

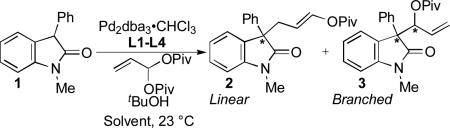
Asymmetric allylic alkylation (AAA)[1] is a powerful and versatile method for the catalytic, asymmetric construction of quaternary stereocenters.[2] In the Mo- and Pd-AAA in particular, a broad range of functionalized electrophiles has been utilized, enabling the rapid synthesis of complex molecules in an atom- and step-economical manner.[3,4] Geminal dicarboxylates (e.g., allylidene dicarboxylates) constitute an under-explored set of electrophiles in this area.[5]
We envisioned a new Pd-AAA reaction employing prochiral nucleophiles and geminal dicarboxylate electrophiles, one that would yield a quaternary stereocenter bearing a linear, three-carbon, enol carboxylate unit. This enol carboxylate could be transformed into a variety of useful functional groups, such as the saturated protected alcohol or the aldehyde arising from a formal Michael addition to acrolein. However, previous research has indicated that geminal dicarboxylate-derived, chiral π-allylpalladium complexes generally undergo intermolecular reaction at the ipso position, affording branched products.[6] In addition to requiring regioselective formation of linear products, the proposed transformation must also proceed with high chemo- and enantioselectivity. When examining prochiral nucleophiles in the Pd-AAA, the latter parameter represents an especially significant challenge, as the nucleophile must approach the π-allylpalladium distal to its chiral information, potentially rendering enantiodiscrimination difficult.[7]
We selected oxindoles as nucleophiles for this transformation as the 3,3-disubstituted oxindole structural motif and its derivatives feature prominently in biologically active natural products and pharmaceutical compounds. Moreover, the enantioselective construction of 3,3-disubstituted oxindoles is a significant synthetic challenge.[8] Further, an asymmetric Michael addition to acrolein represents its own challenge. Possibly owing to the tendencies for acrolein to undergo 1,2- (vs. 1,4-) reaction and to polymerize,[9] only one strategy for the asymmetric Michael addition of an oxindole to acrolein has been reported by Maruoka and co-workers[10] and only for a limited set of N-Boc, 3-Ph oxindoles (Scheme 1).[11]
Scheme 1.

Asymmetric Michael Additions of Oxindoles to Acrolein
Maruoka and co-workers have demonstrated the conversion of these products into valuable alkaloid structural motifs, underscoring synthetic interest in this transformation.[10] Though it stands out as a pioneering result, this process requires cryogenic temperatures, excess base, and dilute conditions. We postulated that our Pd-AAA approach to an enol carboxylate would, via a subsequent hydrolysis stage, enable a formal Michael addition that could be performed at ambient temperature, higher concentration, and with sufficient chemoselectivity to employ unprotected oxindoles.
Our initial discovery efforts focused on the alkylation of oxindole nucleophile 1, in the presence of Pd2dba3•CHCl3 ligated by our DPPBA ligands (Table 1). Drawing on our prior methodology for the allylation of this substrate,[8h] PhMe was used as the reaction solvent, with tBuOH included as an additive. Unsurprisingly, the reaction of this nucleophile with allylidene diacetate led to a complex mixture of branched, linear, aldehydic, and possibly dimeric products. However, switching to the bulkier allylidene dipivalate provided much cleaner reactivity as well as higher selectivity for linear product 2 over branched product 3. Importantly, this electrophile is readily prepared in one step and in very good yield (81%) on gram-scale, from the reaction of acrolein with pivalic anhydride (see Supporting Information).[12]
Table 1.
Selected Optimization Experiments[a]
| |||||
|---|---|---|---|---|---|
| Entry | Solvent | Ligand | % Conv. / % Yield (2 + 3)b | l:b (2:3)b | % ee (2)f |
| 1 | PhMe | L1 | 68 / 65 | 4.9:1 | 60 |
| 2 | PhMe | L2 | 81 / 80 | > 19:1 | 91 |
| 3 | PhMe | L3 | 9 / 8 | -- | -- |
| 4 | PhMe | L4 | < 5 / < 5 | -- | -- |
| 5 | Dioxane | L2 | 66 / 65 | > 19:1 | 76 |
| 6 | DCE | L2 | 53 / 52 | 5.8:1 | 83 |
| 7 | THF | L2 | > 95 / > 95 | > 19:1 | 92 |
| 8c | THF | L2 | > 95 / 91d | > 19:1 | 92 |
| 9 | tBuOH | L2 | > 95 / 85 | > 19:1 | 76 |
| 10e | PhMe | L2 | 75 / 65 | > 19:1 | 90 |
| 11e | THF | L2 | > 95 / 75 | > 19:1 | 92 |
|
|
||||
|
|
||||
Unless specified otherwise, reactions were performed with 0.05 mmol 1, 5.0 mol % Pd2dba3•CHCl3, 15.0 mol % L1-L4, 1.5 equiv electrophile, and 5.0 equiv tBuOH at 0.20 M for 24 h.
