM sleep after sleep deprivation. The integral of the power spectra was taken for both wake and non-REM states and the percentage for each frequency band was plotted in a histogram (repeated measures two-way analysis of variance; Bonferroni posttest; P < 0.05; n = 7).
Immunostaining of c-Fos
Animals were anesthetized with isoflurane and they were immediately decapitated in ice-cold phosphate buffered saline solution following a 6-h sleep deprivation challenge or a 3-h sleep recovery period. The brains were removed and immersed in 4% paraformaldehyde for 2 days, placed in 30% sucrose solution for cryoprotection, and cut into 40-μm coronal sections. Sections were washed in 0.05 M potassium phosphate buffered saline and incubated for 2 days at 4°C antic-Fos (1:500,000) polyclonal antibody (Oncogene Sciences, Boston, MA). Then sections were washed and probed with a biotinylated antirabbit secondary antibody (1:600, Vector Labs, Burlingame, CA). Nickel-enhanced diaminobenzidine (DAB) reaction was done by using the Vectastain ABC kit (45 μL of reagents A and B, Vector Labs, Burlingame, CA) and then allowed to incubate for 20 min in a chromogen solution containing nickel sulfate, DAB (Sigma, St Louis, MO), sodium acetate (Amresco, Solon, OH), 3% hydrogen peroxide (Sigma). After the nickel-DAB staining, the sections were washed again and then were incubated for 2 days at 4°C with the second primary antibody anti-Kv2.2 (0.3 μg/mL) polyclonal antibody (Alomone-APC120, Jerusalem, Israel). Immunoreactivity was detected using Alexa dye-conjugated secondary antibodies (Invitrogen, Carlsbad, CA). Bright field and fluorescence images were taken with a cooled charge coupled device (CCD) camera installed on an Axiovert-200M microscope (Carl Zeiss, Thornwood, NY), with 10×/0.5 NA, 20×/0.8 NA, 40×/1.3 NA, and 63×/1.4 NA lenses, using Axiovision software (Carl Zeiss). Only minor corrections of brightness and contrast were performed.
Dual Immunofluorescence Labeling of Kv2.2 and Parvalbumin
Free-floating sections were blocked and permeabilized with 10% normal goat serum (Chemicon, Billerica, MA) in a phosphate buffer containing 0.3% Triton X-100 for 1 h, then incubated overnight at 4°C with the rabbit anti-Kv2.2 antibody (0.1 μg/mL) and mouse anti-parvalbumin antibody (1:10,000, Sigma). Immunoreactivity was detected using Alexa dyeconjugated secondary antibodies (Invitrogen).
Double Labeling of GAD-67 mRNA and c-Fos protein
The procedure for double labeling of mRNA and protein has been previously described in detail.21,22 Briefly, 30-μm tissue sections obtained from every sixth section of the forebrain, which encompass the MCPO, were subjected to hybridization with the biotinylated glutamic acid decarboxylase 67 (GAD67) probe. The probe was prepared from GAD67 complementary DNA construct (provided by Dr. N.J. Tillakaratne). The probe (final concentration was 600 ng/kbp/mL) was mixed with yeast transfer RNA (yeast tRNA, Ambion, Austin, TX) at concentration of 0.25 mg/mL, denatured by heating to 85-90°C for 3 min, then cooled immediately on ice for 5 min before use. The GAD67 riboprobe was then mixed with hybridization buffer (750 μL deionized formamide, 300 μL 50% dextran sulfate, 90 μL 5M NaCl, 12 μL 500 mM ethylenediaminetetraacetic acid (EDTA), 30 μL 50× Denhardt solution, and 250 μL diethylpyrocarbonate-treated water), warmed to 50°C, placed into microbeakers with the sections and remained undisturbed overnight. After the treatment with RNAse and extensive washing, the biotinylated probe was detected using the ABC peroxidase method with nickel-DAB.23 Following the development of the mRNA signal, the tissue sections were immunostained with the c-Fos antibody and DAB.
Cell Counting
For the immunolabeling of Kv2.2 and c-Fos, three consecutive coronal sections spanning 120 μm of the basal fore-brain from three to four WT mice were used for the analysis. These sections were immunostained with anti-c-Fos and anti-Kv2.2 antibodies. Cells that were positive for these antigens were counted within a region of 1 mm2 starting at the indentation of the ventral surface of the basal forebrain bilaterally. The percentages of Kv2.2-expressing neurons positive for c-Fos immunoreactivity were averaged among three to four animals. For the evaluation of c-Fos expression in the ventral lateral preoptic area (VLPO), the number of c-Fos-positive neurons was counted per square millimeter area. VLPO was identified based on surrounding anatomical landmarks.
For the double-labeling of GAD67 mRNA and c-Fos protein, neurons positive for GAD67 mRNA were counted at 200× magnification and scored for the presence of c-Fos. Only cells with a visible nucleus (stained or unstained) were counted. Slides were coded so that the observer was blind to the geno-type of the animal.
Biological Rhythms and Analysis
Circadian behavior was measured by wheel-running activity in 12:12 h LD cycle and in constant darkness (DD).24 Periods were obtained from 7 days in the LD cycle and 14 days in DD by using chi-squared periodogram analysis (ClockLab Software, Actimetrics, running in Matlab v6.1, Mathworks, Natick, MA). Activity levels were presented from DD.
Statistical Analysis
Data are presented as mean ± standard error of the mean. Statistical analyses were made using Student t-test, Mann-Whitney U test, or analysis of variance (ANOVA) with post hoc analysis. Most data was analyzed using the GraphPad Prism (GraphPad Software, La Jolla, CA). The cumulative probability plots were generated and analyzed using the ‘R’ statistical software with the Kolmogorov-Smirnov test.
