

Abstract
Estrogen is an important regulator of dermal fibroblast functions, including extracellular matrix (ECM) synthesis. Estrogen mediates its effects through estrogen receptors (ERs), ERα and ERβ; however, regulation of ERs in dermal fibroblasts remains poorly understood. Friend leukemia integration factor 1 (Fli1), a member of the Ets transcription factor family, has been shown to play a pivotal role in regulation of the ECM genes in dermal fibroblasts. The aim of this study was to examine a possible interaction between Fli1 and estrogen pathways, focusing on ERα. We show that treatment of human dermal fibroblasts with transforming growth factor-β (TGF-β) increases ERα protein and mRNA levels. Similarly, ERα expression was increased in response to small interfering RNA (siRNA)-mediated depletion of Fli1, suggesting that Fli1 is a mediator of the TGF-β effects on ERα expression. Accordingly, we showed that Fli1 binds to the most proximal region of the ERα promoter, and dissociates from the promoter upon TGF-β treatment. An inverse correlation between Fli1 and ERα expression levels was confirmed in cultured skin fibroblasts obtained from Fli1+/− mice and in the skin of Fli1+/− mice in vivo. This study supports a role of Fli1 as a negative regulator of the ERα gene in dermal fibroblasts.
INTRODUCTION
Friend leukemia integration factor 1 (Fli1) is a member of the Ets transcription factor family characterized by the presence of the evolutionarily conserved DNA-binding domain (ETS domain) that recognizes the purine-rich GGA(A/T) core sequence (Ets-binding site (EBS)). Fli1 is highly expressed in hematopoietic cell lineages and in vascular endothelial cells (Hollenhorst et al., 2004). It plays an important role in megakaryocytic differentiation (Spyropoulos et al., 2000) and in vascular homeostasis (Asano et al., 2010). Specifically, in endothelial cells Fli1 regulates genes involved in vessel maturation and stabilization (Asano et al., 2010). Although Fli1 expression in dermal fibroblasts is relatively low, recent studies have shown that Fli1 plays a pivotal role in the regulation of the extracellular matrix (ECM) genes, including type I collagen (Czuwara-Ladykowska et al., 2001; Kubo et al., 2003; Asano et al., 2009), tenascin-C (Shirasaki et al., 1999), matrix metalloproteinase 1 (MMP-1) (Jinnin et al., 2005), and the multifunctional matricellular factor CTGF/CCN2 (Nakerakanti et al., 2006). Transforming growth factor-β (TGF-β) regulates transcriptional activity of Fli1 through post-translational modifications, including protein kinase C-δ (PKC-δ)-dependent phosphorylation and subsequent p300-CBP associated factor-dependent acetylation (Asano et al., 2007; Asano and Trojanowska, 2009).
Estrogen affects many aspects of skin physiology. It increases skin thickness and collagen content (Varila et al., 1995; Rittie et al., 2008), decreases fine wrinkles (Creidi et al., 1994), accelerates wound healing (Ashcroft et al., 1999), and changes the quality of scarring (Ashcroft et al., 1997). Estrogen exerts its actions through the well-characterized estrogen receptors (ERs), ERα and ERβ; however, their specific roles in the skin have not been fully defined. Furthermore, the expression profile of the two ERs varies according to location and tissue type, suggesting that each has different, cell-specific roles (Hall and Phillips, 2005). ERs function as ligand-inducible transcription factors, and ligand-bound ERs can bind directly to estrogen response elements in the promoters of target genes, or they can interact with other transcription factor complexes like Jun/Fos (Kushner et al., 2000) or specificity protein 1 (Saville et al., 2000) and influence transcription of genes whose promoters do not harbor estrogen response elements.
Previous studies using various experimental models have shown the inhibitory effects of estrogen on TGF-β signaling through direct interaction between Smad2/3 and ERα (Matsuda et al., 2001; Ito et al. 2010). Because our recent studies have demonstrated the importance of Fli1 in the TGF-β signaling in dermal fibroblasts, in this study we wished to examine the potential relationship between the TGF-β/Fli1 and the estrogen pathways focusing on ERα. We found that Fli1 functions as a negative transcriptional regulator of the ERα gene in dermal fibroblasts both in vivo and in vitro, suggesting that some of the pathogenic effects of Fli1 downregulation in cutaneous fibrosis and chronic wound healing (Kubo et al., 2003; Sakthianandeswaren et al., 2010) could be mediated by the upregulation of estrogen/ERα signaling.
RESULTS
TGF-β upregulates ERα expression in human dermal fibroblasts via Fli1-mediated pathway
The presence of both ER isoforms has previously been documented in dermal fibroblasts in vitro and in the skin in vivo (Thornton et al., 2003; Haczynski et al., 2004; Pelletier and Ren, 2004; Yoo et al., 2007). As according to a recent study ERα represents a predominant isoform expressed in human dermal fibroblasts (Rittie et al., 2008), we focused on this isoform and first confirmed its expression by reverse transcription-PCR (RT-PCR) (Figure 1a) and western blot (Figure 1b). MCF-7 breast cancer cells, which express full-length ERα, as well as 46 and 36 kDa splice variants (Flouriot et al., 2000; Wang et al., 2005), were used as a positive control. As shown in Figure 1b, dermal fibroblasts express full-length and a 46 kDa isoform of ERα, although at much lower level than MCF-7.
