

Abstract
Insufficient cell proliferation has been suggested as a potential cause of age related tissue dysgenesis in mammals. However, genetic manipulation of cell cycle regulators in the germ lines of mice results in changes in animal size but not progeroid phenotypes. Here we increase levels of the cyclin dependent kinase inhibitor Cdkn1b (p27kip1) in adult mice through doxycycline inducible expression and show this results in reduced cell proliferation in multiple tissues. The mice undergo changes resembling aging even in the absence of an elevated DNA damage response or evidence of senescent cells suggesting an altered balance between genetic and tissue aging. In contrast, suppressing cell proliferation by doxycycline treatment of neonates retards growth, but the onset of degenerative changes is delayed during the period of reduced body mass. These results support the hypothesis that many of the most recognizable features of mammalian aging can result from an imbalance between cell production and the mass of tissue that must be maintained.
Introduction
Accumulation of genetic damage in somatic tissue has been postulated to contribute to mammalian aging 1, 2. Further, progeroid phenotypes are observed when mechanisms that maintain genome integrity (e.g. telomerase 3,ErccI 4, BubR15) are compromised. Additional studies suggest that effects of genetic damage are indirect, resulting from induction of DNA damage response pathways. On DNA damage, p53 and/or Rb signaling pathways are induced leading to elevation of the cyclin dependent kinase inhibitors Cdkn1a (p21) and/or Cdkn2a (p16Ink4a) and either temporary or permanent cell cycle arrest 6. One explanation for the progeroid phenotypes of mutations affecting genome integrity is that cell proliferation is suppressed to a level that causes tissue dysfunction 7. In support of this idea, senescent cells accumulate in mammalian tissues with age and both Cdkn1a and Cdkn2a are elevated in their expression 8. Further, deletion of Cdkn1a or Cdkn2a has been shown to rescue progeroid phenotypes resulting from mutations that compromise genome integrity. For example, germline deletion of Cdkn1a delays the progeroid phenotype that occurs in late generation telomerase knockout mice 9. Similarly, germ line deletion of Cdkn2a delays the premature aging of BubR1 mutant mice 10. In the context of otherwise normal mice, Cdkn2a deletion has been shown to result in a partial rescue of the decrease in stem/progenitor cell survival or function that occurs during aging in at least a subset of tissues11–13 and germ line deletion of Cdkn1a has been shown to shown to increase stem and progenitor cell proliferation in a variety of specific tissues in vivo14–16. However, in the sub-ventricular zone of the brain14 and hematopoietic system16, an early elevation in proliferation of NSCs or HSCs in Cdkn1a knockout mice is followed by reduced stem cell numbers later in life, consistent with stem cell exhaustion.
The phenotypes resulting when levels of an additional cyclin dependent kinase, Cdkn1b (p27), are modulated are also relevant to the consequences of modulating cell proliferation for tissue maintenance. Cdkn1b is not directly induced by p53 or Rb and does not show elevated expression during aging. Instead, it is regulated by extracellular anti-proliferative signals including contact inhibition or serum deprivation17–18. Nonetheless transgenic models in which the levels of Cdkn1b are affected do exhibit altered cell proliferation in the in vivo situation. Cdkn1b null mice grow to approximately twice normal size and exhibit organomegaly19, 20. They develop pituitary adenomas by 10 weeks of age with high penetrance, hence the effect on overall longevity is not known. Conversely, it has been possible to determine the effect of elevated Cdkn1b expression since Cdkn1b is a primary target the Culin-RING ubiquitin ligase SCFSkp2 which mediates its degradation. Skp2 null mice exhibit elevated levels of Cdkn1b, reduced proliferation in many tissues, and grow to less than 75% of the size of wild type animals21. Nonetheless, Skp2 null mice do not exhibit premature tissue degeneration. The lack of age related phenotypes in these mice poses a particular challenge to the idea that insufficient cell proliferation accounts for age related tissue dysfunction.
Here the consequences of reduced cell proliferation in multiple tissues of adult mice are assessed through the ectopic expression of Cdkn1b (p27kip1) under doxycycline inducible control. In sharp contrast to the phenotype of germ line Skp2 knockout mice, doxycycline induced over-expression of Cdkn1b in adult animals results in many changes characteristic of advanced age at both the gross phenotypic and histological levels within a few months of treatment.
Results
Cdkn1b expression in TreTight-Cdkn1b transgenic mice
A line of transgenic mice in which Cdkn1b (p27kip1) is expressed from the TreTight promoter (TreTight-Cdkn1b) was constructed and mated with ROSA26-TetON (R26-M2rtTA) transgenic mice to create R26-M2rtTA;TRE-Tight-Cdkn1b bigenic mice. Cdkn1b expression is under tight control of doxycycline in R26-M2rtTA;TRE-tight-Cdkn1b transgenic mice. Figure 1A shows Cdkn1b expression levels in thymus from R26-M2rtTA - and R26-M2rtTA + mice which either do or do not carry the TRE-Tight-Cdkn1b transgene and which have been treated, or are untreated, with 1 mg/ml doxycycline in their drinking water. Cdkn1b expression over endogenous levels is evident only in R26-M2rtTA;TRE-Tight-Cdkn1b mice that have received doxycycline. Additionally, the level of ectopic Cdkn1b expression is doxycycline dosage dependent (Figure 1C) and expression remains at elevated levels during prolonged doxycycline treatment in most tissues (Figure 1B).
Figure 1. Induction of Cdkn1b protein and phenotypic changes in R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice.
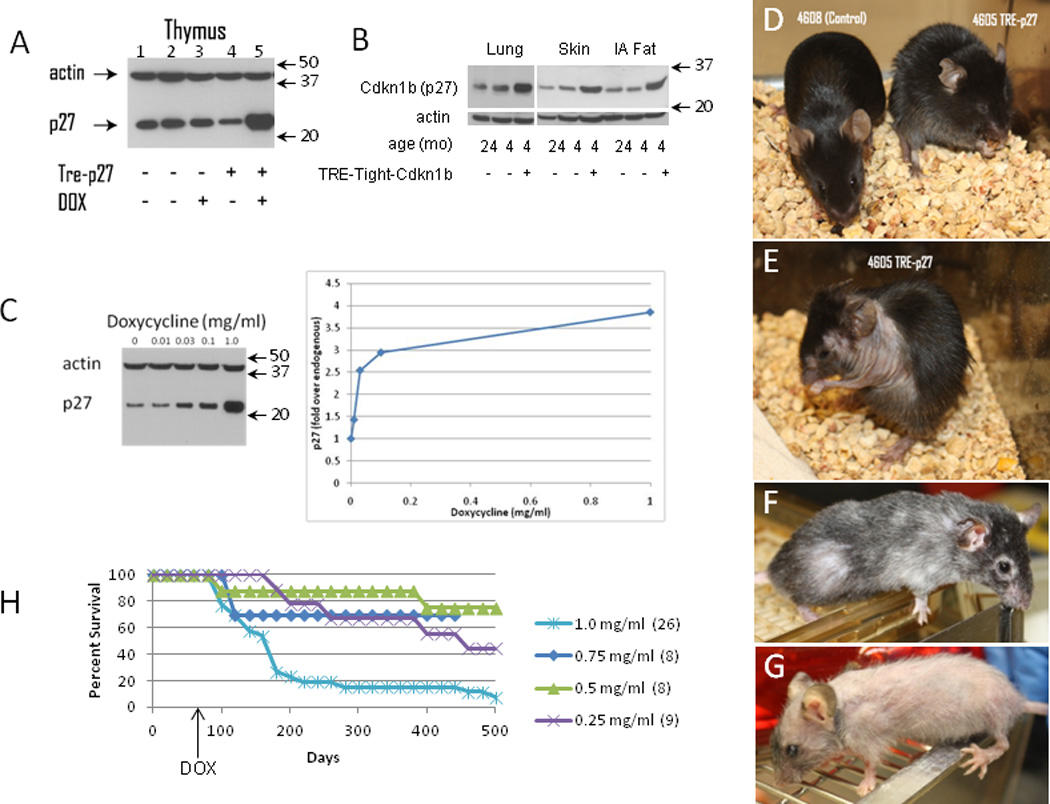
Panel A : Western blot showing Cdkn1b (p27) and actin in thymuses of mice carrying the R26-M2rtTA transgene with or without the TreTight-Cdkn1b transgene or treatment with 1 mg/ml doxycycline (DOX) for 24 hours (a full-length image of this blot is shown in Supplementary Figure 10, panel A). Panel B shows Cdkn1b protein levels in lung, skin and intra-abdominal fat of R26-M2rtTA transgenic mice that either carry or do not carry the TRE-Tight-Cdkn1b transgene where, starting at two months of age, mice were continuously treated with 1 mg/ml doxycycline for two months and assessed at 4 months of age. Additionally, Cdkn1b expression in the same tissues from a control 24 month old mouse is included for comparison. Panel C shows a titration of doxyxycline concentration verses Cdkn1b expression in the thymus where mice were treated with the indicated concentrations of doxycycline for 3 days. A western blot is shown on the left and quantified by densitometry of Cdkn1b levels corrected for loading based on the signal from actin on the right. Arrows to the right of panels A–C indicate the positions of size markers in kDa. Panel D shows gross phenotypic changes in an R26-M2rtTA-control (left, mouse 4608) and R26-M2rtTA;TreTight-Cdkn1b (right, mouse 4605) mice continuously treated with 1 mg/ml doxycycline beginning at 2 months of age for a period of three weeks. The same R26-M2rtTA;TreTight-Cdkn1b mouse is also shown at 6 weeks (panel E) or 5 months (panel F) of continuous doxycycline administration. Panel G shows a mouse treated, beginning at two months of age, continuously for 9 months with 0.75 mg/ml doxycycline. Panel H shows survival in days of R26-M2rtTA;TreTight-Cdkn1b mice treated with various concentrations of doxycycline as indicated in the figure where the number of mice per group is given in parentheses. None of 40 Cdkn1b over-expressing mice that have died during doxycycline treatments ranging from 0.25 to 1.0 mg/ml had overt tumors on necropsy (a rate of <2.5%). This is much lower than the expected rate for this strain (40%). In the present study, one of the 4 control animals that died (25%) succumbed to thymic lymphoma.