RESULTS
Kv2.2-Expressing GABAergic Neurons Are Wake-Active Neurons
In studying the role of the Kv2.2-GABAergic neurons of the BF in the sleep-wake cycle, we first investigated whether these neurons are wake- or sleep-active neurons. To address this, we used c-Fos expression as a marker of neuronal activity. The expression of this immediate early gene has been used to assess neuronal activity and also to correlate changes in neuronal activity in different brain regions with changes in vigilant states.25–28 We adapted a method from Sherin and colleagues,26 whereby wild-type (WT) mice are sleep deprived for 6 h by gentle agitation. During this consolidated wake period, ‘wake-active neurons’ are expected to express c-Fos,26 of which half-life is about 120 min.29 Conversely, in animals that are allowed to obtain 3 h of recovery sleep following the sleep deprivation challenge, we should be able to detect ‘sleep-active neurons’ that accumulate c-Fos during the consolidated sleep period.
The VLPO is a well-established sleep center that expresses c-Fos in sleep-active neurons.26,30,31 To validate the method, we used this area as a positive control. Our analysis revealed significantly more c-Fos positive neurons in animals with recovery sleep than those from sleep deprived animals (100.8 ± 17.5 versus 40.8 ± 8.2, unpaired Student t-test, P = 0.021), a result consistent with previous reports.26,30,31
We then applied this method to the BF to determine whether Kv2.2-GABAergic neurons express more c-Fos in the consolidated wake or sleep state. Cell-counting analysis showed that more Kv2.2 neurons were positive with c-Fos in the MCPO/ HDB of sleep deprived mice than those of sleep recovered animals (unpaired Student t-test, P = 0.0001, Figures 1A and 1B). These results strongly suggest that Kv2.2-GABAergic neurons are primarily ‘wake-active neurons.’
Enhanced c-FOS Expression in BF GABAergic neurons in Kv2.2 KO Mice
The result of the c-FOS experiment led us to the hypothesis that Kv2.2-expressing neurons regulate arousal. To test this, we used Kv2.2 KO mice12,18 to examine whether these mutant mice exhibit altered sleep-wake cycles. The removal or downregulation of the K+ channel is expected to augment the activity of Kv2.2-GABAergic neurons. We therefore predict that the sleep physiology is altered in Kv2.2 KO mice. Homozygous KO and WT littermates were obtained from heterozygous intercrosses. The Kv2.2 KO mice developed normally, where there were no apparent changes in general morphology, fertility, and locomotive behaviors (data not shown).
We then tested whether the removal of Kv2.2 changes the activity of these GABAergic neurons. Because we do not have an alternative marker for this novel population of BF GABAergic neurons, we were not able to target them for efficient electrophysiological recordings in Kv2.2 KO mice. Instead, we again used the expression of c-Fos as an indirect readout of their activity as a population.26 As Kv2.2 is expressed in about 60% of BF GABAergic neurons,12 the predicted augmentation of activity in the neurons, from which Kv2.2 is removed, would be represented as an increase in the number of c-Fos positive neurons in the entire GABAergic population in the MCPO/HDB. We used GAD67 transcripts as a marker of BF GABAergic neurons. Brain sections were prepared from naive Kv2.2 KO and WT mice during the light phase and subjected to in situ hybridization for GAD67. These sections were then immunostained for c-Fos. Our analysis showed that there was a significant increase in the number and the fraction of c-Fos positive GABAergic neurons in the MCPO/HDB of Kv2.2 KO mice than that obtained with WT mice (Figure 1C). There were no changes in the number of c-Fos positive cells without GAD67 (data not shown), indicating that the increase was specific to GABAergic neurons in the MCPO/HDB. We also found a similar increase in c-Fos in GABAergic neurons in the vertical limb of the diagonal band of Broca (14.5 ± 3.3 in WT versus 49.7 ± 7.1 cells in KO, P = 0.0061). It should also be noted that the expression level of GAD67 transcript was significantly increased in the MCPO of the KO mice (31 ± 3% increase in Kv2.2 KO mice, P = 0.0002) (Figure 1D), that is consistent with the activity-dependent expression of the gene.32–34
Kv2.2 KO Mice Exhibit Changes in Their Sleep-Wake Architecture
Given the increased expression of the activity marker c-Fos in BF GABAergic neurons, we next tested the prediction that Kv2.2 KO mice exhibit altered wakefulness by recording EEG/ EMG in freely-moving mice. Kv2.2 KO exhibited apparently normal EEG and EMG waveforms (Figure 2A), where the three vigilance states were readily identified, similar to WT mice. The recordings showed characteristic patterns of EEG/EMG for wake (low-amplitude, high-frequency EEG and high-amplitude EMG), NREM sleep (high-amplitude, low-frequency EEG and low-amplitude EMG) and REM sleep (low-amplitude, high-frequency EEG and muscle atonia, which is indicated by the brackets) (Figure 2A).
Figure 2.

Sleep-wake patterns of Kv2.2 knockout and wild-type mice. (A) Representative electroencephalographic (EEG) and electromyographic (EMG) recordings from wild-type (WT) and Kv2.2 knockout (KO) mice showing each vigilant state. Wake (low-amplitude, high- frequency EEG and high-amplitude EMG; left inset), nonrapid eye movement (non-REM) sleep (high-amplitude, low-frequency EEG and low-amplitude EMG; right inset), and rapid eye movement (REM) sleep (low-amplitude, high-frequency EEG and muscle atonia, brackets). Scales in the insets: 1 sec and 100 μV. (B) During the 12-h dark periods (illustrated by black bars on the x-axis) both groups have an increase in wakefulness (blue line) and a concurrent decrease in non-REM sleep (green line). Conversely, during the 12-h light period, both groups have an increase in non-REM sleep and a decline in wakefulness. There was a slight increase in REM sleep (red line) during the light period as well. Dotted lines represent standard error of the mean for each curve. (C) Average time in waking in each of the light cycles were obtained from B. (D) The ratio of time in waking in the dark period over that in the light period was computed from B for each genotype.