Figure 1. Estrogen receptor α (ERα) mRNA and protein expression is upregulated by transforming growth factor-β (TGF-β) in human dermal fibroblasts.

(a) Reverse transcription-PCR (RT-PCR) analysis of ERα in fibroblasts. (b) Western blot analysis of ERα protein in fibroblasts (50 μg) and MCF-7 cells (30 μg). The blots were reprobed with anti-β-actin antibody. (c) Western blot of ERα protein determined in serum-starved dermal fibroblasts treated with transforming growth factor-β1 (TGF-β1; 2 ng ml−1) for 24 hours. Representative data of three independent experiments are shown with quantitative representation obtained by densitometric analysis (lower left panel). ERα mRNA levels were determined by quantitative RT-PCR (lower right panel). Values are normalized relative to control (arbitrarily set as 1). Means±SD of three independent experiments is shown. *P<0.05 (Student's t-test) versus control.
We next examined the effect of TGF-β1 on expression of ERα in human dermal fibroblasts by western blot and quantitative real-time RT-PCR. TGF-β1 consistently increased the levels of ERα protein and mRNA 24 hours after stimulation (Figure 1c), which correlated with an increase in COL1A1 (collagen, type I, α1) mRNA expression (data not shown).
As Fli1 mediates some of the TGF-β effects in dermal fibroblasts, we examined whether Fli1 also contributes to the ERα expression by depleting Fli1 using small interfering RNA (siRNA). Treatment of fibroblasts with 10 nM of Fli1 siRNA for 72 hours resulted in a reduction of Fli1 mRNA and protein levels by ~90% (Figure 2a). As shown in Figure 2b, depletion of Fli1 resulted in upregulation of both ERα protein (1.82-fold) and mRNA (1.44-fold). The increase was comparable with the TGF-β1-induced ERα levels. These observations suggest that Fli1 might be a primary regulator of ERα expression downstream of TGF-β signaling in human dermal fibroblasts.
Figure 2. Friend leukemia integration factor 1 (Fli1) downregulation induces estrogen receptor α (ERα) expression.

(a) Fli1 protein and mRNA levels and (b) ERα protein and mRNA levels after small interfering RNA (siRNA)-mediated Fli1 depletion in dermal fibroblasts. Cells were transfected with either 10 nm of Fli1 siRNA or corresponding concentration of control siRNA. After 48 hours, cells were serum-starved to remove the effect of serum. Cells were harvested 72 hours after transfection. Protein levels of Fli1 and ERα were determined by western blot. The blots were reprobed with anti-β-actin antibody, and representative data of three independent experiments are shown. mRNA levels of Fli1 and ERα were determined by quantitative RT-PCR. Means±SD of three independent experiments is shown, with values normalized to control (arbitrarily set at 1). *P<0.01 (Student's t-test) versus control.
Fli1 interacts with the ERα promoter in vivo
Transcription of ERα is directed by at least seven different promoters, resulting in mRNAs that differ in 5′ untranslated regions (Figure 3a) (Kos et al., 2001). As promoter usage varies among tissues and cell types (Reid et al., 2002), we examined the promoter usage in dermal fibroblasts. For this experiment, forward primers specific for the 5′ untranslated region of the ERα mRNA produced from each promoter were coupled with a common reverse primer directed toward exon 1 for RT-PCR analyses. As shown in Figure 3b, dermal fibroblasts primarily utilized promoter A; however, all of the promoters were employed to different extents, consistent with previous report in MCF-7 cells (Murphy et al., 2009). Promoter A is the most proximal promoter of the human ERα gene (Murphy et al., 2009). Analysis of the 1,300 bp sequence, which includes both promoters A and B, by Tfsitescan (www.ifti.org) program revealed four potential EBSs (Figure 3c). To determine if Fli1 interacts with the ERα promoter region, we used the chromatin immunoprecipitation (ChIP) assay and the DNA affinity precipitation assay. Crosslinked chromatin was immunoprecipitated with an antibody to Fli1, and the purified genomic DNA was amplified with three sets of primers specific for each putative binding site within promoter A (Figure 3c). As shown in Figure 3d, Fli1 occupies the −681 to −523 and the −458 to −298 regions, but not the −1,300 to −1,090 region of promoter A in the absence of TGF-β1 stimulation. Upon TGF-β1 stimulation, Fli1 binding to these regions remained unchanged at 3 hours, but was no longer detectable at 24 hours (Figure 3d). ChIP without antibody did not yield a significant amount of bound DNA.
Figure 3. Friend leukemia integration factor 1 (Fli1) occupies the estrogen receptor α (ERα) promoter.