Cdkn1b overexpression in adults induces a progeroid phenotype
The gross phenotypic effects of Cdkn1b over-expression are shown in Figure 1D–1G. Phenotypic changes first become apparent at ~2–3 weeks and manifest as a change in posture, ruffled fir and transient diarrhea that resolves over a period of approximately 2–3 weeks (Figure 1D). An obvious nuchal to caudal wave of hair loss occurs at ~ the time of the second telogen (postnatal day 45–70 22) which is consistent with delayed or inefficient re-initiation of hair growth (Figure 1E). Over time, hair growth is re-established to varying degrees but typically is reduced in density and exhibits loss of pigmentation (Figures 1F–G). Additional macroscopic changes occur over time including thinning skin and lesions, loss of corneal transparency and generalized frailty. The time at which phenotypic changes occur is delayed in mice treated with lower doses of doxycycline, at least through 0.25 mg/ml, however the severity of changes is similar. Survival of mice treated with varying doses of doxycycline is shown in Figure 1H. Loss of mice during the first 4–6 wks, particularly at the highest dose, results from gastrointestinal problems (see below).
Histological examinations of several tissues were performed for Cdkn1b over-expressing mice in comparison to controls. Abdominal skin is shown in Figure 2A (R26-M2rtTA+ control that does not carry TRE-Tight-Cdkn1b) and Figure 2B (R26-M2rtTA;TRE-Tight-Cdkn1b) for mice treated with 0.25 mg/ml doxycycline for a period of 5 months and confirms Cdkn1b over-expression results in a reduction in skin thickness to approximately half that of control mice. The majority of this reduction is a consequence of loss of adipose tissue from the hypodermal layer. Gastrocnemius and abdominal muscles are shown in Figure 2C–2F. In each case the diameter of individual fibers is reduced in Cdkn1b over expressing mice (Figures 2G and 2H).
Figure 2. Histological changes in skin and muscle of Cdkn1b over-expressing mice.
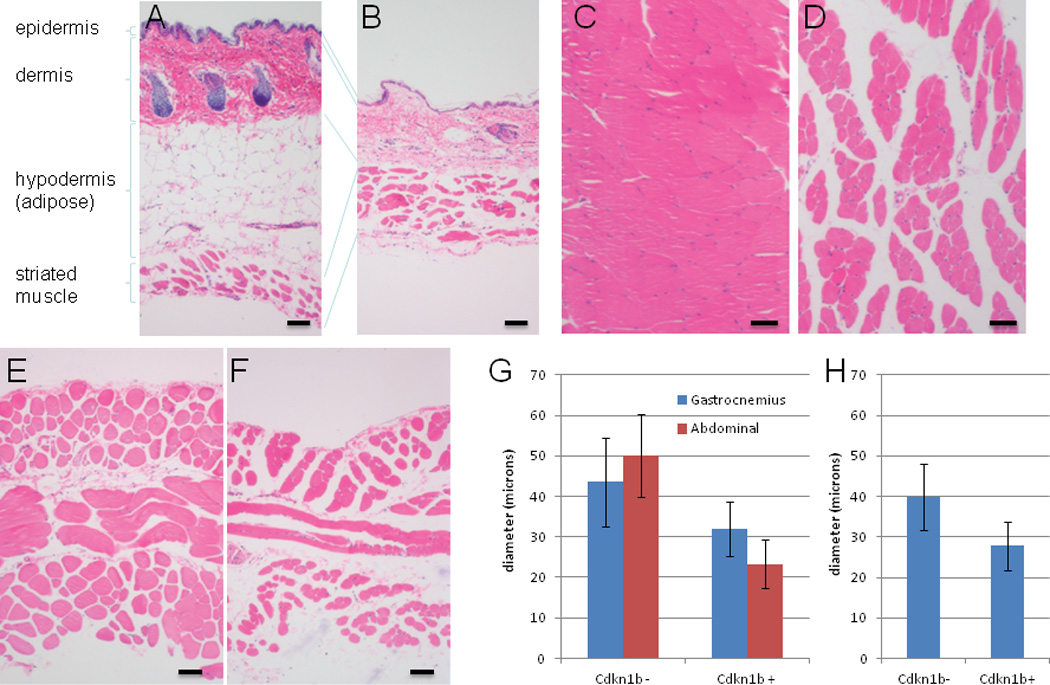
For this set of experiments R26-M2rtTA;TRE-Tight-Cdkn1b (panels B, D and F) and control (R26-M2rtTA+ litermates that do not carry the TRE-Tight-Cdkn1b transgene, panels A, C, and E) mice were treated, beginning at two months of age, with 0.25 mg/ml doxycycline continuously for a period of 5 months. Histological sections were prepared from abdominal skin (panels A and B), gastrocnemius muscle (panels C and D) and abdominal muscle (panels E and F) and stained with hematoxylin and eosin prior to image capture using a 10× objective. Various layers of the skin are indicated for panels A and B. In panel G, diameters were determined for gastrocnemius and abdominal musle fibers (n=50, error bars indicate s.d.) from control and R26-M2rtTA;TRE-Tight-Cdkn1b mice treated, beginning at two months of age, with 0.25 mg/ml doxycycline continuously for a period of 5 months and show a decrease in Cdkn1b over-expressing mice (gastrocnemius ρ = 7.1×10−7; abdominal ρ = 4.8×10−2`1; two-tailed t-test). Panel H shows muscle fiber diameters determined for gastrocnemeus muscle of control and R26-M2rtTA;TRE-Tight-Cdkn1b mice (n=50, error bars indicate s.d.) treated beginning at two months of age with 0.5 mg/ml doxycycline continuously for a period of 8 months and also show a decrease in Cdkn1b over-expressing mice (ρ = 6.2 × 10−2`7; two-tailed t-test).
Stem and progenitor cell cycling in Cdkn1b over-expressing mice
To measure the effect of Cdkn1b expression on cell proliferation, R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice were treated with doxycycline for various periods and administered IdU continuously for 3 days and CldU as a pulse 2 hours prior to sacrifice. Histological sections were prepared for the sub-ventricular zone (SVZ) of the brain, a site of active neural stem cell proliferation in adults, as shown in Figures 3A–3F, and Supplementary Figure S1. Results from this study show that within three days of the start of 1.0 mg/ml doxycycline treatment, cells incorporating nucleoside analogs are reduced to ~1/5th the level of wild type mice and that this reduction in the level of division is maintained throughout at least 5 months of treatment Figure 3G).
Figure 3. Proliferating and replication competent cells in the brain and muscles of R26-M2rtTA;TRE-Tight-Cdkn1b mice.