Baseline recordings of Kv2.2 KO and WT mice were carried out during a 48-h time period (days 1 and 2) to assess the overall pattern of sleep-wake cycle. Both the KO and WT mice exhibited normal diurnal sleep patterns (Figure 2B). During the 12-h dark period, the time spent in wake increased with a concomitant decrease in the time spent in sleep, particularly NREM sleep. Conversely, during the 12-h light period, the time spent in NREM sleep increased with a concomitant decrease in time spent awake. REM sleep was minor but also exhibited a slight increase during the light period in both WT and KO mice. Because Kv2.2 KO mice appeared to have a slight increase in the time spent in waking during the dark period (Figure 2B), we analyzed the average time in waking (Figure 2C) as well as the ratio of time in waking during the dark period over the light periods (Figure 2D). However, there were no significant differences detected in these analyses. The duration of each vigilant state was not significantly different (Figure 3), although there was a weak trend that the duration of REM sleep is decreased during the dark period in Kv2.2 KO mice (P = 0.13). These results indicate that the gross pattern of the sleep-wake cycle is not altered in Kv2.2 KO mice.
Figure 3.
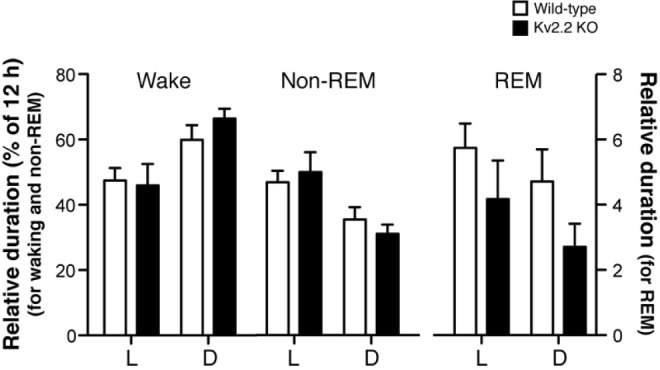
Duration of each vigilant state in wild-type and Kv2.2 knockout (KO) mice. Duration of each vigilant state during either the light or dark cycles is expressed as a percentage of the total duration (12 h). The left Y-axis is for waking and nonrapid eye movement (non-REM) sleep, and the right axis is for rapid eye movement (REM) sleep.
To investigate whether the architecture of the sleep-wake cycle is altered in Kv2.2 KO mice, we developed hypnograms from the baseline recordings (Figure 4A). Based on visual inspection of the hypnograms from the baseline recordings, the KO mice appeared to exhibit longer wake bouts compared to WT mice (Figure 4A). To quantify the difference, we first assessed whether KO mice spend more time in the wake state as compared to WT mice. The duration of every wake bout episode from all animals were plotted and analyzed in a cumulative probability plot (Figure 4B). The results showed that Kv2.2 KO mice have an increased number of longer wake bouts than WT, as evidenced by the rightward shift of the curve (P < 0.05, Kolmogorov-Smirnov test). The average bout duration also showed a slight increase (278 ± 49 sec in WT versus 471 ± 155 sec in KO in the light cycle, and 360 ± 71 sec in WT versus 641 ± 127 sec in KO in the dark cycle) with a trend (P = 0.07 for the dark cycle). We also compared the number of transitions from the wake state to any of the sleep states (REM and NREM) and found that Kv2.2 KO mice transitioned less frequently than their WT counterparts (Figure 4C), indicating that they tend to stay longer in the wake state. The difference was significant in the dark period (P = 0.02, unpaired Student t-test) with a positive trend in the light period (P = 0.05). The number of wake bouts during the dark period (72 ± 5 in WT versus 55 ± 6 in KO, P = 0.04), but not in the light period (81 ± 7 in WT and 62 ± 10 in Kv2.2 KO), was also statistically different. These results indicate that Kv2.2 KO mice exhibit an altered sleep-wake architecture.
Homeostasis of the Sleep-Wake Cycle is Altered in Kv2.2 KO Mice
The sleep-wake cycle reflects a balance of two opponent processes, the homeostatic drive and the circadian drive.35,36 Therefore, it is possible that the phenotype of Kv2.2 KO mice is derived from changes in either one or both of these factors. To test whether the homeostatic drive is affected in the KO animals, we assessed how they respond to changes in sleep homeostasis by mild sleep deprivation. Following the baseline recording in days 1 and 2, a 6-h sleep deprivation was given to the KO and WT animals during the light period of day 3 (starting at 07:00 when lights were turned on). Both WT and Kv2.2 KO mice went to sleep immediately after sleep deprivation to a similar extent (Figure 5A). Although both WT and Kv2.2 KO mice did not exhibit a robust recovery sleep response immediately after sleep deprivation, presumably because of the mildness of sleep deprivation, these results indicate that the homeostatic sleep drive is not largely altered in Kv2.2 KO mice.
Figure 5.