Determined by 1H NMR analysis of the crude reaction mixture, with mesitylene as an internal standard.
Reaction performed with 0.10 mmol 1, 2.5 mol % Pd2dba3•CHCl3, 7.5 mol % L2, 1.5 equiv electrophile, and 5.0 equiv tBuOH at 0.40 M for 24 h.
Isolated yield after chromatography.
Reactions performed without tBuOH.
Determined by chiral HPLC.
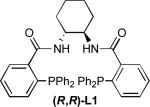
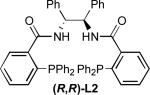
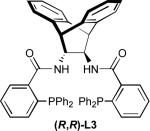
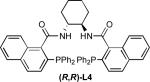
We evaluated this reaction with respect to our standard ligand set, to optimize the formation of 2 (Entries 1-4). Although L1 provided 2 with moderate conversion, regioselectivity, and enantioselectivity, the use of stilbene-derived ligand L2 provided very good conversion, essentially complete selectivity for the desired linear product, and excellent enantioselectivity. Surprisingly, ligands L3 and L4 afforded low conversion. Notably, the addition of exogenous base (e.g., Et3N, Cs2CO3, or tetramethylguanidine) led to poorer regio- and/or enantioselectivity, implicating significant counter-ion effects and suggesting that the hydroxyoxindole tautomer of 1 is the best nucleophile in terms of selectivity.
The effect of the reaction solvent was next explored (Entries 5-7), whereupon it was discovered that, when performed in THF, the reaction was complete within 24 h yet maintained excellent regio- and enantioselectivity. To improve reaction efficiency, the effects of catalyst loading and concentration were also investigated (Entry 8). Pleasingly, halving the catalyst loading (to 2.5 mol % Pd2dba3•CHCl3 and 7.5 mol % L2) while simultaneously doubling the reaction concentration (to 0.40 M) led to essentially identical reactivity (>95% conversion, 91% isolated yield, 92% ee) within the same time period.
The effect of the tBuOH additive was also investigated (Entries 9-11). Reactions performed in PhMe or THF without tBuOH resulted in good conversion and high regio- and enantioselectivity; however, yields of 2 were diminished compared to reactions that included tBuOH. Full conversion occurred when the reaction was performed in pure tBuOH, but the enantioselectivity decreased. These results suggest that the mixed PhMe/tBuOH or THF/tBuOH systems afford high enantioselectivity while minimizing side reactions, potentially including further reactions of 2 or 3.
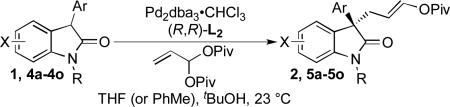
With these optimized conditions in hand, the substrate scope was evaluated. Thus, a number of oxindole nucleophiles were prepared according to standard procedures and subjected to the optimized reaction conditions (Table 2).
Table 2.
Scope of Pd-AAA with Oxindole Nucleophiles[a]
| ||||
|---|---|---|---|---|
| Product | R | Yield (%)b | l:bc | ee (%)d |
|
H (2) p-OMe (5a) p-CF3 (5b) p-Br (5c) p-Cl (5d) m-Me (5e) |
91 (90)e 88 98 82 93 87 |
>19:1 (> 19:1)e >19:1 >19:1 >19:1 >19:1 >19:1 |
92 (90)e 83 90 88 88 88 |
|
89 | > 19:1 | 90 | |
|
88 | > 19:1 | 94 | |
|
91 | > 19:1 | 96 | |
|
96 | > 19:1 | 95 | |
|
82 | > 19:1 | 93 | |
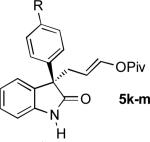
|
H (5k) OMe (5l) F (5m) |
96 (91)f 88 97 |
13:1 (13:1)f 11:1 17:1 |
90 (89)f 84 89 |
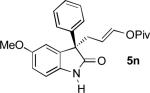
|
75 | 9:1 | 87 | |
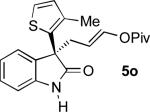
|
93 | 14:1 | 92 | |
Unless specified otherwise, reactions were performed with 0.10 mmol 1 or 4a-4o, 2.5 mol% Pd2dba3•CHCl3, 7.5 mol % L2, 1.5 equiv electrophile, and 5.0 equiv tBuOH at 0.40 M in PhMe or THF for 18 - 72 h.