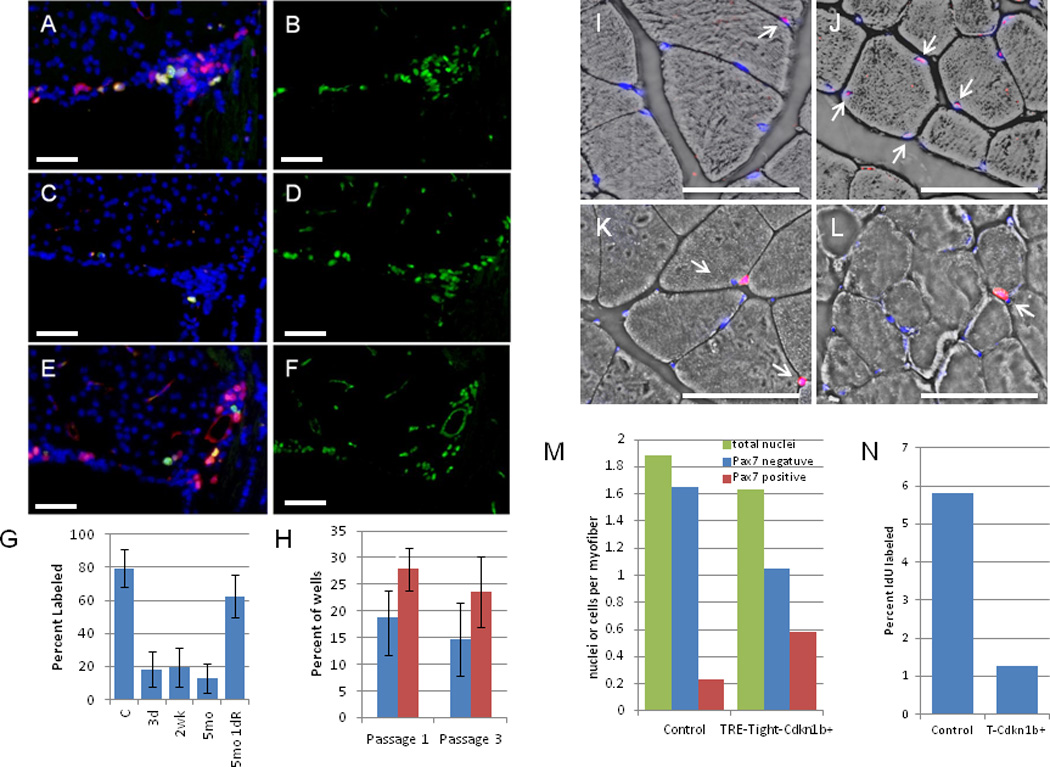
Supraventricular zones (SVZs) from R26-M2rtTA;TRE-Tight-Cdkn1b mice either untreated (A and B) or treated with 1 mg/ml doxycycline between months 2–7 (C and D) or treated between months 2–7 but removed from doxycycline treatment for 24 hours (E and F). IdU was administered for 3 days (A–D) or 1 day (E and F) and CldU 2 hours prior to sacrifice. A, C, and E are stained for IdU (red), CldU (green), and DAPI (blue). B, D, and F are the same sections stained for Mcm2 (green). Images were captured using a 40× objective. G, percent of Mcm2+ cells that were also positive for IdU incorporation in SVZ sections from untreated (n = 4) or treated with 1 mg/ml doxycycline for 3 d (n = 6), 2 wk (n = 5), 5 mo (n = 7), or 5 mo followed by removal from doxyxcline for 1 day (5mo 1dR, n = 6) R26-M2rtTA;TRE-Tight-Cdkn1b mice. Error bars indicate s.d. IdU incorporation is reduced in treated relative to control mice (3 d ρ = 0.00014, 2 wk ρ = 0.00017, 5 mo ρ = 0.00017; two-tailed t-tests) but returns to levels that are not significantly different from control on doxyxcline removal (ρ = 0.067; two-tailed t-tests). H shows clonogenic neurosphere assays where the percent of wells in 96 well plates (n = 4, error bars indicate s.d.), cultured in the absence of doxycycline, that contained neurospheres after 1 or 3 passages is shown for R26-M2rtTA-control (blue bars) and R26-M2rtTA;TRE-Tight-Cdkn1b (red bars) mice that had been continuously treated with 1 mg/ml doxycycline from 2 months through 16 months of age. More neurospheres are recovered from Cdkn1b over expressing mice than controls (passage 1 ρ = 0.032; passage 3 ρ = 0.013; two-tailed t-tests). I–L, muscle fibers from the gastrocnemius muscles of R26-M2rtTA control (I and K) and R26-M2rtTA;TRE-Tight-Cdkn1b (J and L) mice treated with 0.25 mg/ml doxycycline from mo 2–7 and IdU during the final three weeks. I and J, Pax7 (red) and DAPI (blue) overlain on a bright-field image (arrows, Pax7+ nuclei). K and L, IdU (red) and DAPI (blue) (arrows, IdU positive nuclei). N, total, Pax7 negative, and Pax7+ nuclei per myofiber (M) and percent of nuclei labeled with IdU (N) were quantified.
To determine whether the reduction in cell division evidenced by reduced nucleoside analog incorporation resulted from loss of stem and progenitor cells, the presence of Mcm2 expressing cells (Mcm2 marks cells which are competent for replication23) was compared for control and 1 mg/ml doxycycline treated R26-M2rtTA;TRE-Tight-Cdkn1b mice (Figures 3B and 3D and Supplementary Figure S1). These studies demonstrate that, despite reduced proliferation, Mcm2 expressing cells remain present in the SVZ during prolonged doxycycline treatment at levels that are similar to those found in untreated control animals. To confirm that these cells remain capable of entering the cell cycle, R26-M2rtTA;TRE-Tight-Cdkn1b mice that had been administered 1 mg/ml doxycycline continuously for 5 months were removed from doxycycline for a period of 24 hours and simultaneously labeled with IdU. Mice were additionally labeled with CldU during the final 2 hours following doxycycline removal (Figures 3E and 3F). These studies demonstrate that Mcm2 expressing cells resume cycling within 24 hours following doxycycline removal, to nearly the level observed in untreated control animals (Figure 3G). To confirm that stem cell levels in the brain are unaffected by prolonged Cdkn1b over expression, neurosphere cultures were prepared in micro-titre wells at limiting dilution from control ((R26-M2rtTA + littermates that do not carry TRE-Tight-Cdkn1b) and R26-M2rtTA;TRE-Tight-Cdkn1b mice following continuous 1.0 mg/ml doxycycline treatment for a period of 14 months. Cultures were maintained in the absence of doxycycline and the number of neurospheres present following 10 days of culture, or following two re-passages, were determined (Figure 3H). For both the initial cultures and following re-passaging, the number of neurospheres recovered from mice that had been continuously treated with doxycyline for over 1 year was similar to or higher than that for controls. Together, these results show that, although the rate of proliferation of neural stem/progenitor cells in Cdkn1b over expressing mice is reduced, these cells remain present at levels similar to control animals.
The frequency of muscle stem and progenitor cells was also assessed for the gastrocnemius muscle in R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice treated (beginning at 2 mo of age) with 0.25 mg/ml doxycycline for 5 months and controls (R26-M2rtTA + litermates that do not carry TRE-Tight-Cdkn1b but were treated with doxycycline in parallel) using Pax7 as a stem/progenitor cell marker24 (Figures 3I and 3J). The number of Pax7 positive and negative nuclei per muscle fiber were determined (Figure 3M). The total number of nuclei associated with sections of each fiber is modestly reduced in Cdkn1b over-expressing mice, from 1.9 to 1.6. A larger shift occurs in the distribution between Pax7 negative and positive nuclei where the frequency of Pax7 negative nuclei is reduced by over 1/3, from 1.7 to 1.0 nuclei per fiber, while the frequency of Pax7 positive nuclei per fiber is nearly tripled, shifting from ~0.2 in control animals to ~0.6 in Cdkn1b over expressing mice. Since, in wt mice, Pax7 is known to be expressed in satellite cells and rapidly proliferating transient amplifying progeny, one explanation for this shift is that delayed replication in the transient amplifying population results in cells stalling at this step of differentiation with a resulting reduction in contribution to muscle fibers. Consistent with this observation, long term labeling of muscle with IdU (three weeks) demonstrated that the number of nuclei incorporating IdU decreased to approximately 20% of wt levels in Cdkn1b over expressing mice (Figures 3K, 3L, and 3N).