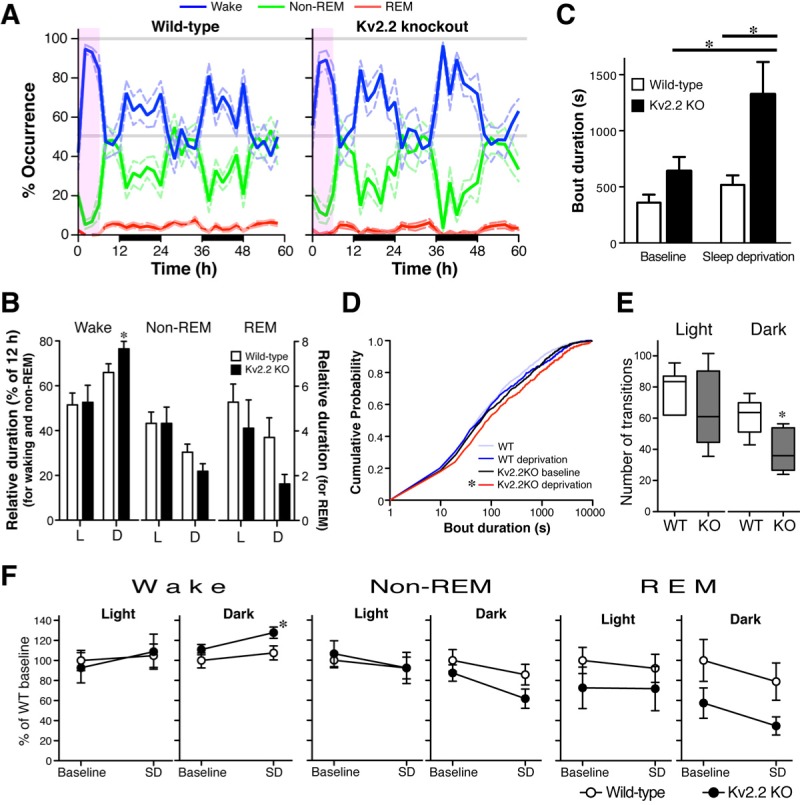
Exacerbated phenotype of Kv2.2 knockout (KO) mice following a mild sleep deprivation challenge. (A) During the next 2 full days following sleep deprivation, the sleep-wake patterns of the Kv2.2 KO mice changed, particularly with increased occurrence of wakefulness (blue line) during the dark periods (illustrated by black bars on the x-axis). The period of sleep deprivation is shown with a light purple box. (B) Duration of each vigilant state in wild-type (WT) and Kv2.2 KO mice. The data from days 4 and 5 were analyzed together for each light cycle (dark cycles, 12∼24 and 36∼48 h; light cycles, 24∼36 and 48∼60 h in A). Duration of each vigilant state during either the light or dark cycles is expressed as a percentage of the total duration (12 h). *Unpaired Student t-test, P < 0.05. (C) The average duration of wake bouts was increased from baseline in the Kv2.2 KO mice during the dark period (paired t-test; P = 0.03; n = 7). Following sleep deprivation, the average wake bout duration of Kv2.2 KO mice was significantly longer than that of WT littermates during the dark period (unpaired Student t-test; P = 0.01; n = 7). Average bout duration of wake episodes was calculated across a 2-h interval for each dark period for 48 h before and after the sleep-deprivation challenge. (D) Cumulative probability distribution of the duration of all the wake bout episodes during the dark period before and after sleep deprivation revealed a more dramatic rightward shift in the Kv2.2 KO mice. (Kolmogorov-Smirnov test; P < 0.05; n = 7.). (E) The average number of transitions from the wake state to any of the sleep states (nonrapid eye movement [non-REM] and rapid eye movement [REM]) during the light and dark periods after the sleep deprivation. Kv2.2 KO mice exhibited less transitions than WT mice in the dark state (unpaired Student t-test; P = 0.04; n = 7). (F) Relative changes in the duration of each vigilant state after the sleep deprivation (SD). Duration is expressed as a percentage to the baseline value of wild-type mice. *Unpaired Student t-test, P < 0.05.
However, after this recovery period, we observed a marked change in the maintenance of the wake state in the next 2 full days (day 4, 12-36 h; and day 5, 36-60 h in Figure 5A). Kv2.2 KO mice showed a significant increase (P = 0.02 with unpaired Student t-test) in the duration of wake in the dark periods (Figures 5B and 5F). There was also a reduction in the duration of REM sleep, but the difference was not statistically significant (P = 0.06). Consistently, the average duration of wake bouts in the KO mice became significantly longer following sleep deprivation compared to the baseline (P = 0.03 with unpaired Student t-test; P < 0.05 with two-way ANOVA and Bonferroni post hoc test) and significantly greater than WT mice following sleep deprivation (P = 0.01 with unpaired Student t-test; P < 0.01 with two-way ANOVA and Bonferroni post hoc test; Figure 5C). This also caused a further rightward shift in the cumulative probability plot in which we plotted the duration of each wake bout episodes from the KO mice before and after sleep deprivation (P < 0.05, Kolmogorov-Smirnov test between plots from the baseline and after sleep deprivation; Figure 5D). Consistently with the extended duration of wake bouts, the number of transitions from the wake state to any of the sleep states was also reduced in the dark period after sleep deprivation (Figure 5E). The difference was significant in the dark period (P = 0.02 with unpaired Student t-test; P < 0.05 with with two-way ANOVA and Bonferroni post hoc test) with a positive trend in the light period (P = 0.05 with unpaired Student t-test). Although the traditional homeostatic response to sleep deprivation was not largely altered in Kv2.2 KO mice, these changes in response to sleep deprivation indicate that the homeostatic regulation of the sleep-wake cycle is somewhat altered in Kv2.2 KO mice (see Discussion).