(a) ERα promoter (Kos et al., 2001). (b) ERα promoter utilization in dermal fibroblasts. Reverse transcription-PCR (RT-PCR) was performed using forward primers specific for each upstream exon and a reverse primer specific for exon 1. (c) ERα promoter A; Ets binding sites (closed ovals). (d) Chromatin immunoprecipitation (ChIP) assay of Fli1 occupancy of the promoter A. (e) Fli1 binding to the Ets sites in the ERα promoter using 5′ end-labeled wild-type (WT) and mutated at the Ets site (MUT) oligonucleotides. (f) Co-transfections of ERα-Luc with empty or Fli1 expression vector in fibroblasts with or without transforming growth factor-β (TGF-β) stimulation (Tb), normalized using pSV-β-galactosidase. The values depicted are means±SE of four experiments, *P<0.05.
The results of the ChIP analyses were further validated by DNA affinity precipitation assay. Biotinylated oligonucleotide sequences representing putative EBSs in the proximal ERα promoter were used in binding reactions with cell extracts enriched with Fli1 protein, which were obtained from cells with ectopic Fli1 expression. As presented in Figure 3e, and consistent with ChIP analysis, we observed strong and specific Fli1 binding to the −375 site. The −681 to −523 region, which showed a weak binding in the ChIP assay, encompasses two potential EBSs. Fli1 showed specific binding to the −577 site, but not the −630 site. A weak binding was also observed to the −1,223 site. Taken together, these data suggest that the −375 site in the proximal ERα promoter may represent a principal response element for Fli1 in this promoter region.
To study the functional role of Fli1 in regulating ERα promoter activity, we performed co-transfections of Fli1 and the 3,000 bp fragment of the ERα promoter linked to the luciferase reporter gene. As shown in Figure 3f, overexpression of Fli1 abrogated ERα promoter activity by 50%, thus supporting the role of Fli1 as a negative regulator of the ERα gene. Consistent with the mRNA data, TGF-β treatment enhanced ERα promoter activity in the presence of control vector. However, unexpectedly, overexpression of Fli1 enhanced the effects of TGF-β, suggesting that the additional indirect effects of Fli1 overexpression may influence the effects of TGF-β in this assay.
Expression of ERα is elevated in Fli1+/− mouse fibroblasts
To further investigate the role of Fli1 in the ERα expression, we used mice carrying a targeted disruption of Fli1. As Fli1−/− mouse embryos die by day 12.5 (Hart et al., 2000; Spyropoulos et al., 2000), Fli1+/− mice were used. First, dermal fibroblasts were isolated from female Fli1+/+ and Fli1+/− mice, and the effect of Fli1 downregulation on ERα expression in cultured dermal fibroblasts was determined by western blot and quantitative RT-PCR analyses. As expected, the levels of Fli1 protein and mRNA in fibroblasts from Fli1+/− mice were reduced by ~50% relative to fibroblasts from Fli1+/+ mice (Figure 4a), whereas ERα expression was upregulated at both the protein (2.5-fold) and mRNA (1.9-fold) levels in Fli1+/− fibroblasts (Figure 4b). These data are consistent with the results obtained from Fli1 siRNA-treated human dermal fibroblasts, indicating an inverse relationship between expression of Fli1 and ERα in dermal fibroblasts.
Figure 4. Estrogen receptor α (ERα) levels are elevated in Fli1+/− mouse fibroblasts and in the skin in vivo.
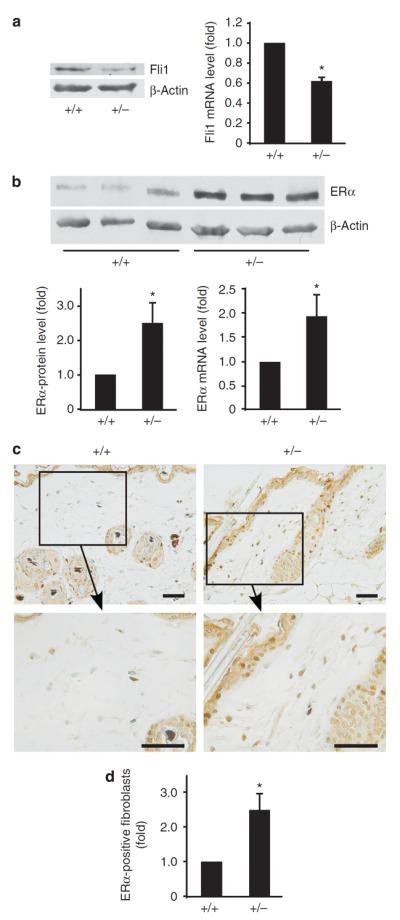
Protein and mRNA levels of (a) friend leukemia integration factor 1 (Fli1) and (b) ERα in dermal fibroblasts from female Fli1+/+ and Fli1+/− mice. The value was normalized relative to that of Fli1+/+ mouse fibroblasts (arbitrarily set as 1) and means±SD of three experiments are shown. *P<0.05. (c) Immunodetection of ERα in skin samples from Fli1+/+ and Fli1+/− mice. Original magnification is 200-fold (upper panels) and 400-fold (lower panels). Scale bar=0.02 mm. (b) The relative proportion of ERα-positive fibroblasts in dermis. At least 100 fibroblasts were counted for each specimen. The value was normalized relative to that of Fli1+/+ mouse skin (arbitrarily set as 1) and means±SD of three mice are shown. *P<0.05.