The effect of Cdkn1b over expression on cell proliferation in the small intestine was also examined. R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice were treated with doxycycline for various periods and administered IdU (3 days) and CldU (2 hours) prior to sacrifice. The number of crypt nuclei incorporating CldU during a two hour labeling period is reduced to approximately 2% of wild type values at 3 days of doxycycline administration in R26-M2rtTA;TRE-Tight-Cdkn1b mice (from 23.1 s.d. 3.3 to 0.45 s.d. 0.6; Figures 4A vs. 4B, green). In these animals no crypts were observed to contain more than 3 labeled nuclei and over 60% of the crypts contained no labeled nuclei. Additionally, all crypts exhibit evidence of Cdkn1b over expression (Figure 4B, red). Nonetheless, IdU administration over a 3 d period results in a high proportion of labeled cells demonstrating that the effect of Cdkn1b over expression is to slow, rather than block, cell cycling (Figure 4E, red).
Figure 4. CldU incorporation and Cdkn1b expression in the ileums of doxycycline treated R26-M2rtTA;TRE-Tight-Cdkn1b mice.

M2rtTA;TRE-Tight-Cdkn1b mice were either untreated (panel A), or treated at two months of age with 1 mg/ml doxycycline continuously for 3d (panels B and E), 2 weeks (panels C and F) or 5 months (panel D). Paraffin sections were prepared and stained for CldU incorporation (green) and Cdkn1b (red) in panels A–D and F, or CldU (green) and IdU (red) incorporation in panel E, and images were captured using a 20× objective. In panel G the frequency with which Mcm2+ nuclei (see Supplementary Figure S2) at various positions from the bases of the crypts label with CldU following a 2 hour pulse is quantified for control (blue squares) and Cdkn1b over-expressing (red triangles) mice. In panels H–K the distribution of crypts containing different numbers of nuclei incorporating CldU is plotted for untreated (Panel H) and 3d (Panel I), two week (Panel J) and 5 month (Panel K) doxycycline treated mice.
Prior studies have shown that the base of the small intestinal crypt contains two types of cells, crypt basal columnar (CBC) cells (all of which express the wnt-responsive G protein-coupled receptor Lgr525) and Paneth cells. CBC cells have been shown to be the predominant active stem cells within the small intestine and clonal tracing data supports that these cells undergo frequent symmetric divisions to give rise to either two transient amplifying cells, which move up the crypt and following several rapid divisions differentiate to epithelial lineages, or two basal columnar cells that replace stem cells lost to differentiation26. The replication licensing factor Mcm2 is expressed in all crypt basal columnar cells and transit amplifying cells but not Paneth cells (ref 27 and Supplementary figure S2, panel A). Measurement of the proportion of Mcm2+ cells incorporating CldU at different positions within Cdkn1b over-expressing crypts of 3 day and 2 week doxycycline treated mice demonstrates that proliferation is reduced in both CBC stem cells at the crypt base and progenitor cells at higher positions within the crypts (Figure 4G). This observation demonstrates that Cdkn1b over-expression suppresses cell division in both stem and transit amplifying cells. Additionally, these studies show that, as for transit amplifying cells, the number of Mcm2+ CBC stem cells is progressively reduced during Cdkn1b over-expression (Supplementary Figures S2B–S2J). Depletion of stem cells is not expected if these cells undergo invariant asymmetric division but is consistent with a symmetric, neutral drift, model of stem cell division26.
Following longer periods of doxycycline treatment, CldU labeling demonstrates that a subset of crypts exhibit more active cell cycling. At two weeks of doxycycyline treatment (Figure 4B), approximately 8% of the crypts contain as many or more labeled nuclei than crypts of control animals (Figures 4C, 4F, and 4I). These crypts exhibit morphology consistent with crypts undergoing fission (Figures 4C and 4F; green). Additionally, crypts containing rapidly cycling cells fail to exhibit Cdkn1b over expression (Figures 4C and 4F, red). Following 5 months of continuous doxycycline treatment, most crypts contain a high proportion of CldU labeled cells, similar to controls, and fail to exhibit Cdkn1b over-expression, although evidence of Cdkn1b over expression and a reduced rate of cycling are still found in ~1/4 of the crypts (Figures 4D and 4J). These results are consistent with prior studies demonstrating that the intestine has an exceptional ability to eliminate stem cells that divide at suboptimal rates27–32. It is likely that survival of Cdkn1b over expressing mice depends on replacement of the majority of the intestinal epithelia with cells in which the transgene is no longer expressed and that the early loss of a fraction of Cdkn1b over expressing mice, particularly at the highest doses of doxycycline, results from insufficient intestinal function.
DNA damage response and cellular senescence
To address the possibility that Cdkn1b over expression caused tissue degeneration through increase genetic damage and induction of a DNA damage response, ser18 phosphorylation of p53, a key event in the DNA damage response was measured. The levels of pp53 (ser18) were determined in several tissues of 4 month old Cdkn1b over expressing mice and compared to levels in 4 and 24 month old controls (Figure 5A). Rather than showing elevated levels, as might be expected if Cdkn1b expression results in increased genetic damage, pp53 (ser18) levels are reduced in many different tissues of Cdkn1b over expressing mice. Consistent with reduced pp53(ser18) levels, the frequency of cells with γ-H2AX and TP53bp1 foci is reduced in the epidermis of Cdkn1b over expressing mice (Figures 5B–5G; similar results were obtained for γ-H2AX foci in SVZ cells, Supplementary Figure S3). The possibility that elevated Cdkn1b levels induce cellular senescence was assessed by measuring the level of senescence associated β-galactosidase (SAβgal) activity in abdominal fat, a tissue in which SAβgal activity is known to increase during aging and in progeroid mouse models of aging5. Even though total abdominal fat is reduced in Cdkn1b over expressing mice, senescence associated β-galactosidase activity is not elevated in this tissue relative to controls (Figure 5H). Il-6, a prominent cytokine expressed in DNA damage induced senescent cells33, is also not elevated in Cdkn1b over-expressing mice (Supplementary Figure S4), consistent with the lack of accumulation of senescent cells. Finally, prior studies of Skp2 null mice, in which Cdkn1b is over expressed to levels similar to those achieved here, have shown that nuclear enlargement and polyploidy occur within a subset of tissues including the tubular epithelia of the kidney21. However, Skp2 null mice also over express cyclin E21 and polyploidy within these tissues may result from cyclin E, rather than Cdkn1b, over expression. Nuclear enlargement is not observed in kidney tubular epithelial cells in Cdknb1 over expressing mice (Supplementary Figure S5) demonstrating that Cdkn1b over expression, alone, is not responsible for this phenotype of Skp2 null mice.
Figure 5. DNA damage response and expression of senescence markers in Cdkn1b over-expressing mice.