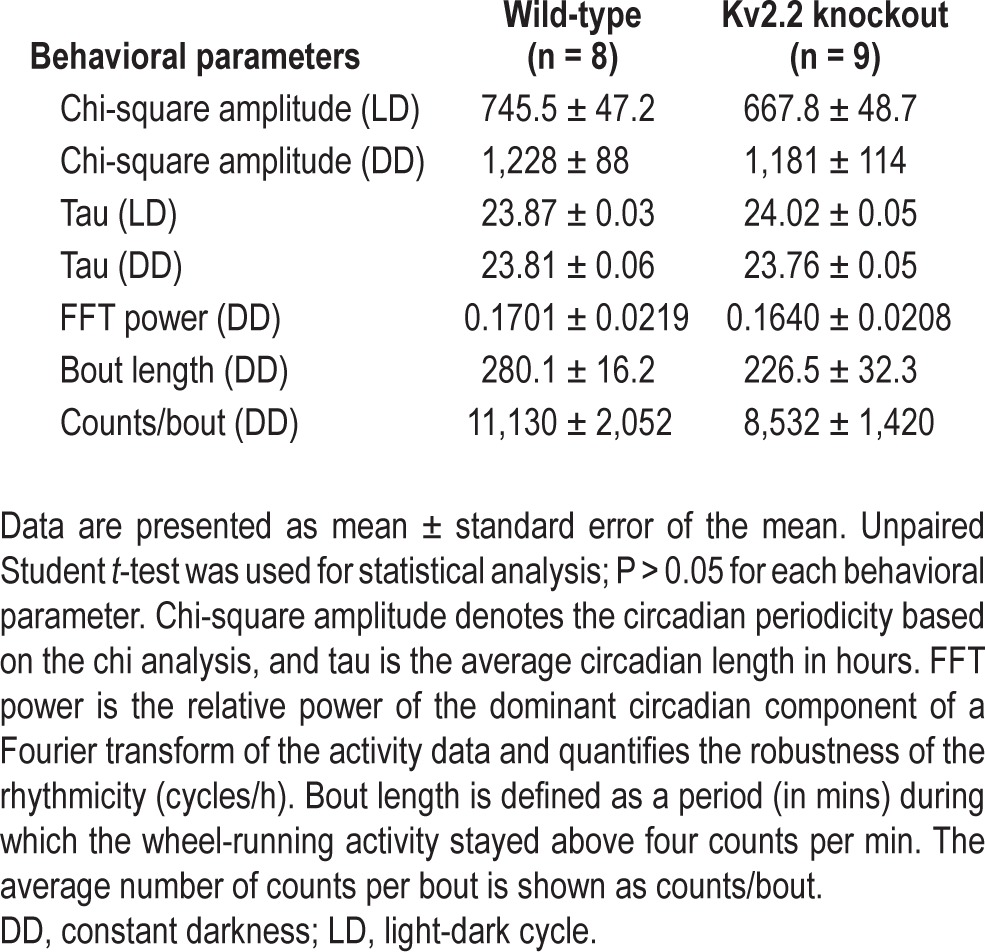
To test whether the circadian drive is affected in Kv2.2 KO mice, we monitored wheel-running activity to assess possible changes in the circadian rhythms. WT and Kv2.2 KO mice were subjected to a 12:12 h LD cycle for 7 days and released into DD for 14 days, during which their activity was recorded in actograms (Figure 6). Both genotypic groups exhibited similar behavioral patterns of consolidated locomotive activity during the active period of the LD cycle. During DD where intrinsic circadian regulation is assessed, normal free-running circadian rhythms were observed in both WT and KO mice. A chi-squared periodogram analysis revealed no statistical differences in the overall circadian amplitude during LD and DD conditions (Table 1). Therefore, the altered sleep-wake architecture does not result from changes in the overall circadian regulation.
Figure 6.
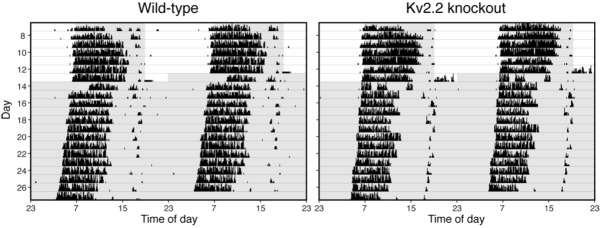
No changes in circadian rhythmicity as measured by wheel-running activity. Representative actograms from wild-type and Kv2.2 knockout mice for 7 days in light-dark (LD) conditions and 14 days in constant darkness (DD); the shaded region. Recordings following the sixth day are shown. These actograms are double-plotted, and the shaded areas represent the 12-h dark period during LD conditions and constant darkness during DD.
Table 1.
Circadian parameters of wild-type and Kv2.2 knockout mice
Altered Sleep EEG in Kv2.2 KO Mice
The observed increase in waking in Kv2.2 KO mice indicates that either sleep states may be disrupted or that the wake state may frequently override the sleep states in these animals. To obtain insight into how the behavioral phenotype of Kv2.2 KO arises, we analyzed cortical EEG signals in detail. First, 30 min worth of wake EEG and sleep EEG were extracted from 3-h recordings of EEG (Figure 7A), based on the total power of EEG (for sleep EEG) and EMG (for wake EEG), but not on the frequencies of EEG, using thresholding (see Methods). Because this procedure would ignore low-amplitude REM sleep EEG, sleep EEG is likely to represent NREM sleep EEG. Both WT and KO mice exhibited similarities in the overall power spectra (Figure 7B). There was a peak in the delta frequency range in the NREM sleep EEG. In contrast, in the wake EEG, there was a dramatic decrease in the power of the delta frequency signals with a concomitant increase in the power of the gamma frequency signals.
However, we noticed that the difference in the power spectra between NREM and wake EEGs was much smaller in KO mice as compared to WT littermates (Figure 7B). In fact, when we compared the relative power of the discrete frequency bands within the power spectra between the genotypic groups, there was a significant reduction in the delta power of the NREM sleep EEG in Kv2.2 KO mice (P < 0.05; repeated-measures two-way ANOVA; Figure 7B). The significant difference was observed only after sleep deprivation, but not in the baseline data (not shown). Also, there was no significant difference in the wake EEG either in the baseline or after sleep deprivation (Figure 7C). These results indicate that the behavioral phenotype of Kv2.2 KO mice after sleep deprivation may be due to altered activity patterns of cortical neurons during NREM sleep.
Expression of Parvalbumin in Kv2.2-GABAergic Neurons
As indicated in our previous study, Kv2.2-GABAergic neurons exhibit a similar morphology to that of the putative cortical projecting GABAergic neurons of the BF.2,12,37,38 Therefore, Kv2.2-GABAergic neurons may regulate the activity of cortical neurons via direct projection. To test this, we used a calcium-binding protein, parvalbumin, as a marker of projecting GABAergic neurons because of projecting GABAergic neurons in the BF have been shown to express parvalbumin.8 Immunohistochemical analysis (Figure 8) revealed that less than 4% of Kv2.2-GABAergic neurons were positive for parvalbumin (3.76 ± 0.41% of the Kv2.2-GABAergic neurons were parvalbumin-positive within the MCPO/HDB). These results indicate that Kv2.2-GABAergic neurons are not the known populations of projecting GABAergic neurons and may not regulate cortical activity directly.