We next examined the expression pattern of ERα in mouse skin by immunohistochemistry. Sections from dorsal skin of female Fli1+/+ and Fli1+/− mice were immunostained with a specific antibody against mouse ERα. As reported previously (Thornton et al., 2003; Pelletier and Ren, 2004), high expression of ERα protein was detected in the sebaceous gland of skin from both Fli1+/+ and Fli1+/− mice (Figure 4c). Relatively high expression of ERα was also detected in the epidermis and hair follicles. Significant difference in ERα expression pattern was observed in the dermis. In Fli1+/− mice, the majority of dermal cells with fibroblastic morphology expressed ERα, whereas only moderate expression of ERα was observed in the dermis of Fli1+/+ mice (Figure 4c). To quantify these differences, ERα-positive fibroblasts were counted in the dermis of Fli1+/+ and Fli1+/− mice. The relative proportion of positive ERα fibroblasts was 2.5-fold higher in Fli1+/− mice than in Fli1+/+ mice (Figure 4d). These results support the conclusion that Fli1 is a functional negative regulator of ERα gene in dermal fibroblasts.
To determine whether changes in ERα expression levels have functional consequences with respect to matrix synthesis, collagen mRNA and protein levels were examined in Fli1+/+ and Fli1+/− fibroblasts after treatment with the ERα activator, 17β-estradiol, or the ERα inhibitor, tamoxifen. As shown in Figure 5, treatment with tamoxifen decreased basal COL1A1 mRNA and protein levels, suggesting that ER signaling contributes to collagen expression and synthesis in dermal fibroblasts. In agreement with previous reports, Fli1+/− fibroblasts produced elevated levels of collagen (Kubo et al., 2003). Furthermore, fibroblasts obtained from Fli1+/− mice showed more pronounced responses to both tamoxifen and 17β-estradiol. For example, tamoxifen reduced basal levels of COL1A1 mRNA by ~15% in wild-type (WT) male fibroblasts versus ~31% in Fli1+/− male fibroblasts (Figure 5a). The response to tamoxifen was greater in female fibroblasts with ~39% reduction in WT fibroblasts and ~54% reduction in Fli1+/− fibroblasts (Figure 5b). On the other hand, response to 17β-estradiol was in general stronger in male fibroblasts with ~66% increase in Col1A1 in WT fibroblasts and ~112% increase in Fli1+/− fibroblasts (Figure 5a). In WT female fibroblasts, the increase of COL1A1 mRNA was ~83 versus ~49% in Fli1+/− fibroblasts (Figure 5b). Similar responses were also observed at the collagen protein level. Together, these experiments support a profibrotic role for ERα in dermal fibroblasts.
Figure 5. Fli1+/− fibroblasts show enhanced fibrotic response to tamoxifen and 17β-estradiol stimulation.
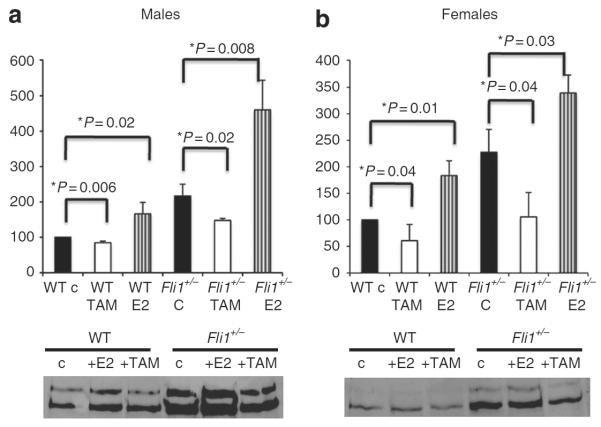
Dermal fibroblasts from (a) male and (b) female Fli1+/+ and Fli1+/− mice were stimulated for 24 hours with 17β-estradiol (10−10 m) (E2) or tamoxifen (10−7 m) (TAM). Collagen α1(I) mRNA levels were determined by quantitative reverse transcription-PCR (qRT-PCR). Means±SD of three independent experiments is shown, with values normalized to control (arbitrarily set at 100). Representative western blot of collagen protein levels in the conditioned media is shown in the bottom panels. WT, wild type.