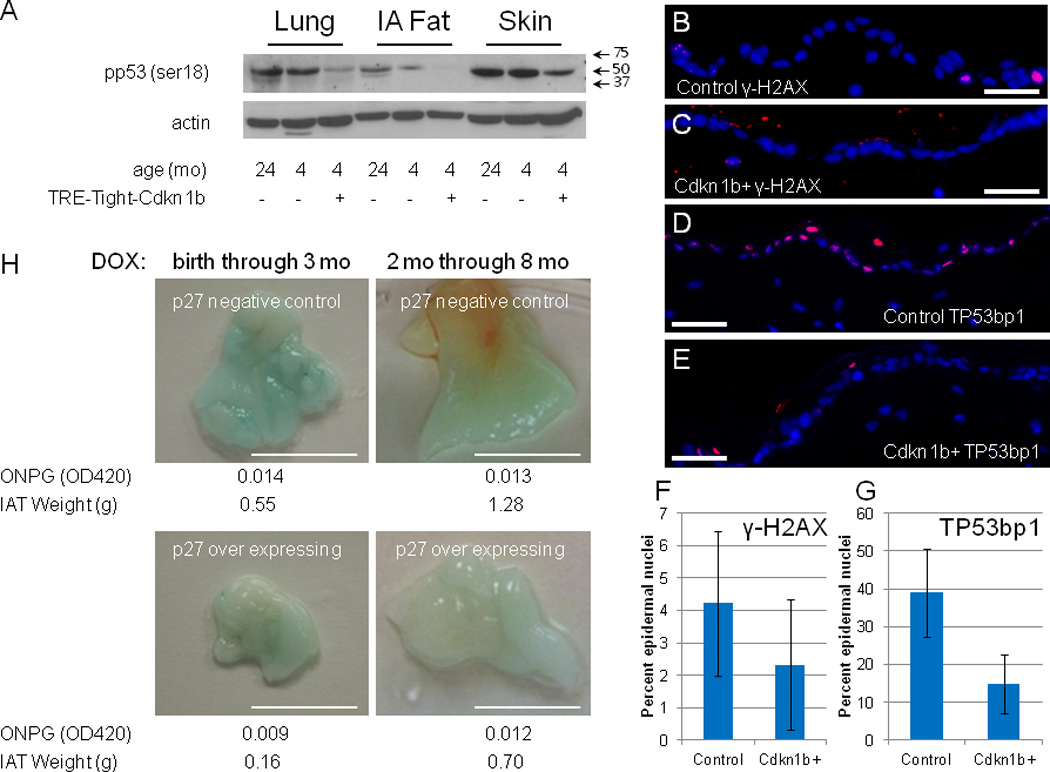
Panel A shows western blots for pp53(ser18) (a full-length image of this blot is shown in Supplementary Figure 10, panel B) and actin in lung, intra-abdominal fat (IA Fat), and skin of R26-M2rtTA transgenic mice that either carry or do not carry the TRE-Tight-Cdkn1b transgene as indicated in the figure where, starting at two months of age, mice were continuously treated with 1 mg/ml doxycycline for two months and assessed at 4 months of age. Additionally, Cdkn1b expression in the same tissues from a control 24 month old mouse is included for comparison. Arrows indicate the positions of size markers in kDa. Panels B and C show immune-fluorescence staining for γ-H2AX in epidermal sections of control (B) and Cdkn1b over-expressing (C) mice. Panels D and E show immune-fluorescence staining for TP53BP1 in epidermal sections of control (D) and Cdkn1b over-expressing (E) mice. Panels F and G demonstrate reduced frequency of γ-H2AX (n = 9, error bars indicate s.d., ρ = 0.04, two-tailed t-test) and TP53BP1 (n = 10, error bars indicate s.d , ρ = 3.7 × 10−5, two-tailed t-test) foci, respectively, in epidermal sections from Cdkn1b over-expressing mice relative to controls. Panel H shows SA-β galactosidase staining of intra-abdominal fat (IAT) for R26-M2rtTA control and R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice that were treated with 1 mg/ml doxycycline either from birth through 3 months of age (left panels) or starting at 2 months of age and through 8 months of age (right panels). Bars = 5 mm. Quantitative assessment of SA-β galactosidase activity was made using ONPG as a substrate and results from this assay are shown below each panel. Additionally, the weight of total IAT recovered from each animal is given in grams (g). Qualitatively similar SA-β galactosidase staining was observed in a two additional sets of control and R26-M2rtTA;TRE-Tight-Cdkn1b mice treated with 0.25 mg/ ml doxycycline from months 2–7 although quantitative measures were not performed.
Reversibility of the Cdkn1b over expression phenotype
The observations above demonstrate that, despite degenerative changes, adult mice over expressing Cdkn1b for long periods of time retain neural stem/progenitor and muscle satellite cells. The continued presence of these cells and the lack of evidence for accumulation of senescent cells raise the possibility that many of the phenotypic changes resulting from Cdkn1b over expression might be reversed on return of Cdkn1b to wt levels following removal of doxycycline. To test this possibility, R26-M2rtTA;TRE-Tight-Cdkn1b mice that had been continuously treated with doxycycline for 8 or more months were removed from doxycycline and monitored for reversal of phenotypic changes. Surprisingly, these mice showed little improvement at a gross phenotypic level and some aspects of their condition declined more rapidly when removed from doxycycline than observed for mice that were continued on doxycycline for similar or longer periods (Figure 6A). In particular, removal of Cdkn1b over expressing mice from doxycycline resulted in exacerbation of skin lesions that limited the time over which the mice could be followed.
Figure 6. Effect of doxycycline removal on the gross phenotype and skin of R26-M2rtTA;TRE-Tight-Cdkn1b mice.
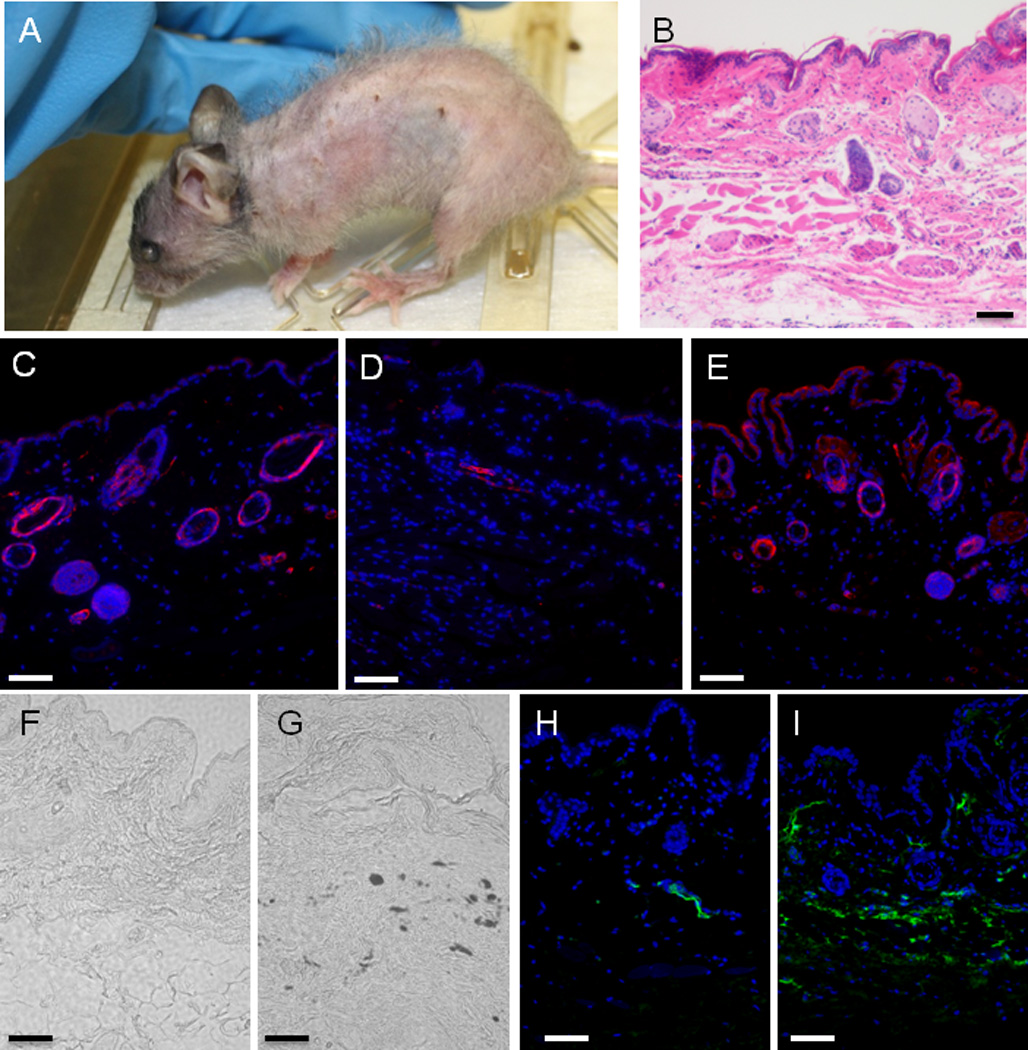
The phenotype of an R26-M2rtTA;TRE-Tight-Cdkn1b mouse treated with 0.75 mg/ml doxycycline from 2 through 9 months of age followed by removal from doxycycline for 3 additional months is shown in Panel A (where the same animal is shown prior to removal from doxycycline in Figure 1, panel G). Panels B–I show images of histological sections from abdominal skin of this mouse where B is HE stained and taken with a 10× objective, D–E are immuno-fluorescence images stained for smooth muscle actin (red) and DAPI (blue) and taken with a 20 × objective. Fand G are unstained phase contrast images taken with a 20× objective. H and I are immuno-fluorescence images stained for IgG (green) and DAPI (blue) and taken with a 20 × objective. Panels C, F and H are controls prepared from an R26-M2rtTA mouse treated with doxycycline, panel D is an R26-M2rtTA;TRE-Tight-Cdkn1b mouse treated continuously with doxycycline and panels E, G, and I are from an R26-M2rtTA;TRE-Tight-Cdkn1b mouse following treatment with and removal from doxycycline as described above.