Figure 8.
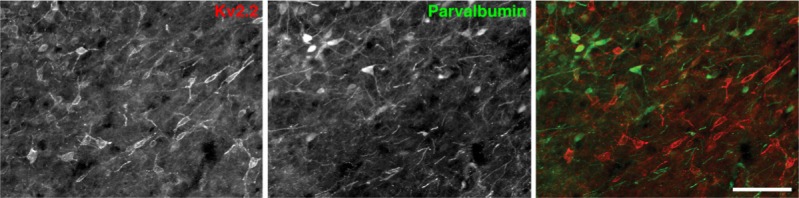
Lack of detectable immunolabeling of parvalbumin in Kv2.2-GABAergic neurons. Coronal sections were double immunolabeled with anti-Kv2.2 and antiparvalbumin antibodies. Scale bar, 100 μm.
DISCUSSION
In the current study, we demonstrated that Kv2.2-expressing GABAergic neurons in the BF express c-Fos specifically during the wake state, indicating that they are ‘wake-active neurons.’ This characteristic is similar to the population of GABAergic neurons that were also shown to be active during waking.10 This is also somewhat comparable to BF cholinergic neurons, which maximally discharge during wake, although we do not know whether Kv2.2-GABAergic neurons are also active in the REM state as are BF cholinergic neurons.10,39 Based on our finding and previous studies, we hypothesized that Kv2.2-expressing neurons regulate wakefulness and cortical activation. To test the hypothesis, we investigated the sleep-wake cycle of Kv2.2 KO mice.
Kv2.2 has been shown to be highly expressed in the medial nucleus of the trapezoid body neurons in addition to GABAergic neurons in the basal forebrain. It has been suggested that in these high-frequency firing neurons Kv2.2 contributes to maintain action potential amplitude by regulating the interspike potential and by relieving Na+ channels from inactivation.17 Therefore, removing Kv2.2 could potentially decrease neuronal firing by reducing the availability of Na+ channels. Conversely, blocking the Kv2.1 channel, which is very similar to Kv2.2 in biophysical properties, in hippocampal neurons has been shown to increase action potential firing.40 In the current study, we found that the BF GABAergic neurons exhibited augmented expression of c-Fos in Kv2.2 KO mice. Although c-Fos expression may reflect changes in Ca2+ and other signal messengers rather than changes in the firing rate per se, previous studies have shown that c-Fos expression is tightly correlated with action potential firing and synaptic activity.41,42 Therefore, we hypothesize that the removal of Kv2.2 from these particular neurons results in the augmentation of their activity.
We found that the architecture of the sleep-wake cycle is significantly altered in Kv2.2 KO mice, whereas these animals exhibited somewhat normal sleep-wake cycles. Particularly, these mice had an increased number of long wake bouts (> 100 sec) than WT littermates. This altered sleep-wake architecture was not attributable to changes in the circadian rhythm per se, as we did not detect any significant differences in the circadian activity of Kv2.2 KO mice. Because the sleep-wake cycle is still under the strong regulation of the circadian clock, this may explain why we observed the increased wakefulness preferentially during the dark period.
Then, what is affected in these animals? To address this question, we took advantage of the classic sleep deprivation, which has been used to assess the homeostatic aspect in the regulation of the sleep-wake cycle.43 If an animal (in normal conditions) were deprived of sleep for some period of time, there would be a subsequent increase in the amount of sleep due to an increase in the homeostatic drive. Therefore, analyzing the response to sleep deprivation would provide insight into whether and how the homeostatic regulation is altered. We did not find any significant differences in the recovery sleep between WT and Kv2.2 KO mice. This is presumably because, at least in part, the sleep deprivation challenge we used was mild and did not elicit a significant increase in recovery sleep as compared to baseline.
However, the sleep deprivation challenge caused a significant increase in the average length of wake bouts and the duration of the wake state during the dark period in Kv2.2 KO mice in the following 2 full days. This was not observed in WT animals, suggesting that there is a form of homeostasis that maintains the normal sleep-wake cycle following sleep deprivation. The increased maintenance of waking in Kv2.2 KO mice after sleep deprivation therefore indicates that this type of homeostasis is altered in these animals. Considering that Kv2.2-GABAergic neurons are active during waking and that the c-Fos expression is increased in the absence of Kv2.2, we speculate that their propensity of activation is augmented in Kv2.2 KO mice, particularly during the dark cycle. This presumably makes them prone to disturbances such as sleep deprivation, thereby resulting in the exacerbated maintenance of waking in the dark cycle after sleep deprivation.
To obtain further insight into how the changes in the sleep architechture occur, we performed in-depth analysis of cortical EEG signals from NREM sleep and wake states. The analysis revealed that EEG of NREM sleep is altered in Kv2.2 KO mice. Although it still exhibited the increased power in the delta frequency range, a characteristic of NREM sleep EEG, the amplitude of the delta power was significantly decreased as compared to the NREM sleep EEG of WT mice. Interestingly, the wake EEG was not significantly altered in the KO mice. Considering that Kv2.2-expressing neurons are almost strictly active in the wake state in WT mice, we speculate that the neurons lacking this somatic delayed rectifier become active even during periods of NREM sleep in KO mice. This might then affect the NREM sleep-EEG signals and interrupt the sleep state in Kv2.2 KO mice. We hope that we can address this finding in the near future, as we obtain a tool such as Kv2.2-GFP (green fluorescent protein) mice or an alternative marker for these GABAergic neurons for electrophysiology and c-Fos analysis.
