

Abstract
Purpose.
Accumulation of Alu RNA transcripts due to DICER1 deficiency in the retinal pigmented epithelium (RPE) promotes geographic atrophy. Recently we showed that Alu RNA activated the NLRP3 inflammasome, leading to RPE cell death via interleukin-18 (IL-18)-mediated MyD88 signaling. However, the molecular basis for NLRP3 inflammasome activation by Alu RNA is not well understood. We sought to decipher the key signaling events triggered by Alu RNA that lead to priming and activation of the NLRP3 inflammasome and, ultimately, to RPE degeneration by investigating the roles of the purinoreceptor P2X7, the transcription factor NF-κB, and the Toll-like receptors (TLRs) in these processes.
Methods.
Human and mouse RPE cells were transfected with a plasmid encoding an Alu element (pAlu) or an in vitro-transcribed Alu RNA. Inflammasome priming was assessed by measuring NLRP3 and IL18 mRNA levels by real-time quantitative PCR. Using immunoblotting, we assessed NF-κB activation by monitoring phosphorylation of its p65 subunit, and inflammasome activation by monitoring caspase-1 cleavage into its active form. RPE degeneration was induced in mice by subretinal transfection of pAlu or Alu RNA. The NF-κB inhibitor BAY 11-7082, the P2X7 receptor antagonist A-740003, and the NLRP3 inflammasome inhibitor glyburide were delivered by intravitreous injections. We studied wild-type (WT) C57Bl/6J, P2rx7−/−, Nfkb1−/−, and Tlr23479−/− mice. RPE degeneration was assessed by fundus photography and zonula occludens-1 (ZO-1) staining of mouse RPE.
Results.
Alu RNA-induced NF-κB activation, independent of TLR-1, -2, -3, -4, -6, -7, and -9 signaling, was required for priming the NLRP3 inflammasome. Nfkb1−/− and P2rx7−/− mice and WT mice treated with the pharmacological inhibitors of NF-κB, P2X7, or NLRP3, were protected against Alu RNA-induced RPE degeneration.
Conclusions.
NF-κB and P2X7 are critical signaling intermediates in Alu RNA-induced inflammasome priming and RPE degeneration. These molecules are novel targets for rational drug development for geographic atrophy.
Keywords: AMD, inflammasome, NLRP3
Our studies provide comprehensive analysis of critical signaling pathways regulating Alu RNA-induced inflammasome activation and RPE degeneration. Our data demonstrate that NF-κB and P2X7 are critical signaling intermediates in Alu RNA-induced inflammasome priming and RPE degeneration.
Introduction
Geographic atrophy (GA) is an advanced form of age-related macular degeneration characterized by central loss of vision due to confluent areas of retinal pigmented epithelium (RPE) loss and overlying photoreceptor degeneration.1–3 To date, there is no approved therapy available for this disease, due largely to lack of clear understanding of its molecular pathogenesis. Recently, we showed that the microRNA (miRNA) processing enzyme DICER1 is specifically reduced in the RPE of GA eyes. The reduced DICER1 levels in the RPE result in an increased abundance of Alu RNA transcripts, which in turn promotes RPE cell death.4,5 Under healthy conditions, DICER1-mediated enzymatic processing metabolizes these Alu RNAs into innocuous cleavage fragments; consequently a deficit in DICER1 abundance results in an increased accumulation of toxic Alu RNA transcripts and RPE degeneration.4
Alu RNAs are noncoding transcripts belonging to the Alu family of retrotransposons, an abundant repetitive DNA sequence in the human genome. Typically, Alu RNA is an ∼300 nucleotide (nt) transcript with a double-stranded dimeric secondary structure consisting of right and left arms separated by an A-rich linker.6 Accumulation of these noncoding Alu RNA transcripts due to DICER1 deficiency induced human RPE cell death and RPE degeneration in mice.4 More recent studies identified that Alu RNA cytotoxicity in RPE is mediated by activation of the inflammasome NLRP3 and ensuing interleukin-18 (IL-18) and MyD88 signaling.5
NLRP3, an intracellular pattern recognition receptor (PRR) of the nod-like receptor (NLR) family forms large multiprotein complexes called inflammasomes. A diverse class of signals including cytosolic DNA, RNA, bacteria, and viruses stimulates the NLRP3 inflammasome leading to activation of caspase-1 and secretion of IL-18 and IL-1β.7,8 NLRP3 inflammasome activation models posit the requirement of at least two signals, “priming” and “activation” (Fig. 1A). Priming involves the upregulation of the inflammasome gene expression via various transcriptionally active signaling receptors; activation involves assembly of a multiprotein inflammasome complex and proteolytic processing of caspase-1, IL-18, and IL-1β.7,8
Figure 1.