Histological sections of skin from 12 month old control (R26-M2rtTA+ mice that do not carry TRE-Tight-Cdkn1b but which were treated with doxycycline) and R26-M2rtTA;TRE-Tight-Cdkn1b continuously treated with doxycycline for 9 months and removed from doxycycline for 3 months mice are shown in Figures 6B–6I. Thinning of the hypodermal layer is not reversed in mice removed from doxycycline (Figure 6B). Nonetheless, cell density within dermal and epidermal layers is increased. Hair follicles and sebaceous glands are more prominent in the dermal layer and many of the new cells stain for smooth muscle actin expression in both the epidermal and dermal layers (Figures 6C–6E). Unstained phase contrast images are shown in Figures 6F and 6G, and demonstrate the presence of pockets of melanocytes in mice removed from doxycycline (brown spots are also observed in the skin of live mice, Figure 6A). Deposition of IgG is also observed (Figures 6H and 6I). The effect of Cdkn1b over-expression on CD34+ putative stem cells within the hair follicle bulge was also assessed (Supplementary Figure S6). CD34+ cells are reduced to ~1/4 of control numbers in mice continuously over-expressing Cdkn1b; however, within the hair follicles that remain, the frequency of these cells recovers to near control levels in mice that have been removed from doxycycline.
The effect of doxycycline removal on the ratio of lymphoid to myeloid cells within the peripheral blood was also determined. Mice treated continuously with doxycycline show a reduction in this ratio from ~8.4 to ~2.1 (Supplementary Figure S7, panels A and B). Removal of doxycycline does not result in a reversal of the effect through at least 7 weeks (Supplementary Figure S7, panel C).
Despite failure to reverse lymphoid/myeloid ratios in the blood and the worsened condition of the skin, there is evidence of recovery in some tissues of Cdkn1b mice following removal from doxycycline. Both the gastrocnemius and abdominal muscles show substantial recovery of muscle fiber diameter following doxycycline removal (Supplementary Figure S8). There is, additionally, an increase in nuclei associated each muscle fiber. However, these improvements are accompanied by increased infiltration of adipose tissue. The infiltration of muscle with adipose tissue and an increased presence of melanocyctes and myofibroblasts within the skin are changes that frequently accompany normal aging of these tissues. Although such changes may occur in response to tissue degeneration in normally aging tissues, they may be suppressed by reduced proliferation in animals continuously over-expressing Cdkn1b. Over growth of subsets of cells on release from this proliferation block could account for the more rapid decline of Cdkn1b over expressing mice that have been removed from doxycycline relative to those that are continued on doxycycline treatment. These observations suggest that loss of tissue organization and irreversibility of compensatory changes may contribute significantly to degeneration of some tissues during aging.
Cdkn1b over expression in neonatal mice
Prior studies have shown that Cdkn1b is over expressed throughout development in most tissues of Skp2 null mice21. Although these mice are smaller they do not exhibit progeroid phenotypes. To examine the effect of Cdkn1b over expression on immature mice, nursing mothers with new born litters of neonatal R26-M2rtTA mice, either carrying or not carrying the TRE-Tight-Cdkn1b transgene, were placed on 1 mg/ml doxycycline and weights of the animals were determined weekly beginning at three weeks and through 15 weeks of age (Figure 7A). Cdkn1b over expressing mice show a reduced rate of growth during the period between 4 and 10 weeks of age during which non-transgenic animals grow nearly exponentially to more than double in weight. In contrast, Cdkn1b over expressing animals exhibit a more linear weight gain such that the greatest differences between control and Cdkn1b over expressing mice occur between weeks 4–6, during which the weight of control animals is nearly double that of Cdkn1b over expressing animals. Morphologically, Cdkn1b over expressing mice exhibit an obvious developmental delay during this period (Figures 7B, 7D, and 7E). Despite the developmental delay, mice in which Cdkn1b is over expressed during neonatal development show a significant reduction in degenerative phenotypes relative to mice that were initially placed on doxycycline as 2 month old adults for similar durations (compare Figures 7D – 7G with Figures 7H – 7K). The improved phenotypes include reduced loss of hair and pigmentation, thicker skin with fewer lesion and greater corneal transparency. Improvements are also apparent in histological sections of skin and muscle relative to age matched controls (Supplementary Figure S9). Tissue from control and Cdkn1b over-expressing mice are shown for the gastrocnemius muscle in panels A and B and the abdominal muscles in panels C and D. Fiber diameters were determined (panel E) and demonstrate no significant difference between control and Cdkn1b over expressing mice for either of these muscles. Skin thickness is also similar between 8 week old control and Cdkn1b over expressing mice, (Supplementary Figures S9F and S9G). Nonetheless, the number of cells incorporating nucleoside analog is reduced (Supplementary Figures S9H–S9J) consistent with the delayed growth of these animals. R26-M2rtTA;TRE-Tight-Cdkn1b transgenic mice treated with doxycycline as neonates eventually achieve body mass similar to that of controls at which point degenerative phenotypes become apparent (Figure 7C). These results are consistent with the idea that a key determinate of whether degenerative phenotypes are observed at the tissue level is the ratio between cell proliferation and the mass of the tissue that must be maintained.
Figure 7. Phenotypic effects of Cdkn1b over-expression in neonatal versus adult animals.
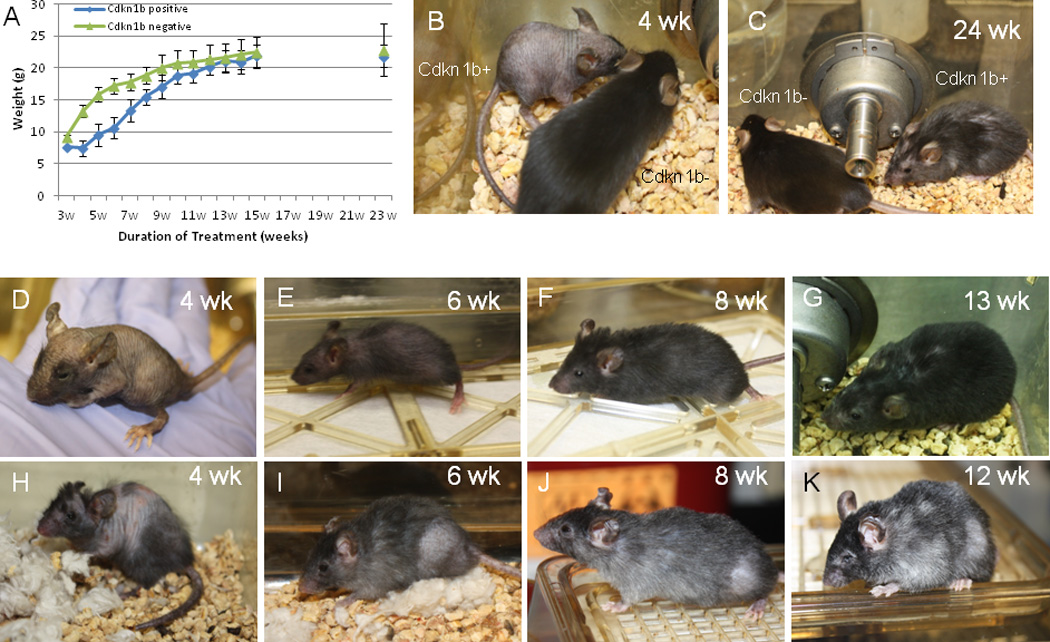
Panel A shows weight in grams of R26-M2rtTA;TRE-Tight-Cdkn1b (n = 22) and R26-M2rtTA control (n=21) littermates during neonatal development when mothers were placed on 0.25 mg/ml doxycycline on birth of the pups (error bars indicate s.d.). Panel B shows comparison of an R26-M2rtTA;TRE-Tight-Cdkn1b (Cdkn1b+) with an R26-M2rtTA (Cdkn1b−) control littermate at 4 weeks of development following continuous treatment with 0.25 mg/ml doxyxcycline. Panel C shows the same pair of animals following 24 weeks of continuous doxycycline administration. Panels D–K compare mice treated for various times with 0.25 mg/ml doxycycline either as neonates (panels D–G) or when treatment was begun at 2 months of age (panels H–K) where panels D and H are 4 weeks of treatment, panels E and I are 6 weeks of treatment, panels F and J are 8 weeks of treatment, and panels G and K are 12–13 weeks of treatment.
Discussion
Cdkn1b is a key regulator of cell cycle progression that was initially identified based on its induction by extracellular anti-proliferative signals including contact inhibition or serum deprivation. Like the other members of this family, Cdkn1a (p21) and Cdkn1c (p57), Cdkn1b has the ability to bind to the cyclin-CDK holoenzyme and inhibit CDK complexes required for G1 progression and S phase entry. It is known to inhibit cell cycle progression when over expressed in essentially all cell types in culture. Consistent with results from cultured cells, adult mice in which over expression of Cdkn1b is broadly induced demonstrate reduced levels of cell proliferation in all of the tissues examined (sub-ventricular zone, skeletal muscle, skin and transiently in the intestine) in the present study. Many of the degenerative changes observed in Cdkn1b over expressing mice are likely to be a consequence of reduced cell proliferation.