DISCUSSION
Fli1 has emerged as an important regulator of ECM synthesis in the skin (Asano et al., 2009) and other organs (Elkareh et al., 2009). Although it is well established that Fli1 is a transcriptional repressor of the interstitional collagen genes, other functions of Fli1 in dermal fibroblasts have not been fully examined. Herein, we show that in human dermal fibroblasts Fli1 works as a transcriptional repressor of the ERα gene via binding to the proximal region of the ERα promoter. In response to TGF-β, Fli1 dissociates from the ERα promoter, suggesting that analogous to the collagen gene, ERα gene expression might be regulated through the TGF-β/PKC-δ/Fli1 pathway (Asano and Trojanowska, 2009). Relevant to our findings, upregulation of PKC-δ was shown to contribute to the antiestrogen resistance in mammary tumor cells, and PKC-δ was shown to be involved in activation and nuclear translocation of ERα in those cells (De Servi et al., 2005; Nabha et al., 2005). Additionally, our study demonstrates that reduced Fli1 expression correlates with elevated expression of ERα in mouse dermal fibroblasts in vitro and in vivo. Interestingly, the effects of Fli1 downregulation on the ERα expression were more pronounced in vivo than in cultured fibroblasts, suggesting that additional indirect effects of Fli1 on ERα expression levels may contribute to these effects, as well.
There is a complex cross-talk between the estrogen/ERα and TGF-β pathways. In many cell types, estrogen/ERα potently inhibits Smad2/3, as well as Smad1 signaling through the molecular interactions between Smads and the ERα protein (Matsuda et al., 2001; Yamamoto et al., 2002; Ito et al., 2010), whereas a positive effect on Smad signaling was observed in prolactin-producing cells (Giacomini et al., 2009). Less is known about the reciprocal effects of TGF-β on estrogen signaling, but an enhancement of ER-mediated transcription by TGF-β was described in mesangial cells (Matsuda et al., 2001). In this study we observed upregulation of ERα in response to TGF-β, suggesting that signaling through this receptor might be involved in the downstream effects of TGF-β, including ECM production in dermal fibroblasts. However, in spite of a large number of studies documenting stimulatory effects of topical estrogen on collagen accumulation in the skin (Rittie et al., 2008), the mechanistic aspects of collagen gene regulation by estrogen remain poorly understood. Estrogen was also shown to induce collagen and other matrix proteins in cultured dermal fibroblasts (Surazynski et al., 2003; Soldano et al., 2010). Interestingly, the stimulatory effects of estrogen on ECM synthesis were more pronounced in fibroblasts obtained from the skin of patients with scleroderma (Soldano et al., 2010). This may be a result of an augmented PKC-δ/Fli1 (Jimenez et al., 2001; Asano and Trojanowska, 2009) signaling present in scleroderma fibroblasts. Although further studies are needed to confirm this observation and to further explore enhanced sensitivity of scleroderma fibroblasts to estrogen stimulation, these observations suggest that estrogen may enhance the fibrotic process in scleroderma possibly as a part of an activated TGF-β/PKC-δ/Fli1 axis.
MATERIALS AND METHODS
Reagents
Recombinant human TGF-β1 was obtained from Peprotech (Locky Hill, NJ). Tamoxifen and E2-17β-estradiol were purchased from EMD4Biosciences (Gibbstown, NJ). The polyclonal rabbit anti-Fli1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA); goat anti-type I collagen was from Southern Biotech (Birmingham, AL).
Cell culture
Human dermal fibroblast culture was established from skin biopsies from dorsal forearm of healthy donors, upon informed consent and in compliance with the institutional review board for human studies. The study was conducted according to the Declaration of Helsinki Principles. All healthy donors were adult young women (range 19–34 years old) to exclude differences based on sex. Human and mouse skin tissue was dissociated enzymatically by 0.25% collagenase (Sigma, St Louis, MO) and 0.05% DNase (Sigma) in DMEM (Gibco BRL, Grand Island, NY) with 20% fetal bovine serum (HyClone, Logan, UT). Digested tissue was placed in a six-well plate in 2 ml of DMEM with 10% fetal bovine serum and grown for 3–5 days. The resulting confluent culture was subsequently passaged in DMEM with 10% fetal bovine serum.
Western blot analysis
Cells were lysed in Lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 3 mm MgCl2, 1 mm CaCl2, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride (EMD, Gibbstown, NJ), and Protease inhibitor cocktail set III, EDTA free (EMD)). Protein concentration was determined by BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Equal amounts (50 μg) of total protein samples from dermal fibroblasts were separated via 10% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated with Tris-buffered saline containing 3% non-fat dry milk (Bio-Rad, Hercules, CA) for 1 hour at room temperature, followed by incubation with rabbit anti-ERα antibody (G-20, Santa Cruz Biotechnology) for detection of human ERα and MC-20 (Santa Cruz) for detection of mouse ERα in a dilution of 1:500 or rabbit anti-Fli1 antibody in a dilution of 1:2,000 overnight at 4 °C. To control for protein loading, blots were probed for β-actin expression using monoclonal anti-β-actin antibody (Sigma). After incubation with horseradish peroxidase-conjugated secondary antibody (GE Health-care, Piscataway, NJ) in a dilution of 1:5,000, the signals were visualized using enhanced chemiluminescence reagents (Thermo Scientific).