(A) Two-signal model of the NLRP3 inflammasome is shown: NLRP3 activation requires two signals called “priming” and “activation.” Priming involves induction of inflammasome genes (NLRP3, IL-18, and IL-1β), and activation involves the assembly of a multiprotein scaffold consisting of NLRP3, ASC, and pro-caspase-1, which executes the proteolytic activation of caspase-1. Subsequently, the activated caspase-1 cleaves pro-IL-18 and pro-IL-1β into mature cytokines. Human RPE cells preincubated with vehicle (DMSO) or the NF-κB inhibitor Bay 11-7082 (20 μM) were exposed to Alu RNA via transfection of a plasmid encoding Alu RNA (pAlu) or an empty vector control plasmid (pNull). Alu RNA-induced priming of NLRP3 was assessed by real-time qPCR for (B) NLRP3 mRNA abundance and (C) IL-18 mRNA abundance. n = 3. (D) Protein levels of NLRP3 and IL-18 were induced in human RPE cells exposed to Alu RNA; right panel shows densitometric quantification of the bands on the immunoblot. (E) NF-κB is activated (phospho p65) in human RPE cells exposed to Alu RNA; bar chart shows densitometric quantification of the immunoreactive phospho p65 bands. Gene expression results are expressed as means ± SEM, with P < 0.05 considered statistically significant. n represents the number of experiments from which the data was obtained. Representative immunoblots from 3 independent experiments are shown.
Priming of the NLRP3 inflammasome is regulated by NF-κB activation by various proinflammatory signals emanating from Toll-like receptor (TLR) activation and production of reactive oxygen species (ROS).6,8 The mechanisms regulating the activation step of the NLRP3 inflammasome are ambiguous, although it is clear that P2X7 and ROS are major contributors to this process in multiple systems.7,8 Also it is clear that there is an interplay between P2X7 and ROS processes; for example, P2X7 signaling leads to ROS generation-dependent inflammasome priming.9–11 Interestingly, as we demonstrated, Alu RNA activation of the NLRP3 inflammasome occurred via ROS intermediates.5 Therefore, we investigated whether P2X7 signaling was also involved in Alu RNA-induced inflammasome activation.
Here, we demonstrate that NF-κB signaling and P2X7 activation play key roles in Alu RNA-induced inflammasome priming and activation and RPE degeneration. We also show that Alu RNA-induced NF-κB activation is independent of TLR signaling, suggesting sensing of Alu RNA by an unknown intracellular pattern recognition receptor.
Materials and Methods
Mice
All animal experiments were approved by institutional review committees and carried out in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research. Wild-type (WT) C57BL/6J, Nfkb1−/− (C57BL/6J mice backcrossed for 12 generations), and P2rx7−/− (C57BL/6J mice backcrossed for 7 generations) mice were purchased from Jackson Laboratory (Bar Harbor, ME). “Quintuple knockout” mice simultaneously deficient for TLR2, TLR3, TLR4, TLR7, and TLR9 (Tlr23479−/− mice, C57BL/6J, backcrossed for 10 generations) have been previously described.12 For all procedures, anesthesia was achieved by intraperitoneal injection of 100 mg/kg ketamine hydrochloride (Ft. Dodge Animal Health) and 10 mg/kg xylazine (Phoenix Scientific), and pupils were dilated with topical 1% tropicamide (Alcon Laboratories).
Fundus Photography
Retinal photographs of dilated mouse eyes were taken with a model TRC-50 IX camera (Topcon) linked to a digital imaging system (Sony).
Subretinal Injection
Subretinal injections (1 μL) in mice were given using a Pico-Injector (PLI-100; Harvard Apparatus). In vivo transfection of plasmids encoding Alu sequences (pAlu) or empty control vector (pNull) was carried out by using a DNA transfection reagent (10% NeuroPORTER; Genlantis) as previously described.4,5,13,14 In vitro-transcribed Alu RNA (0.3 μg/μL) was injected as described earlier.4,5
Drug Treatments
Glyburide, glipizide (Sigma-Aldrich), and P2X7 inhibitors (Santa Cruz Biotechnology) dissolved in PBS or dimethyl sulfoxide (DMSO) were injected into the vitreous humor in a total volume of 1 μL with a 33-gauge microsyringe (Exmire; Ito Corp.). WT mice received the inhibitors or vehicle delivered into the vitreous 24 hours before and again 72 hours after exposure to Alu RNA.