Additional functions for Cdkn1b have been described. Cytoplasmic Cdkn1b has been shown to regulate the actin cytoskeleton and affect cell migration by modulation of Rho34. This function of Cdkn1b could contribute to some of the phenotypes observed in the present study (e.g. failure of SVZ progenitors to migrate to the olfactory bulb or failure of muscle satellite cells to fuse to myofibrils). Cdkn1b has also been shown to interact in a transcriptional repressor complex35 and loss of Cdkn1b results in elevated Sox2 expression in multiple tissues including fibroblasts, lung, retina and brain36. Sox2 is known to be expressed in and contribute to the stem cell phenotype of a number of somatic stem cells37–39 and reduced Sox2 expression could result in loss of adult stem cells. However, it is unclear that wt levels of Cdkn1b are limiting for Sox2 repression and elevating Cdkn1b levels over wt may not result in a further reduction. The increase in SVZ and muscle stem and progenitor cells is also not consistent with this role.
The number of CBC stem cells per crypt is reduced in the small intestine following inhibition of cell division by Cdkn1b over-expression. While it is possible this loss is a direct result of a transcriptional repressor function of Cdkn1b, the mode of stem cell division in this tissue could also explain the observation. Prior studies have shown that intestinal crypt homeostasis is maintained by competition between symmetrically dividing stem cells26 such that over time individual stem cell lineages become fixed or extinct. Cdkn1b reduces the rate of division of CBC stem cells and, unlike the case for an invariant asymmetric mode of division, reduced division of the stem cell pool is expected to result in the observed reduction of these cells. A smaller pool of stem cells is expected to increase the rate at which individual stem cell lineages become fixed or undergo extinction within a crypt. Further, the likelihood that all stem cells within a crypt are lost to differentiation is increased and would result in a reduction in the number of intestinal crypts were it not for replaced by crypt fission. A reduced rate of cell proliferation of stem cell within hair follicles may also account for the effects of Cdkn1b over-expression within this tissue. Similar to the intestine, the number of Cd34+ stem cells present in the bulge of each follicle decreases when cell proliferation is suppressed by Cdkn1b over-expression. Loss of hair follicles due to a complete loss of stem cells within a follicle could account for alopecia in these mice. On removal of doxycycline, the number of Cd34+ stem cells recovers in the remaining follicles; however, very little reversal of alopecia is observed consistent with a lack of generation of new follicles by fission. Additional studies will be necessary to determine if the mode of division of lymphoid or myeloid biased hematopoietic stem cells and the architecture of the hematopoietic stem cell niche play a role in the lymphoid to myeloid shift resulting from Cdkn1b over-expression or the failure to recover wild type ratios following doxycycline removal.
A variety of mouse models in which genome stability is compromised have been shown to exhibit a progeroid phenotype where the phenotype is thought to result from activation of the DNA damage response and the accumulation of senescent cells. For example, loss of telomerase results in impaired tissue maintenance and shorted life-span where these consequences are thought to result from elevated genetic damage caused by telomere dysfunction40. However, the effect of genetic damage on the aging phenotype may be indirect and result from reduced cell proliferation and induction of a senescent phenotype mediated by the DNA damage response. Consistent with this idea, Cdkn1a (p21) deletion improves stem cell function and partially rescues the lifespan of mice with dysfunctional telomeres9. Further, progeroid mice in which Trp53 is constitutively hyperactive (p53+/m) exhibit reduced cell cycling, which may be a consequence of an elevated DNA damage response to normal rates of genetic damage41,42. The number of senescent cells is also increased in tissues of p53+/m mice compared to wild-type42. BubR1 is a key inhibitor of the E3 ubiquitin ligase APC/CCdc20 which, among other functions, regulates chromosome segregation. BubR1 hypomorphic mice exhibit elevated levels of aneuploidy and, in a variety of tissues, degeneration at early ages5. Conversely, strengthening the mitotic checkpoint through elevated levels of BubR1 has been shown to reduce genetic damage accumulation and to extend the healthy lifespan of mice43. Within a subset of tissues of BubR1 hypomorphic mice (including skeletal muscle, fat, and eye) p16Ink4a is expressed at chronically high levels presumably in response to DNA damage. These tissues exhibit elevated levels of SAβ-galactosidase and other markers of senescence and reduced levels BrdU incorporating cells. Further, BubR1 hypomorphic mice lacking p16Ink4a, and mice in which p16Ink4a expressing cells are continuously ablated, ameliorate degenerative changes specifically in these tissues and exhibit increased numbers of BrdU incorporating cells10, 44. It has been hypothesized that degenerative changes are a consequence of the effects of secretory changes associated with senescent cells on the tissue microenvironment44. The observation here that reduced cell proliferation, even in the absence of the accumulation of senescent cells, is sufficient to result in age related degenerative changes suggests that many of the effects that senescent cells exert on the tissue microenvironment could result from suppression of cell proliferation.
pp53(ser18) levels and DNA damage foci are reduced in proliferating tissues of Cdkn1b over expressing mice, relative to wt animals, suggesting that replication related errors are responsible for much of the genetic damage that occurs during aging. Consistent with this result cancer incidence is greatly reduced in animals in which Cdkn1b is over expressed as adults. Further, unlike p53+/m, telomere deficient mice, or BubR1 hypomorphic mice, senescent cells do not accumulate to elevated levels in Cdkn1b over-expressing mice. Nonetheless, these mice are less robust than wild type and exhibit degenerative tissue changes consistent with an aged phenotype. The apparent improvement in genome maintenance but reduction in tissue homeostasis suggest that Cdkn1b over expression in adult animals alters the balance between genetic and tissue aging.
Cdkn1b expression is not increased during aging in wt animals. Further, in skeletal muscle a sub-set of satellite cells has been shown to break quiescence and increase proliferation in aging mice due to changes in growth factor signaling in the aged niche45. However, it is not necessary that genes contributing to an aged phenotype show a change in their level of activity during aging; rather, simply that their basal level of activity is incompatible with complete tissue maintenance over time46. Loss of Cdkn1b function in knockout mice results in a preferential increase in progenitor cell division relative to stem cells47, suggesting that endogenous Cdkn1b functions to suppress division specifically in progenitor cells but not stem cells. Hence, the modulation of stem and progenitor cell proliferation rates achieved by over-expressing Cdkn1b in the present study is unlikely to reflect a normal role for this gene in control of stem cell division during aging of wild type mice. Nonetheless, to the extent that reduced Cdkn1b expression allows increased division of progenitors without a corresponding increase in stem cell division, the present studies suggest that partial suppression of Cdkn1b activity in aging adult animals could improve tissue maintenance without loss of genome integrity within stem cells.
Methods
Plasmid and Transgenic Mouse Construction
pTRE-Tight-Cdkn1b was constructed by insertion of a 1261bp fragment from pCMV-Sport6-Cdkn1b (Open Biosystems) containing the Cdkn1b coding sequence into multiple cloning site of pTRE-Tight (Invitrogen). Transgenic animals were produced by pronuclear injection of a 1.8 kbp XhoI fragment isolated from pTRE-Tight-Cdkn1b and containing the TRE promoter, Cdkn1b gene and SV40 pA into C567BL6 zygotes. One founder was identified and maintained on a C57Bl6 background using primers designed with the forward primer (TgTRE-F: 5’ GAGTTTACTCCCTATCAGTGATAGAGAACG) in the TRE promoter and the reverse primer (TgTRE-R: CTCCAGGCGATCTGACGGTTC) immediately downstream of the SV40 pA. The TRE-Tight-Cdkn1b mouse line is available to the research community through The Jackson Laboratory (JAX Stock Number 017631).
Mouse crosses and treatments
B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J (Jackson Laboratories, referred to as R26-M2rtTA) mice were crossed with TRE-Tight-Cdkn1b mice to generate bigenic R26-M2rtTA;TRE-Tight-Cdkn1b mice. Bigenic mice were administered doxycycline (Sigma) in their drinking water at the concentrations and durations indicated in the text.
In cases where mice were labeled with IdU and CldU, IdU was administered in the drinking water at 0.5 mg/ml for 3 days or 3 weeks in different experiments and mice were injected with 100 µl of 10 mg/ml CldU in isotonic saline 2 hr prior to the end of the experiment23. All procedures involving mice were approved by the RPCI IACUC.