Isolated yield of both regioisomers after chromatography.
Determined by 1H NMR analysis.
Determined by chiral HPLC.
Reaction performed on 1.00 mmol scale.
Reaction performed on 0.50 mmol scale.
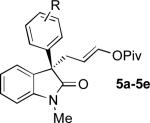
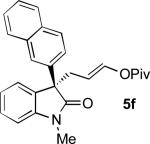
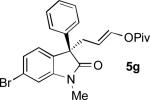
The Pd-AAA reaction was compatible with significant structural variation on both the aryl group and the oxindole core. Notably, electron-neutral (2), electron-rich (5a), and electron-deficient (5b-5d) aryl substituents were all well-tolerated.[13,14] These included aryl halides, which underwent the Pd-AAA in high yield and without competitive oxidative addition reactions. Further, the steric demands of the nucleophile could be increased without ill effect, as meta-substituted product 5e and 2-naphthyl product 5f were obtained in high yield and with excellent selectivity. Halogen substitution on the oxindole core was also tolerated (5g), again without competitive oxidative addition.
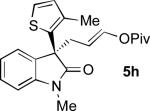
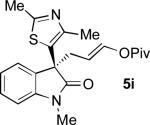
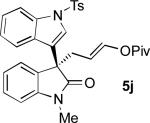
Oxindoles bearing heteroaryl substituents at the 3-position proved among the best substrates for this reaction, affording very high yields and enantioselectivities (5h-5j). Especially notable is the synthesis of compound 5j, as 3-indole oxindoles are established precursors to biologically active indole alkaloids.[15] Specifically, the 3-indole, 3-enol pivalate compound 5j bears similarity to synthetic intermediates toward gliocladin C.[8m,15]
To our delight, unprotected oxindoles underwent the Pd-AAA with high chemoselectivity for alkylation at the 3-position. Thus, products 5k-5o were obtained in high yield and enantioselectivity, with only slight decreases in regioselectivity.[16] Importantly, this compatibility oxindoles also extended to a 3-heteroaryl nucleophile, as evidenced by the synthesis of 5o with high yield and selectivity. Furthermore, 5-substitution on the oxindole core was tolerated (5n).
Although small in magnitude, an interesting substituent effect was observed in the unprotected oxindole series: while yields and enantioselectivities were generally consistent for these substrates, a more electron-deficient substrate (5m) afforded slightly greater regioselectivity than an electron-neutral substrate (5k), which in turn afforded slightly greater selectivity than electron-rich substrates (5l, 5n). This suggests that a subtle electronic influence may exist wherein the oxindole enolate is destabilized by electron-donating substituents, resulting in a slightly greater propensity to attack the π-allylpalladium at its more electrophilic ipso position, leading to more branched product.[17]
Having demonstrated the utility of this method for the synthesis of enantioenriched enol pivalates, we investigated its application toward the synthesis of functionalized products (Scheme 2). Focusing first on conversion to the corresponding aldehyde, we were pleased to find that simply treating 2 with methanolic KOH cleanly provided the desired aldehyde 6 in a gratifying 84% yield, without competitive aldol or retro-Michael reactions. Theorizing that these reaction conditions would not be incompatible with those from the Pd-AAA reaction, we investigated a one-pot transformation of 1 to 6. In this event, the Pd-AAA reaction was performed according to the standard procedure. After complete consumption of 1, the reaction mixture was directly treated with the basic solution under ambient conditions, efficiently providing 6 in only slightly decreased yield, validating our original proposal for this application of the subject Pd-AAA reaction.
Scheme 2.