Assessment of RPE Degeneration
Alu RNA-mediated RPE degeneration was induced by exposing mice to Alu RNA via subretinal injection of a plasmid encoding an Alu element (pAlu) (or a control plasmid) or an in vitro-transcribed Alu RNA.4,5 Seven days later, RPE health was assessed by fundus photography and immunofluorescence staining of zonula occludens-1 (ZO-1) on RPE flat mounts (whole mount of posterior eye cup containing RPE and choroid layers). Mouse RPE/choroid flat mounts were fixed with 4% paraformaldehyde or 100% methanol, stained with rabbit polyclonal antibodies against mouse ZO-1 (1:100, Invitrogen) and visualized with Alexa594 (Invitrogen). All images were obtained by microscopy (model SP-5, Leica; or Axio Observer Z1, Zeiss).
Cell Culture
All cells were maintained at 37°C in a 5% CO2 environment. Primary mouse and human fetal RPE cells were isolated as previously described.15,16 Primary mouse RPE cells were isolated from 6- to 8-week-old WT C57BL/6J mice. Mouse RPE samples were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 20% fetal bovine serum (FBS) and standard antibiotics concentrations, and primary human RPE cells were maintained in DMEM supplemented with 10% FBS and antibiotics. For examining the role of NF-κB in NLRP3 priming, we preincubated the specific NF-κB inhibitor Bay 11-7082 (Sigma-Aldrich) for 1 hour with RPE cells before stimulating them with Alu RNA.
In Vitro Transcription of Alu RNAs
T7 promoter containing Alu expression plasmid was linearized and used for making in vitro–transcribed Alu RNA by using an in vitro transcription kit (T7-Flash transcription kit; AmpliScribe; Epicenter) following the manufacturer's instructions. The resulting Alu RNA was treated with DNase and purified (MEGAclear; Ambion), and integrity was monitored by gel electrophoresis.
Transient Transfection
Human or mouse RPE cells were transfected with pNull, pAlu, or in vitro–transcribed Alu RNA by using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
Western Blotting
Cells lysed in radioimmunoprecipitation assay (Sigma-Aldrich) lysis buffer supplemented with protease cocktail inhibitor were homogenized by sonication. Protein concentrations were determined using a Bradford assay kit (Bio-Rad) with bovine serum albumin as a standard. Equal amounts of protein samples (20–40 μg) prepared in Laemmli buffer were resolved by SDS-PAGE on Tris-glycine gels (Novex; Invitrogen) and transferred onto polyvinylidene difluoride membranes (Immun-Blot; Bio-Rad). The transferred membranes were blocked for 1 hour at room temperature and incubated with antibodies against caspase-1 (1:1000 dilution; Invitrogen), IL-18 (1:1000 dilution; MBL International), NLRP3 (1:1000 dilution; Enzo Life Sciences) and NF-κB (1:1000; Cell Signaling) at 4°C overnight. Immunoreactive bands were developed by enhanced chemiluminescence reaction. Band intensities on the immunoblots were quantified using ImageJ software (National Institutes of Health).
Real-Time PCR
Human/mouse RPE cells were exposed to Alu RNA via transfection of a plasmid encoding an Alu element (pAlu); 24 hours later, we analyzed inflammasome priming by examining the mRNA levels of pro-IL-18 and NLRP3 by using real-time quantitative RT-PCR (RT-qPCR). Total RNA was extracted from cells using TRIzol reagent (Invitrogen) following the manufacturer's instructions. An equal quantity of DNase-treated RNA was reverse transcribed using a reverse transcription kit (QuantiTect; Qiagen). The RT products (cDNA) were amplified by real-time qPCR (7900 HT Fast real-time PCR system; Applied Biosystems) with SYBR Green detection system. At the end of amplification, melting curve analysis was applied using the dissociation protocol from the sequence detection system to exclude contamination with nonspecific PCR products. For negative controls, no RT products were used as templates in the qPCR. Relative expressions of target genes were determined by the 2−DDCt method using the 18S rRNA housekeeping gene. Oligonucleotide primers specific for human IL18 (forward, 5′-ATCACTTGCACTCCGGAGGTA-3′, and reverse, 5′-AGAGCGCAATGGTGCAATC-3′), human NLRP3 (forward, 5′-GCACCTGTTGTGCAATCTGAA-3′, and reverse, 5′-TCCTGACAACATGCTGATGTGA-3′), human 18S rRNA (forward, 5′-CGCAGC TAGGAATAATGGAATAGG-3′, and reverse, 5′-GCCTCAGTTCCGAAAACCAA-3′), mouse Nlrp3 (forward, 5′-ATGCTGCTTCGACATCTCCT-3′, and reverse, 5′-AACCAATGCGAGATCCTGAC-3′), mouse Il18 (forward, 5′-GACAGCCTGTGTT CGAGGAT-3′, and reverse, 5′-TGGATCCATTTCCTCAAAGG-3′), and mouse 18S rRNA (forward, 5′-TTCGTATTGCGCCGCTAGA-3′, and reverse, 5′-CTTTCGCTCTGGTCCGTCTT-3′) were used.