Quantitative RT-PCR analysis
Total RNA was isolated using Tri reagent (MRC, Cincinnati, OH). Then, 2 μg of total RNA was reverse transcribed with random hexamers using a Transcriptor First Strand complementary DNA Synthesis kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer's protocol. Real-time PCR assays were performed using the StepOnePlus Real-Time PCR system (Applied Biosystems, Foster City, CA). The amplification mixture (10 μl) contained 1 μl of complementary DNA, 0.5 μm of each primer, and 5 μl of SYBR Green PCR Master Mix. The primers are listed in the Supplementary Table S1 online. To quantify mRNA expression of human ERα, real-time TaqMan PCR was performed. Validated primer/probe set directed toward the exon 3–4 boundaries (Hs00174860_m1; Applied Biosystems) was used to amplify the target gene. Complementary DNA (2 μl) was added to 10 μl of TaqMan Gene Expression Master Mix in the amplification mixture (20 μl). All samples were analyzed in parallel for gene expressions of either human glyceraldehyde-3-phosphate dehydrogenase or mouse β2-microglobin as an internal control by SYBR Green method. The primers are listed in the Supplementary Table S1 online. Cycling condition consisted of an initial incubation at 50 °C for 2 minutes and 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds and 58 °C for 1 minute. The relative change in the levels of genes of interest was determined by the 2−ΔΔCT method.
Suppression of Fli1 by siRNA
Cells were transfected with either siRNA specific to human Fli1 (ON-TARGETplus SMART pool; Thermo scientific) or negative control siRNA (Qiagen, Valencia, CA) at the concentration of 10 nm using HiPerfect reagent (Qiagen) according to the manufacturer's protocol. After 72 hours, total protein and RNA were extracted. To remove the effect of serum, cells were serum-starved for the last 24 hours.
RT-PCR analysis
For analysis of ERα transcripts, PCR was performed with Taq DNA polymerase (New England Biolabs, Ipswich, MA) using the primers specific to human ERα (Figure 1a) or exon-specific primers (Figure 3b). Reaction without reverse transcription served as a control for genomic DNA contamination. The primers specific to human ERα are listed in the Supplementary Table S2A online. As the primer set is directed toward exon 1–2 boundaries and the splicing variants of human ERα (46 and 36 kDa isoforms) lack exon 1, mRNAs of the splicing variants should not be detected in this system. The forward primers for the ERα mRNAs produced by transcription from promoters A, B, C, D, E2, and F are listed in the Supplementary Table S2B online. The reverse primer (Supplementary Table S2B online) was from exon 1. Cycling conditions were as follows: an initial 5 minute denaturation at 95 °C followed by 45 cycles consisting of 30 seconds at 95 °C, 30 seconds at 57 °C, and 1 minute at 72 °C. A final extension for 10 minutes at 72 °C concluded the reactions.
ChIP assay
ChIP assay was carried out as described previously (Nakerakanti et al., 2006). Briefly, cells were treated with 1% formaldehyde (Sigma) for 10 minutes. The crosslinked chromatin was then collected and sheared by sonication to yield an average size of 300–500 bp. The DNA fragments were immunoprecipitated overnight with or without polyclonal anti-Fli1 antibody at 4 °C. After reversal of crosslinking, the immunoprecipitated chromatin was amplified by regular PCR. The primers are listed in the Supplementary Table S3 online. The amplified DNA products were resolved by agarose gel electrophoresis.
DNA affinity precipitation assay
The assay was carried out as described previously (Pannu et al., 2008). Briefly, cell extracts were prepared in RIPA buffer from confluent dishes of Hek293T cells transduced with Fli1 adenovirus at the multiplicity of infection of 5 for 24 hours. The extracts were precleared with streptavidin-coated agarose beads (Invitrogen, Carlsbad, CA), and the supernatants were incubated with 500 pmol of 5′ biotinylated double-stranded oligonucleotide, either WT or mutated (at EBS) (MUT) ERα promoter. After overnight incubation at 4 °C with constant rotation, streptavidin-coated agarose beads were added to each tube, and the samples were rotated for an additional 2 hours at 4 °C. Beads were recovered by centrifugation and washed three times with Tris–EDTA followed by two washes with RIPA buffer and two washes with 1 × phosphate-buffered saline. The beads were suspended in 2 × sample loading buffer at 95 °C for 5 minutes. The supernatants were separated by 10% SDS-PAGE and transferred onto nitrocellulose membrane. Western blotting was performed with antibody against Fli1 (Invitrogen), and the bands were visualized using enhanced chemiluminescence reagent (Pierce, Rockford, IL). The sequences of the forward and reverse oligonucleotides are shown in the Supplementary Table S4 online.