The EEG signals we obtained should originate mainly from the ensemble activity of cortical neurons. Because Kv2.2 is expressed in a subset of cortical pyramidal neurons at very low levels,44 it is possible that the changes in cortical EEG arise from the absence of Kv2.2 in these neurons. However, considering the low-level expression of Kv2.2 compared to that of Kv2.1 in the cortical neurons12,44 and that changes were observed only in the delta frequency in the NREM sleep EEG rather than in the overall EEG signal in any vigilance state, it may be more reasonable to attribute the changes of EEG to a specific regulatory circuit to the cerebral cortex. As Kv2.2-GABAergic neurons do not have the immunochemical marker for the known populations of cortically projecting GABAergic neurons of the BF, we currently do not know how these neurons could affect the activity of cortical neurons. Studies have shown that the generation of delta oscillations in cortical EEG can occur within the cortex via a local circuit in the interplay between interneurons and pyramidal cells, or through the thalamocortical circuit.45 To address by which pathway these neurons affect the activity of cortical neurons in the sleep-wake cycle, extensive tract tracing studies of Kv2.2-GABAergic neurons in the future will be necessary.
In summary, we provide evidence that Kv2.2-expressing neurons, particularly those in the BF, are involved in the regulation of sleep-wake cycle in mice. Further studies aiming at the firing properties, innervation patterns, and hormonal regulation of these unique neurons would provide opportunities for the development of novel therapeutic treatments for sleep disorder.
DISCLOSURE STATEMENT
This was not an industry supported study. Dr. Mong contracted with Karo Bio (Novum S-141 57 Huddinge, Sweden) to conduct preclinical experiments on a compound at the University of Maryland School of Medicine. This study was unrelated to the experiments described in this manuscript. Support was limited to direct research expenses and provision of the compound to be tested. The authors do not presently, nor have they in the past, maintained any financial interest in Karo Bio. This research was supported by NSF IOS-0956237 and NHLBI R01-HL102758 to Dr. Meredith, and NHLBI R01-HL088088 to Dr. Mong. The other authors have indicated no financial conflict of interest.
ACKNOWLEDGMENTS
The authors thank Kaori Misono, Shawn Viechweg, Kazuko Mizutani, and Michael Lai for their technical assistance.
REFERENCES
- 1.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 2.Szymusiak R. Magnocellular nuclei of the basal forebrain: substrates of sleep and arousal regulation. Sleep. 1995;18:478–500. doi: 10.1093/sleep/18.6.478. [DOI] [PubMed] [Google Scholar]
- 3.Jones BE. From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci. 2005;26:578–86. doi: 10.1016/j.tips.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Kaur S, Junek A, Black MA, Semba K. Effects of ibotenate and 192IgG-saporin lesions of the nucleus basalis magnocellularis/substantia innominata on spontaneous sleep and wake states and on recovery sleep after sleep deprivation in rats. J Neurosci. 2008;28:491–504. doi: 10.1523/JNEUROSCI.1585-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuller PM, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol. 2011;519:933–56. doi: 10.1002/cne.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berntson GG, Shafi R, Sarter M. Specific contributions of the basal forebrain corticopetal cholinergic system to electroencephalographic activity and sleep/waking behaviour. Eur J Neurosci. 2002;16:2453–61. doi: 10.1046/j.1460-9568.2002.02310.x. [DOI] [PubMed] [Google Scholar]
- 7.Gritti I, Henny P, Galloni F, Mainville L, Mariotti M, Jones BE. Stereological estimates of the basal forebrain cell population in the rat, including neurons containing choline acetyltransferase, glutamic acid decarboxylase or phosphate-activated glutaminase and colocalizing vesicular glutamate transporters. Neuroscience. 2006;143:1051–64. doi: 10.1016/j.neuroscience.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gritti I, Manns ID, Mainville L, Jones BE. Parvalbumin, calbindin, or calretinin in cortically projecting and GABAergic, cholinergic, or glutamatergic basal forebrain neurons of the rat. J Comp Neurol. 2003;458:11–31. doi: 10.1002/cne.10505. [DOI] [PubMed] [Google Scholar]
- 9.Manns ID, Lee MG, Modirrousta M, Hou YP, Jones BE. Alpha 2 adrenergic receptors on GABAergic, putative sleep-promoting basal forebrain neurons. Eur J Neurosci. 2003;18:723–7. doi: 10.1046/j.1460-9568.2003.02788.x. [DOI] [PubMed] [Google Scholar]
- 10.Hassani OK, Lee MG, Henny P, Jones BE. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep-wake cycle. J Neurosci. 2009;29:11828–40. doi: 10.1523/JNEUROSCI.1259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manns ID, Alonso A, Jones BE. Discharge properties of juxtacellularly labeled and immunohistochemically identified cholinergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J Neurosci. 2000;20:1505–18. doi: 10.1523/JNEUROSCI.20-04-01505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermanstyne TO, Kihira Y, Misono K, Deitchler A, Yanagawa Y, Misonou H. Immunolocalization of the voltage-gated potassium channel Kv2.2 in GABAergic neurons in the basal forebrain of rats and mice. J Comp Neurol. 2010;518:4298–310. doi: 10.1002/cne.22457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan D, Tkatch T, Surmeier DJ, Armstrong WE, Foehring RC. Kv2 Subunits underlie slowly inactivating potassium current in neocortical pyramidal neurons. J Physiol. 2007;581:941–60. doi: 10.1113/jphysiol.2007.128454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci. 2005;25:9185–94. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–62. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- 16.Misonou H. Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist. 2010;16:51–64. doi: 10.1177/1073858409341085. [DOI] [PubMed] [Google Scholar]