Statistical Analyses
Real-time RT-qPCR gene expression results and immunoblot densitometry values expressed as means ± standard error of the mean (SEM) were analyzed using two-tailed Student's t-test. The binary readouts of RPE degeneration (i.e., presence or absence of RPE degeneration on fundus and ZO-1-stained flat mount images) were analyzed using χ2 test. P values < 0.05 were deemed statistically significant.
Results
NF-κB Mediates Alu RNA-Induced Inflammasome Priming
Recently, we demonstrated that Alu RNA accumulation due to DICER1 deficiency induced human RPE cell death and RPE degeneration in mice through activation of caspase-1 via NLRP3 inflammasome activation.5 Activation of NLRP3 requires a priming signal to induce adequate levels of NLRP3, IL-18, and IL-1β and an activating signal to promote assembly of an inflammasome multiprotein complex.7,8 In most cases, priming is initiated by activation of additional pattern recognition receptors such as TLRs or through inflammatory cytokines. Importantly, these diverse signaling events converge to activate NF-κB transcription factors. The NF-κB family of transcription factors regulates many cellular responses including inflammation and cell death. Here we examined whether NF-κB signaling is critical for Alu RNA-induced inflammasome priming and RPE degeneration. The expression of Alu RNA through transfection of an Alu expression plasmid (pAlu) induced priming of the NLRP3 inflammasome as indicated by induction of the NLRP3 (Fig. 1B) and IL-18 genes (Fig. 1C). In support of gene expression data, human RPE cells exposed to Alu RNA also showed an increased abundance of pro-IL-18 and NLRP3 proteins (Fig. 1D). Furthermore Alu RNA-induced NLRP3 inflammasome priming was suppressed by an NF-κB inhibitor (Bay 11-7082) (Figs. 1B, 1C). In support of these findings, immunoblotting of protein lysates of human RPE cells exposed to Alu RNA also demonstrated activation of NF-κB, as indicated by increased phosphorylation of its p65 subunit (Fig. 1E). Furthermore, Alu RNA did not induce priming of the NLRP3 inflammasome in RPE cells isolated from Nfkb1−/− mice (Figs. 2A, 2B). Taken together, these findings implicate NF-κB as the key transcription factor regulating Alu RNA-induced NLRP3 inflammasome priming.
Figure 2.
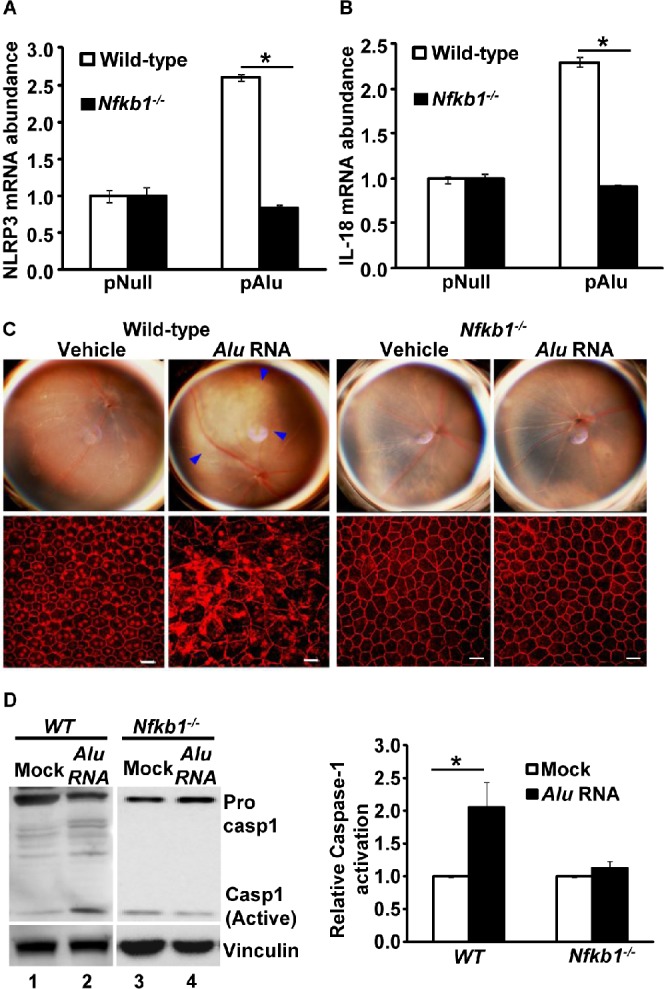
Priming of NLRP3 was analyzed by examining the mRNA abundance of (A) NLRP3 and (B) IL-18 in WT and Nfkb1−/− mouse RPE cells transfected with pAlu or empty vector (pNull). n = 8. (C) Alu RNA causes RPE degeneration in WT mice but not in Nfkb1−/− mice: WT and Nfkb1−/− mice were exposed to Alu RNA by subretinal injection. n = 8. Alu RNA-induced RPE degeneration was assessed by fundus examination and ZO-1 staining of RPE flat mount. Disrupted RPE cell boundaries shown by ZO-1 staining of RPE flat mounts indicate RPE degeneration. Blue arrowheads in the fundus picture indicate areas of RPE degeneration (P = 0.0002, χ2 test). (D) WT and Nfkb1−/−mouse RPE cells were exposed to Alu RNA, and NLRP3 activation was assessed by examining the activation of caspase-1 by Western blotting; the densitometric quantification of active caspase-1 (p20) bands in the immunoblots is presented in the bar graph.