Transient transfections and luciferase reporter assay
ERα promoter-luciferase plasmid construct was described previously (deConinck et al., 1995) and was a gift from Dr Ronald J Weigel. Transient transfection of the ERα promoter plasmid (2 μg) and pSG5 control vector of Fli1 pSGF vector into foreskin fibroblasts was carried out using Amaxa (Walkersville, MD) electroporation device according to the manufacturer's instructions. After overnight incubation at 37 °C, some cells were further stimulated with TGF-β for 24 hours. The cells were harvested and Luciferase activity of the promoter was assayed using Promega Luciferase assay kit as described previously (Nakerakanti et al., 2006).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded skin tissue sections using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Briefly, sections (5-μm thick) were mounted on APES (aminopropyltriethoxy silane solution)-coated slides, deparafinized with Histo-Clear (National Diagnostics, Atlanta, GA), and rehydrated through a graded series of ethanol. Endogenous peroxidase was blocked by incubation in 3% hydrogen peroxide for 30 minutes, followed by incubation with normal blocking serum for 1 hour. The sections were then incubated overnight at 4 °C with antibody against ERα (MC-20), diluted 1:100 in blocking buffer, followed by incubation for 30 minutes with biotinylated secondary antibody solution. The immunoreactivity was visualized with diaminobenzidine (Vector Laboratories), and the sections were counterstained with methylgreen (Vector Laboratories). Images were collected using a microscope (BH-2; Olympus, Center Valley, PA).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health grants R01 AR44883 and R01 AR42334.
Abbreviations
- ChIP
chromatin immunoprecipitation
- EBS
Ets-binding site
- ECM
extracellular matrix
- ER
estrogen receptor
- Fli1
Friend leukemia integration factor 1
- PKC-δ
protein kinase C-δ
- RT-PCR
reverse transcription-PCR
- siRNA
small interfering RNA
- TGF-β
transforming growth factor-β
- WT
wild type
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
REFERENCES
- Asano Y, Czuwara J, Trojanowska M. Transforming growth factor-beta regulates DNA binding activity of transcription factor Fli1 by p300/CREB-binding protein-associated factor-dependent acetylation. J Biol Chem. 2007;282:34672–83. doi: 10.1074/jbc.M703907200. [DOI] [PubMed] [Google Scholar]
- Asano Y, Markiewicz M, Kubo M, et al. Transcription factor Fli1 regulates collagen fibrillogenesis in mouse skin. Mol Cell Biol. 2009;29:425–34. doi: 10.1128/MCB.01278-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Stawski L, Hant F, et al. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176:1983–98. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Trojanowska M. Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300/CREB-binding protein-associated factor and subsequent acetylation in response to transforming growth factor beta. Mol Cell Biol. 2009;29:1882–94. doi: 10.1128/MCB.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft GS, Dodsworth J, van Boxtel E, et al. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med. 1997;3:1209–15. doi: 10.1038/nm1197-1209. [DOI] [PubMed] [Google Scholar]
- Ashcroft GS, Greenwell-Wild T, Horan MA, et al. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am J Pathol. 1999;155:1137–46. doi: 10.1016/S0002-9440(10)65217-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creidi P, Faivre B, Agache P, et al. Effect of a conjugated oestrogen (Premarin) cream on ageing facial skin. A comparative study with a placebo cream. Maturitas. 1994;19:211–23. doi: 10.1016/0378-5122(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Czuwara-Ladykowska J, Shirasaki F, Jackers P, et al. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem. 2001;276:20839–48. doi: 10.1074/jbc.M010133200. [DOI] [PubMed] [Google Scholar]
- De Servi B, Hermani A, Medunjanin S, et al. Impact of PKCdelta on estrogen receptor localization and activity in breast cancer cells. Oncogene. 2005;24:4946–55. doi: 10.1038/sj.onc.1208676. [DOI] [PubMed] [Google Scholar]
- deConinck EC, McPherson LA, Weigel RJ. Transcriptional regulation of estrogen receptor in breast carcinomas. Mol Cell Biol. 1995;15:2191–6. doi: 10.1128/mcb.15.4.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkareh J, Periyasamy SM, Shidyak A, et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol. 2009;296:F1219–26. doi: 10.1152/ajprenal.90710.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flouriot G, Brand H, Denger S, et al. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000;19:4688–700. doi: 10.1093/emboj/19.17.4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini D, Paez-Pereda M, Stalla J, et al. Molecular interaction of BMP-4, TGF-beta, and estrogens in lactotrophs: impact on the PRL promoter. Mol Endocrinol. 2009;23:1102–14. doi: 10.1210/me.2008-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haczynski J, Tarkowski R, Jarzabek K, et al. Differential effects of estradiol, raloxifene and tamoxifen on estrogen receptor expression in cultured human skin fibroblasts. Int J Mol Med. 2004;13:903–8. [PubMed] [Google Scholar]