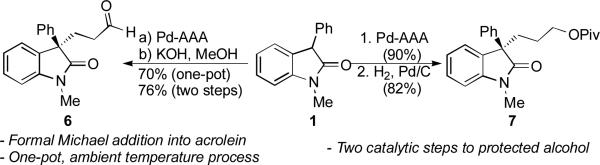
Elaboration of Enol Pivalate 2
In addition, the catalytic hydrogenation of enol pivalate 2 could be accomplished in good yield, affording protected alcohol 7. Related 3,3-disubstituted oxindoles featuring a three-carbon alcohol have attracted attention from the pharmaceutical industry and in natural products synthesis,[18] and this strategy of two ambient temperature, Pd-catalyzed steps provides a practical alternative to, for example, an allylation/hydroboration/oxidation/protection sequence. This application of the present Pd-AAA is especially significant, as the enantioselectivity of this reaction is markedly greater than that of the simple allylation of similar nucleophiles.[8h]
The hydrolysis of N-unprotected enol pivalate 5k was also accomplished, smoothly affording aldehyde 8 in very good yield (Scheme 3). Initial attempts to convert this product to known N-Boc aldehyde 9 were complicated by competitive N-Boc and enol-Boc formation. However, chemoselective N-Boc protection could be accomplished by treating 8 with NaH at low temperature and subsequently quenching the mixture with Boc2O. Aldehyde 9 was thus obtained in good yield, revealing the absolute stereochemistry of enol pivalate 5k and demonstrating our ability to selectively functionalize the oxindole nitrogen.[19]
Scheme 3.
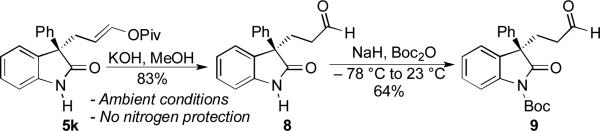
Elaboration and Stereochemical Analysis of 5k
In conclusion, we report a new Pd-AAA reaction of oxindole nucleophiles with allylidene dipivalate, one which proceeds under mild, practical conditions and which regioselectively provides enantioenriched products bearing a synthetically useful linear enol pivalate unit. This reaction tolerates a variety of substitution patterns, including 3-heteroaryl substitution and unprotected oxindoles. The synthetic utility of the enol pivalate product has been illustrated by the one-pot, ambient temperature conversion of the nucleophile to the aldehyde arising from Michael addition to acrolein and by the conversion to a protected alcohol unit. Additional applications of this new Pd-AAA are the subject of continued study in our laboratories, and these results will be reported in due course.
Experimental Section
Representative Procedure for Pd-AAA: A reaction vial was charged with a stir bar, oxindole 1 (223.0 mg, 1.0 mmol, 1 equivalent), Pd2dba3•CHCl3 (25.9 mg, 0.025 mmol, 0.025 equivalent), and (R,R)-L2 (59.2 mg, 0.075 mmol, 0.075 equivalent). The vial was sealed with a septum-lined cap and evacuated and backfilled with N2 three times. THF (2.5 mL) was added, followed by tBuOH (478 μL, 370 mg, 5.0 mmol, 5.0 equivalents). The reaction mixture was stirred until it was homogeneous and an orange color persisted (ca. 10 min), then allylidene dipivalate (387 μL, 364 mg, 1.5 mmol, 1.5 equivalents) was added. The gas inlet was removed, and the vial was sealed thoroughly. The reaction mixture was stirred at room temperature for 24 h, then the septum cap was removed and the reaction mixture was poured into a mixture of Et2O (50 mL) and saturated aqueous NaHCO3 (50 mL). The phases were separated, and the aqueous phase was extracted with Et2O (50 mL). The pooled organic phases were washed with water (2 × 25 mL) then brine (25 mL), dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (6:1 to 4:1 hexanes:EtOAc), affording 2 (327 mg, 90%, >19:1 linear:branched, 90% ee) as a viscous, light yellow oil.
Supplementary Material
Footnotes
We thank the National Institutes of Health (R01GM033049) and the National Science Foundation for their generous support of our programs. J.T.M. is supported by an Abbott Laboratories Stanford Graduate Fellowship. We also thank Johnson-Matthey for their generous gift of palladium salts.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For reviews, see: Trost BM, van Vranken DL. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804.Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w.Trost BM, Machacek MR, Aponick A. Acc. Chem. Res. 2006;39:747. doi: 10.1021/ar040063c.Lu Z, Ma S. Angew. Chem. 2008;120:264.Angew. Chem. Int. Ed. 2008;47:258.Trost BM. Org. Process Res. Dev. 2012;16:185. doi: 10.1021/op200294r.