NF-κB-Mediated Signaling Is Required For Alu RNA-Induced RPE Degeneration
Our cell culture studies with genetic and pharmacological approaches suggested that NF-κB is critical in regulating NLRP3 inflammasome priming. Next, using Nfkb1−/− mice, we tested the role of NF-κB in mediating Alu RNA-induced mouse RPE degeneration. Recently we established a model for in vivo testing of Alu RNA-induced RPE degeneration4,5 by subretinal delivery of Alu RNA expression plasmid (pAlu) or of in vitro-transcribed Alu RNA. Whereas Alu RNA induced RPE degeneration in WT mice, it did not do so in Nfkb1−/− mice (Fig. 2C). To further confirm our rationale that loss of Alu RNA-induced RPE degeneration in Nfkb1−/− mice was indeed due to impaired NLRP3 activation in the absence NF-κB-mediated priming signals, we assessed the ability of Alu RNA to induce caspase-1 activation in Nfkb1−/− mouse RPE cells. As anticipated, Alu RNA was unable to activate caspase-1 in Nfkb1−/− mouse RPE cells (Fig. 2D, compare active casp1 in lane 3 versus that in lane 4) compared to marked caspase-1 activation in WT RPE cells (Fig. 2D, compare active casp1 in lane 1 versus that in lane 2). Collectively, these findings demonstrate that intact NF-κB signaling is a critical regulatory checkpoint in the Alu RNA-induced inflammasome activation and RPE degeneration.
Alu RNA-Induced NF-κB Activation and Inflammasome Priming Are Independent of TLR Signaling
Because Alu RNA-induced NLRP3 inflammasome activation and RPE degeneration were regulated by NF-κB, we next examined the upstream signaling pathways involved in mediating these events. TLRs are an important set of cellular PRRs that detect various structural motifs, including intracellular double-stranded RNA (dsRNA).17 We tested whether Alu RNA, with its double-stranded dimeric secondary structure, activates TLR signaling leading to NF-κB-mediated priming of the NLRP3 inflammasome. To answer this question, we examined activation of NF-κB and inflammasome priming in Tlr23479−/− mouse RPE cells. Surprisingly, NF-κB activation following Alu RNA treatment persisted in TLR signaling-deficient mice (Fig. 3A). Corroborating these data, induction of the IL-18 gene in mice lacking multiple TLRs (Tlr23479−/− mice) was similar to that in WT mice (Fig. 3B). Collectively, these findings demonstrate that Alu RNA-induced NF-κB activation and NLRP3 inflammasome priming occur in a manner independent of TLR activation, suggesting the existence of noncanonical, TLR-independent NF-κB activation and priming of the NLRP3 inflammasome.
Figure 3.