- Hall G, Phillips TJ. Estrogen and skin: the effects of estrogen, menopause, and hormone replacement therapy on the skin. J Am Acad Dermatol. 2005;53:555–68. doi: 10.1016/j.jaad.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Hart A, Melet F, Grossfeld P, et al. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–77. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, Jones DA, Graves BJ. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004;32:5693–702. doi: 10.1093/nar/gkh906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I, Hanyu A, Wayama M, et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J Biol Chem. 2010;285:14747–55. doi: 10.1074/jbc.M109.093039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez SA, Gaidarova S, Saitta B, et al. Role of protein kinase C-delta in the regulation of collagen gene expression in scleroderma fibroblasts. J Clin Invest. 2001;108:1395–403. doi: 10.1172/JCI12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Mimura Y, et al. Matrix metalloproteinase-1 up-regulation by hepatocyte growth factor in human dermal fibroblasts via ERK signaling pathway involves Ets1 and Fli1. Nucleic Acids Res. 2005;33:3540–9. doi: 10.1093/nar/gki648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos M, Reid G, Denger S, et al. Minireview: genomic organization of the human ERalpha gene promoter region. Mol Endocrinol. 2001;15:2057–63. doi: 10.1210/mend.15.12.0731. [DOI] [PubMed] [Google Scholar]
- Kubo M, Czuwara-Ladykowska J, Moussa O, et al. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163:571–81. doi: 10.1016/S0002-9440(10)63685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–7. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Yamamoto T, Muraguchi A, et al. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–14. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- Murphy AJ, Guyre PM, Wira CR, et al. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS One. 2009;4:e5539. doi: 10.1371/journal.pone.0005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabha SM, Glaros S, Hong M, et al. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24:3166–76. doi: 10.1038/sj.onc.1208502. [DOI] [PubMed] [Google Scholar]
- Nakerakanti SS, Kapanadze B, Yamasaki M, et al. Fli1 and Ets1 have distinct roles in connective tissue growth factor/CCN2 gene regulation and induction of the profibrotic gene program. J Biol Chem. 2006;281: 25259–69. doi: 10.1074/jbc.M600466200. [DOI] [PubMed] [Google Scholar]
- Pannu J, Asano Y, Nakerakanti S, et al. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58:2528–37. doi: 10.1002/art.23698. [DOI] [PubMed] [Google Scholar]
- Pelletier G, Ren L. Localization of sex steroid receptors in human skin. Histol Histopathol. 2004;19:629–36. doi: 10.14670/HH-19.629. [DOI] [PubMed] [Google Scholar]
- Reid G, Denger S, Kos M, et al. Human estrogen receptor-alpha: regulation by synthesis, modification and degradation. Cell Mol Life Sci. 2002;59:821–31. doi: 10.1007/s00018-002-8470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittie L, Kang S, Voorhees JJ, et al. Induction of collagen by estradiol: difference between sun-protected and photodamaged human skin in vivo. Arch Dermatol. 2008;144:1129–40. doi: 10.1001/archderm.144.9.1129. [DOI] [PubMed] [Google Scholar]
- Sakthianandeswaren A, Curtis JM, Elso C, et al. Fine mapping of Leishmania major susceptibility Locus lmr2 and evidence of a role for Fli1 in disease and wound healing. Infect Immun. 2010;78:2734–44. doi: 10.1128/IAI.00126-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville B, Wormke M, Wang F, et al. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem. 2000;275:5379–87. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- Shirasaki F, Makhluf HA, LeRoy C, et al. Ets transcription factors cooperate with Sp1 to activate the human tenascin-C promoter. Oncogene. 1999;18:7755–64. doi: 10.1038/sj.onc.1203360. [DOI] [PubMed] [Google Scholar]
- Soldano S, Montagna P, Brizzolara R, et al. Effects of estrogens on extracellular matrix synthesis in cultures of human normal and scleroderma skin fibroblasts. Ann NY Acad Sci. 2010;1193:25–9. doi: 10.1111/j.1749-6632.2009.05296.x. [DOI] [PubMed] [Google Scholar]
- Spyropoulos DD, Pharr PN, Lavenburg KR, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–52. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surazynski A, Jarzabek K, Haczynski J, et al. Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. Int J Mol Med. 2003;12:803–9. [PubMed] [Google Scholar]
- Thornton MJ, Taylor AH, Mulligan K, et al. Oestrogen receptor beta is the predominant oestrogen receptor in human scalp skin. Exp Dermatol. 2003;12:181–90. doi: 10.1034/j.1600-0625.2003.120209.x. [DOI] [PubMed] [Google Scholar]
- Varila E, Rantala I, Oikarinen A, et al. The effect of topical oestradiol on skin collagen of postmenopausal women. Br J Obstet Gynaecol. 1995;102:985–9. doi: 10.1111/j.1471-0528.1995.tb10906.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Shen P, et al. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336:1023–7. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Saatcioglu F, Matsuda T. Cross-talk between bone morphogenic proteins and estrogen receptor signaling. Endocrinology. 2002;143:2635–42. doi: 10.1210/endo.143.7.8877. [DOI] [PubMed] [Google Scholar]
- Yoo HG, Won CH, Lee SR, et al. Expression of androgen and estrogen receptors in human scalp mesenchymal cells in vitro. Arch Dermatol Res. 2007;298:505–9. doi: 10.1007/s00403-006-0721-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.