- 17.Johnston J, Griffin SJ, Baker C, Skrzypiec A, Chernova T, Forsythe ID. Initial segment Kv2.2 channels mediate a slow delayed rectifier and maintain high frequency action potential firing in medial nucleus of the trapezoid body neurons. J Physiol. 2008;586:3493–509. doi: 10.1113/jphysiol.2008.153734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinert JR, Robinson SW, Tong H, Haustein MD, Kopp-Scheinpflug C, Forsythe ID. Nitric oxide is an activity-dependent regulator of target neuron intrinsic excitability. Neuron. 2011;71:291–305. doi: 10.1016/j.neuron.2011.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadjimarkou MM, Benham R, Schwarz JM, Holder MK, Mong JA. Estradiol suppresses rapid eye movement sleep and activation of sleep-active neurons in the ventrolateral preoptic area. Eur J Neurosci. 2008;27:1780–92. doi: 10.1111/j.1460-9568.2008.06142.x. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz MD, Mong JA. Estradiol suppresses recovery of REM sleep following sleep deprivation in ovariectomized female rats. Physiol Behav. 2011;104:962–71. doi: 10.1016/j.physbeh.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berghorn KA, Le WW, Sherman TG, Hoffman GE. Suckling stimulus suppresses messenger RNA for tyrosine hydroxylase in arcuate neurons during lactation. J Comp Neurol. 2001;438:423–32. doi: 10.1002/cne.1325. [DOI] [PubMed] [Google Scholar]
- 22.Koban M, Le WW, Hoffman GE. Changes in hypothalamic corticotropin-releasing hormone, neuropeptide Y, and proopiomelanocortin gene expression during chronic rapid eye movement sleep deprivation of rats. Endocrinology. 2006;147:421–31. doi: 10.1210/en.2005-0695. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman GE, Le WW, Sita LV. The importance of titrating antibodies for immunocytochemical methods. Curr Protoc Neurosci. 2008 doi: 10.1002/0471142301.ns0212s45. Chapter 2:Unit 2.12. [DOI] [PubMed] [Google Scholar]
- 24.Meredith AL, Wiler SW, Miller BH, et al. BK calcium-activated potassium channels regulate circadian behavioral rhythms and pacemaker output. Nat Neurosci. 2006;9:1041–9. doi: 10.1038/nn1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagar SM, Sharp FR, Curran T. Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science. 1988;240:1328–31. doi: 10.1126/science.3131879. [DOI] [PubMed] [Google Scholar]
- 26.Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–9. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- 27.Greco MA, Lu J, Wagner D, Shiromani PJ. c-Fos expression in the cholinergic basal forebrain after enforced wakefulness and recovery sleep. Neuroreport. 2000;11:437–40. doi: 10.1097/00001756-200002280-00002. [DOI] [PubMed] [Google Scholar]
- 28.Modirrousta M, Mainville L, Jones BE. Gabaergic neurons with alpha2-adrenergic receptors in basal forebrain and preoptic area express c-Fos during sleep. Neuroscience. 2004;129:803–10. doi: 10.1016/j.neuroscience.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 29.Curran T, Miller AD, Zokas L, Verma IM. Viral and cellular fos proteins: a comparative analysis. Cell. 1984;36:259–68. doi: 10.1016/0092-8674(84)90219-8. [DOI] [PubMed] [Google Scholar]
- 30.Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998;803:178–88. doi: 10.1016/s0006-8993(98)00631-3. [DOI] [PubMed] [Google Scholar]
- 31.Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–31. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- 32.Patz S, Wirth MJ, Gorba T, Klostermann O, Wahle P. Neuronal activity and neurotrophic factors regulate GAD-65/67 mRNA and protein expression in organotypic cultures of rat visual cortex. Eur J Neurosci. 2003;18:1–12. doi: 10.1046/j.1460-9568.2003.02702.x. [DOI] [PubMed] [Google Scholar]
- 33.Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- 34.Akhtar ND, Land PW. Activity-dependent regulation of glutamic acid decarboxylase in the rat barrel cortex: effects of neonatal versus adult sensory deprivation. J Comp Neurol. 1991;307:200–13. doi: 10.1002/cne.903070204. [DOI] [PubMed] [Google Scholar]
- 35.Daan S, Beersma DG, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–83. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 36.Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saper CB, Akil H, Watson SJ. Lateral hypothalamic innervation of the cerebral cortex: immunoreactive staining for a peptide resembling but immunochemically distinct from pituitary/arcuate alpha-melanocyte stimulating hormone. Brain Res Bull. 1986;16:107–20. doi: 10.1016/0361-9230(86)90018-3. [DOI] [PubMed] [Google Scholar]
- 38.Gritti I, Mainville L, Mancia M, Jones BE. GABAergic and other noncholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J Comp Neurol. 1997;383:163–77. [PubMed] [Google Scholar]
- 39.McKenna JT, Cordeira JW, Jeffrey BA, et al. c-Fos protein expression is increased in cholinergic neurons of the rodent basal forebrain during spontaneous and induced wakefulness. Brain Res Bull. 2009;80:382–8. doi: 10.1016/j.brainresbull.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luckman SM, Dyball RE, Leng G. Induction of c-fos expression in hypothalamic magnocellular neurons requires synaptic activation and not simply increased spike activity. J Neurosci. 1994;14:4825–30. doi: 10.1523/JNEUROSCI.14-08-04825.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fields RD, Eshete F, Stevens B, Itoh K. Action potential-dependent regulation of gene expression: temporal specificity in ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci. 1997;17:7252–66. doi: 10.1523/JNEUROSCI.17-19-07252.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huber R, Deboer T, Tobler I. Effects of sleep deprivation on sleep and sleep EEG in three mouse strains: empirical data and simulations. Brain Res. 2000;857:8–19. doi: 10.1016/s0006-8993(99)02248-9. [DOI] [PubMed] [Google Scholar]
- 44.Kihira Y, Hermanstyne TO, Misonou H. Formation of heteromeric Kv2 channels in mammalian brain neurons. J Biol Chem. 2010;285:15048–55. doi: 10.1074/jbc.M109.074260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steriade M. The corticothalamic system in sleep. Front Biosci. 2003;8:d878–99. doi: 10.2741/1043. [DOI] [PubMed] [Google Scholar]