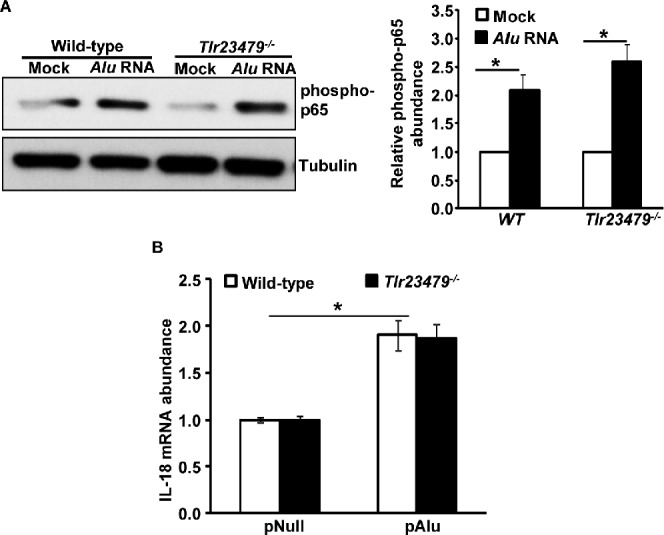
(A) Alu RNA-induced NF-κB activation is independent of signaling via TLR2/3/4/7/9; bar chart at right shows densitometric quantification of the phospho p65 bands on the immunoblot. (B) Alu RNA-induced induction of the IL-18 gene is independent of TLR2/3/4/7/9 signaling. A representative immunoblot from three independent experiments is shown.
P2X7 Mediates Alu RNA-Induced RPE Degeneration/Inflammasome Activation
NLRP3 inflammasome induction is tightly regulated, with multiple signaling pathways controlling priming and assembly of the multiprotein inflammasome complex. Although the signaling pathways regulating priming are well understood, pathways orchestrating the assembly of the multiprotein complex leading to caspase-1 activation remain nebulous. We tested whether P2X7, a well-known inflammasome activator in other systems, is critical for Alu RNA-induced activation of the NLRP3 inflammasome and RPE degeneration. RPE cells isolated from WT and P2rx7−/− mice were transfected with Alu RNA, and activation of caspase-1 was assessed by immunoblotting. Alu RNA induced the proteolytic cleavage of pro-caspase-1 into its active p20 subunit in WT mouse RPE (mRPE) cells (Fig. 4A, compare lanes 1 and 2) but not in P2rx7−/− mouse RPE cells (Fig. 4A, compare lanes 3 and 4). We found that intravitreous administration of the sulfonylurea glyburide, an NLRP3 inflammasome inhibitor acting downstream of P2X7 and upstream of NLRP3 inflammasome assembly,18 protects against RPE degeneration induced by Alu RNA in WT mice (Fig. 4B). In contrast, glipizide, a related sulfonylurea that does not inhibit NLRP3, did not inhibit Alu RNA-induced RPE degeneration. We also observed that, intravitreous delivery of P2X7 inhibitor confers protection against the RPE degeneration induced by Alu RNA (Fig. 4C). Corroborating these findings, we also observed that Alu RNA did not induce RPE degeneration in P2rx7−/− mice (Fig. 4D). Collectively these findings suggest that P2X7 is an essential intermediate in Alu RNA-induced activation of NLRP3 inflammasome and consequent RPE degeneration.
Figure 4.

(A) WT and P2rx7−/− mouse RPE cells were exposed to Alu RNA, and NLRP3 activation was assessed by examining the activation of caspase-1 by Western blotting; densitometric quantification of active caspase-1 (p20) bands in the immunoblot is shown in the bar graph. (B) The NLRP3 inflammasome inhibitor glyburide, which acts downstream of P2X7 receptors, suppressed Alu RNA-induced RPE degeneration in mice. The structurally similar control inhibitor glipizide, which is not an inhibitor of NLRP3, did not suppress Alu RNA-induced RPE degeneration. n = 12 (P = 0.0001, χ2 test). (C) The P2X7 inhibitor suppressed Alu RNA (pAlu)-induced RPE degeneration in WT mice. n = 8 (P = 0.0001, χ2 test). (D) P2X7-deficient mice are not susceptible to Alu RNA-induced RPE degeneration. n = 8 (P = 0.0001, χ2 test). Immunoblots shown in the figure are representative of 3 independent experiments. Blue arrowheads in the fundus picture indicate areas of RPE degeneration. The fundus and ZO-1-stained flat mount images were statistically analyzed using the χ2 test. n represents the number of experiments or animals from which the data was obtained.
Discussion
Recently we reported that Alu RNA accumulation due to DICER1 deficiency induced RPE cell death via activation of the NLRP3 inflammasome.5 The present study expands those findings and provides insight into the signaling pathways that regulate priming and activation of the NLRP3 inflammasome in the context of GA. Using both cell culture and mouse models, we demonstrated the critical importance of NF-κB and P2X7 in mediating Alu RNA-induced RPE degeneration.