Immunoblotting
Protein extracts from tissues were prepared using pre-chilled solutions. Tissues were harvested to PBS and transferred to Ependorf centrifuge tubes containing RIPA Buffer: (20 mM Trish (pH 8.0); 150 mM NaCl; 1% NP-40; 1% sodium deoxycholate) containing protease and phosphatase inhibitors (SigmaFast S8820 plus Phosphatase Inhibitors, Sigma, and PhosStop, Roche Cat No 04906845001). Tissues were homogenized in this solution and the mixture was incubated on ice for 1 hour. Tubes were spun in a microcentrifuge for 40 min at 4 °C at maximum speed (14k rpm). The supernatant was collected and protein concentrations were established by BCA assays (Thermo Scientific #23227). The protein concentrations were equalized between different extracts with RIPA buffer. Following immunoblotting (western blot) Cdkn1b, pp53(ser18), and actin were detected using anti-p27(C-19) (1/1000, Santa Cruz Biotechnology Cat No sc-528), anti-pp53(ser18) (1/1000, Cell Signaling Cat No 9284S), or anti-actin (1/1000, Sigma Cat No A2066) primary antibodies and HRP conjugated anti-rabbit secondary antibody (1/2000, Mllipore AP132P GtxRb IgG (H+L) HRP).
Immunohistochemistry
Tissues were paraffin embedded and 6 µm histological sections were prepared using standard techniques. Immunostaining for Cdkn1b, Pax7, myosin, and Mcm2 were performed, following antigen recovery23, using rabbit anti-p27(C-19) (1/200, Santa Cruz Biotechnology Cat No sc-528), rabbit anti-Pax7 (1/200, Santa Cruz Biotechnology Cat No sc-25409), mouse MF20 (1/200, Developmental Studies Hybridoma Bank) and mouse BM28 (1/200, Transduction Laboratories) primary antibodies followed by Alexaflor 594 conjugated donkey anti-rabbit and Alexaflor 486 conjugated donkey anti-mouse secondary antibodies (1/500, Santa Cruz Biotechnology). IdU and CldU were detected for the same locations following stripping of the initial antibodies and DNA denaturation as described previously23 Briefly, slides were treated with 1.5 M HCl for 1 hr at room temperature, washing with PBSA, treated with 0.05–0.25% trypsin for 1–15 min, depending on the tissue, and stained with mouse monoclonal anti-BrdU antibodies (1:200; cross-reactivity with IdU; Becton Dickinson, Mountain View, CA) and rat monoclonal anti-BrdU antibodies (1:200; cross-reactivity with CldU; Accurate Chemicals, Westbury, NY) at 12°C overnight. Following wash with PBSA, secondary anti-mouse Alexaflor 594 and anti-rat Alexaflor 486 antibodies (1:500; Santa Cruz Biotechnology) were then applied for 2 hr at room temperature.
Peripheral Blood Analysis
Blood was collected from mice by retro-orbital bleeding and prepared for FACS analysis using mixture of CD4-Pacific Blue, CD8-Pacific Blue, B220-Pacific Blue, B220-PeCy7, Mac1-PeCy7, and Gr-1-PeCy7 (1/100, all from eBioscience) as described previously48. Briefly, red blood cells were lysed and samples were incubated with antibody mixture for 20 min on ice prior to washing and analysis by FACS using an LSRII (Becton Dickinson).
Supplementary Material
Acknowledgments
This work was supported by grants from the Ellison Medical Foundation, the NIH-NIA (R01AG041854) and the NIH-NCI (CA130995) to SCP. Cost of animal maintenance was supported in part by an NCI-CCS grant to RPCI.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Szilard L. On the nature of the aging process. Proc Natl Acad Sci USA. 1959;45:30–45. doi: 10.1073/pnas.45.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 3.Holt SE, Shay JW, Wright WE. Refining the telomere-telomerase hypothesis of aging and cancer. Nat Biotechnol. 1996;14(7):836–839. doi: 10.1038/nbt0796-836. [DOI] [PubMed] [Google Scholar]
- 4.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 5.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36(7):744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 6.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 7.Schoppy DW, Ruzankina Y, Brown EJ. Removing all obstacles: a critical role for p53 in promoting tissue renewal. Cell Cycle. 2010;9(7):1313–1319. doi: 10.4161/cc.9.7.11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther. 2013;93(1):105–116. doi: 10.1038/clpt.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T, Ganser A, Schirmacher P, Nakauchi H, Rudolph KL. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39(1):99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 10.Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10(7):825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 12.Molofsk AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443(7110):448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 14.Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19(6):756–767. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooke PS, Holsberger DR, Cimafranca MA, Meling DD, Beals CM, Nakayama K, Nakayama KI, Kiyokawa H. The F box protein S phase kinase-associated protein 2 regulates adipose mass and adipocyte number in vivo. Obesity. 2007;15(6):1400–1408. doi: 10.1038/oby.2007.168. [DOI] [PubMed] [Google Scholar]
- 16.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 17.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78(1):59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 18.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8(1):9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 19.Nakayama K, Ishida N, Shirane M, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 20.Kiyokawa H, Kineman RD, Manova-Todorova KO, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Nakayama K, Hatakeyama S. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19(9):2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plikus MV, Chuong CM. Complex hair cycle domain patterns and regenerative hair waves in living rodents. J Invest Dermatol. 2008;128(5):1071–1080. doi: 10.1038/sj.jid.5701180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24(7):1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):898–899. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 25.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 26.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143(1):134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Pruitt SC, Freeland A, Kudla A. Cell cycle heterogeneity in the small intestinal crypt and maintenance of genome integrity. Stem Cells. 2010;28(7):1250–1259. doi: 10.1002/stem.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, Cole AM, Gregorieff A, de Alboran IM, Clevers H, Clarke AR. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol. 2006;26(22):8418–8426. doi: 10.1128/MCB.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenow KR, Clarke AR, Jones RH. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene. 2009;28(11):1443–1453. doi: 10.1038/onc.2008.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1(1):113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Flier LG, van Gijn ME, Hatzis P, Kujala P, Haegebarth A, Stange DE, Begthel H, van den Born M, Guryev V, Oving I, van Es JH, Barker N, Peters PJ, van de Wetering M, Clevers H. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136(5):903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 32.Begus-Nahrmann Y, Lechel A, Obenauf AC, Nalapareddy K, Peit E, Hoffmann E, Schlaudraff F, Liss B, Schirmacher P, Kestler H, Danenberg E, Barker N, Clevers H, Speicher MR, Rudolph KL. p53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice. Nat Genet. 2009;41(10):1138–1143. doi: 10.1038/ng.426. [DOI] [PubMed] [Google Scholar]
- 33.Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signaling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18(8):862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pippa R, Espinosa L, Gundem G, García-Escudero R, Dominguez A, Orlando S, Gallastegui E, Saiz C, Besson A, Pujol MJ, López-Bigas N, Paramio JM, Bigas A, Bachs O. p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene. 2012;31(38):4207–4220. doi: 10.1038/onc.2011.582. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Cañamero M, Rizzoti K, Carneiro C, Martínez G, Vidal A, Lovell-Badge R, Serrano M. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell. 2012;11(6):845–852. doi: 10.1016/j.stem.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131(15):3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 38.Marqués-Torrejón MÁ, Porlan E, Banito A, Gómez-Ibarlucea E, Lopez-Contreras AJ, Fernández-Capetillo O, Vidal A, Gil J, Torres J, Fariñas I. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell. 2013;12(1):88–100. doi: 10.1016/j.stem.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sachs PC, Francis MP, Zhao M, Brumelle J, Rao RR, Elmore LW, Holt SE. Defining essential stem cell characteristics in adipose-derived stromal cells extracted from distinct anatomical sites. Cell Tissue Res. 2012;349(2):505–515. doi: 10.1007/s00441-012-1423-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96(5):701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 41.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415(6867):45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 42.Hinkal GW, Gatza CE, Parikh N, Donehower LA. Altered senescence, apoptosis, and DNA damage response in a mutant p53 model of accelerated aging. Mech Ageing Dev. 2009;130(4):262–271. doi: 10.1016/j.mad.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013;15(1):96–102. doi: 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–2366. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490(7420):355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120(4):437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 47.Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1) Nat Med. 2000;6(11):1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- 48.Mayle A, Luo M, Jeong M, Goodell MA. Flow cytometry analysis of murine hematopoietic stem cells. Cytometry. 2013;83(1):27–37. doi: 10.1002/cyto.a.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.