NF-κB transcription factors play an important role in orchestrating host inflammatory and immune responses and modulate cellular growth properties by regulating the expression of specific set of cellular genes.19 Similar to inflammasome activation by microbial pathogen-associated molecular patterns (PAMPs), LPS/ATP, toxins, and other triggers, Alu RNA-induced inflammasome priming/activation also depended on the NF-κB-mediated transcriptional activation of NLRP3 and IL-1 cytokines.8,20,21 Assembly of the NLRP3 multiprotein scaffold is influenced by the integration of various proinflammatory signaling pathways. These proinflammatory pathways potentiate inflammasome priming through NF-κB-mediated induction of NLRP3 and IL-1 levels. Furthermore, Alu RNA-induced RPE degeneration was not observed in Nfkb1−/− mice, suggesting that NF-κB inhibition could be an attractive target for GA.
Most experimental protocols studying inflammasome activation use priming with TLR agonists such as LPS, which primes the NLRP3 inflammasome through NF-κB activation.22–27 In the current model, Alu RNA-induced NLRP3 inflammasome priming and RPE degeneration are independent of TLR signaling. Our earlier studies demonstrated that other intracellular RNA sensors such as retinoic acid-inducible gene 1 (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and protein kinase R (PKR) were also not required for Alu RNA-induced RPE degeneration.5 In light of our present study, it appears that further studies are required to elucidate the precise sensor(s) of Alu RNA that enables its induction of NF-κB activation and NLRP3 priming.
The exact mechanism by which NLRP3 is activated remains elusive. Lysosomal destabilization, ROS production, and activation of P2X7 receptor are among the widely supported mechanisms.7,8 Furthermore, the literature suggests that these pathways are not mutually exclusive.7–11 In agreement with those studies, our findings with P2X7, combined with recent studies also implicating ROS in the Alu RNA-induced NLRP3 inflammasome activation, suggest that the complex interplay between ROS and P2X7 is critical for RPE degeneration in the context of Alu RNA cytotoxicity. P2X7 is a ligand-gated ion channel whose activation by high extracellular ATP leads to opening of ion channels that causes a decrease in intracellular K+ levels, which in turn leads to NLRP3 activation.7,28 Future studies will test whether Alu RNA also induces ATP release and K+ efflux via P2X7.
Collectively, our studies provide greater insight into critical signaling pathways regulating Alu RNA-induced inflammasome activation and RPE degeneration (Fig. 5). Identification of NF-κB and P2X7 as critical signaling intermediates regulating Alu RNA-induced RPE degeneration suggests that targeting these pathways might be a useful strategy for rational drug development for treating GA. In addition, our data provide a rationale for testing glyburide, an agent widely used in treatment of type 2 diabetes, in treating GA. Glyburide is a functional inhibitor of ATP-sensitive potassium channels (KATP). Because the KATP channels in pancreatic β cells regulate the secretion of insulin, glyburide's inhibitory effect on KATP is exploited in the treatment of type 2 diabetes.29 Interestingly, glyburide's ability to suppress NLRP3 activation is independent of its inhibitory effect on KATP, as glipizide, a related antidiabetic sulfonylurea drug that also potently blocks KATP, does not inhibit NLRP3.18 Currently it is known only that glyburide acts upstream of NLRP3 and downstream of P2X718; the precise molecular target of glyburide in inhibiting the NLRP3 inflammasome has yet to be identified.
Figure 5.
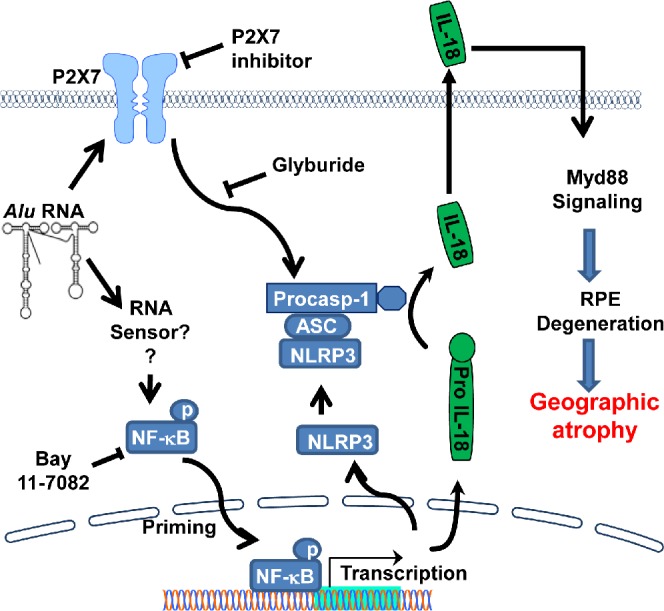
Schematic shows critical signaling pathways regulating NLRP3 priming and activation. Alu RNA-induced NF-κB-mediated transcriptional activation of inflammasome genes (NLRP3 and IL-18) and signaling via P2X7 receptors control NLRP3 inflammasome priming and activation, respectively. Identification of these key signaling events provides critical mechanistic insights into the recent implication of NLRP3 inflammasome activation and IL-18-induced Myd88 signaling in the pathogenesis of GA.5
Acknowledgments
The authors thank Charles Payne, Gary Pattison, Gregory Botzet, Robinette King, Li Xu, Darrell Robertson, Lindsay Toll, and Annette Uittenbogaard for technical assistance.
Supported by National Eye Institute/National Institutes of Health (NIH) Grants R01EY018350, R01EY018836, R01EY020672, and R01EY022238, a Doris Duke Distinguished Clinical Scientist Award, a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research, an Ellison Medical Foundation Senior Scholar in Aging Award, the Dr. E. Vernon Smith and Eloise C. Smith Macular Degeneration Endowed Chair, the Carl Reeves Foundation, and Research to Prevent Blindness departmental unrestricted grant (JA); by Beckman Initiative for Macular Research (NK); by Programme for Advanced Medical Education, sponsored by Fundação Calouste Gulbenkian, Fundação Champalimaud, Ministério da Saúde and Fundação para a Ciência e Tecnologia, Portugal (ABC); by American Heart Association and the National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH, through Grant UL1TR000117 (BDG); by the Arnold and Mabel Beckman Foundation (DRH); and by NIH T32HL091812 and UL1RR033173 (BJF). The content of the article is the sole responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure: N. Kerur, None; Y. Hirano, None; V. Tarallo, None; B.J. Fowler, None; A. Bastos-Carvalho, None; T. Yasuma, None; R. Yasuma, None; Y. Kim, None; D.R. Hinton, None; C.J. Kirschning, None; B.D. Gelfand, None; J. Ambati, P
References
- 1. Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012; 75: 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bird AC. Therapeutic targets in age-related macular disease. J Clin Invest. 2010; 120: 3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013; 13: 438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011; 471: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tarallo V, Hirano Y, Gelfand BD, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012; 149: 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet . 2009; 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012; 13: 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140: 821–832 [DOI] [PubMed] [Google Scholar]
- 9. Bartlett R, Yerbury JJ, Sluyter R. P2X7 receptor activation induces reactive oxygen species formation and cell death in murine EOC13 microglia. Mediators Inflamm. 2013: 271813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007; 282: 2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang B, Sluyter R. P2X7 receptor activation induces reactive oxygen species formation in erythroid cells. Purinergic Signal. 2013; 9: 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oldenburg M, Krüger A, Ferstl R, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science. 2012; 337: 1111–1115 [DOI] [PubMed] [Google Scholar]
- 13. Bennett EA, Keller H, Mills RE, et al. Active Alu retrotransposons in the human genome. Genome Res. 2008; 18: 1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaikh TH, Roy AM, Kim J, Batzer MA, Deininger PL. cDNAs derived from primary and small cytoplasmic Alu (scAlu) transcripts. J Mol Biol. 1997; 271: 222–234 [DOI] [PubMed] [Google Scholar]
- 15. Yang Z, Stratton C, Francis PJ, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engle J Med. 2008; 359: 1456–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang P, Tyrrell J, Han I, Jaffe GJ. Expression and modulation of RPE cell membrane complement regulatory proteins. Invest Ophthalmol Vis Sci. 2009; 50: 3473–3481 [DOI] [PubMed] [Google Scholar]
- 17. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006; 124: 783–801 [DOI] [PubMed] [Google Scholar]
- 18. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009; 187: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perkins ND. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci. 2000; 25: 434–440 [DOI] [PubMed] [Google Scholar]
- 20. Kanneganti TD, Body-Malapel M, Amer A, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006; 281: 36560–36568 [DOI] [PubMed] [Google Scholar]
- 21. Kanneganti TD, Ozören N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006; 440: 233–236 [DOI] [PubMed] [Google Scholar]
- 22. Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008; 105: 9035–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008; 320: 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muruve DA, Pétrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008; 452: 103–107 [DOI] [PubMed] [Google Scholar]
- 25. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008; 9: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006; 440: 237–241 [DOI] [PubMed] [Google Scholar]
- 27. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006; 440: 228–232 [DOI] [PubMed] [Google Scholar]
- 28. Miller CM, Boulter NR, Fuller SJ, et al. The role of the P2X(7) receptor in infectious diseases. PLoS Pathog. 2011; 7: e1002212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005; 115: 2047–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]