- 2.For reviews covering the asymmetric synthesis of quaternary stereocenters, see: Corey EJ, Guzman-Perez A. Angew. Chem. 1998;110:402. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Angew. Chem. Int. Ed. 1998;37:388.Christoffers J, Mann A. Angew. Chem. 2001;113:4725.Angew. Chem. Int. Ed. 2001;40:4591.Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363. doi: 10.1073/pnas.0307113101.Christoffers J, Baro A. Adv. Synth. Catal. 2005;347:1473.Trost BM, Jiang C. Synthesis. 2006;3:369.Christoffers J, Baro A, editors. Quaternary Stereocenters. Wiley-VCH; Weinheim: 2005. Das JP, Marek I. Chem. Commun. 2011;47:4593. doi: 10.1039/c0cc05222a.
- 3.a Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; b Trost BM. Angew. Chem. 1995;107:285. [Google Scholar]; Angew. Chem. Int. Ed. 1995;34:259. [Google Scholar]
- 4.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 5.a Trost BM, Lee CB, Weiss JM. J. Am. Chem. Soc. 1995;117:7247. [Google Scholar]; b Trost BM, Lee CB. J. Am. Chem. Soc. 1998;120:6818. [Google Scholar]; c Trost BM, Lee CB. J. Am. Chem. Soc. 2001;123:3671, 3687. doi: 10.1021/ja003774o. [DOI] [PubMed] [Google Scholar]
- 6.See ref 5a. It has been observed that dimethyl methylmalonate reacts with allylidene diacetate to provide the linear product when PPh3 is used as the ligand; however, the use of ligand L1 with this system favors branched material. See also Trost BM, Vercauteren J. Tetrahedron Lett. 1985;26:131.
- 7.See Trost BM, Radinov R, Grenzer EM. J. Am. Chem. Soc. 1997;119:7879.Trost BM, Ariza X. J. Am. Chem. Soc. 1999;121:10727.Trost BM, Schroeder GM. Chem. Eur. J. 2005;11:174., and references therein.
- 8.For reviews covering the biological significance of 3,3-disubstituted oxindoles and their derivatives as well as the asymmetric synthesis of these compounds, see: Dounay AB, Overman LE. Chem. Rev. 2003;103:2945. doi: 10.1021/cr020039h.Marti C, Carreira EM. Eur. J. Org. Chem. 2003:2209.Galliford CV, Scheidt KA. Angew. Chem. 2007;119:8902. doi: 10.1002/anie.200701342.Angew. Chem. Int. Ed. 2007;46:8748.Steven A, Overman LE. Angew. Chem. 2007;119:5584. doi: 10.1002/anie.200700612.Angew. Chem. Int. Ed. 2007;46:5488.Trost BM, Brennan MK. Synthesis. 2009;18:3003.Zhou F, Liu Y-L, Zhou J. Adv. Synth. Catal. 2010;352:1381.Klein JEMN, Taylor RJK. Eur. J. Org. Chem. 2011:6821. and references therein. For the asymmetric allylic alkylation and benzylation of oxindole nucleophiles using Mo- and Pd-AAA, see: Trost BM, Frederiksen MU. Angew. Chem. 2005;117:312.Angew. Chem. Int. Ed. 2005;44:308.Trost BM, Zhang Y. J. Am. Chem. Soc. 2006;128:4590. doi: 10.1021/ja060560j.Trost BM, Zhang Y. J. Am. Chem. Soc. 2007;129:14548. doi: 10.1021/ja0755717.Trost BM, Zhang Y. Chem. Eur. J. 2010;16:296. doi: 10.1002/chem.200902770.Trost BM, Czabaniuk LC. J. Am. Chem. Soc. 2010;132:15534. doi: 10.1021/ja1079755.Trost BM, Xie J, Sieber JD. J. Am. Chem. Soc. 2011;133:20611. doi: 10.1021/ja209244m.
- 9.a Zweifel GS, Nantz MH. Modern Organic Synthesis. 1st. ed. W.H. Freeman; New York: 2007. [Google Scholar]; b Vicario JL, Badia D, Carrillo L, Reyes E. Organocatalytic Enantioselective Conjugate Addition Reactions. RSC Publishing; Oxford: 2010. RSC Catalysis Series No. 5. [Google Scholar]
- 10.He R, Ding C, Maruoka K. Angew. Chem. 2009;121:4629. doi: 10.1002/anie.200901277.Angew. Chem. Int. Ed. 2009;48:4559. Following our submission of this manuscript, a report from Lu and co-workers on chiral phosphine catalyzed asymmetric Michael additions of N-Boc oxindoles was published online Zhong F, Dou X, Han X, Yao W, Zhu Q, Meng Y, Lu Y. Angew. Chem. Int. Ed. 2012 doi: 10.1002/anie.201208285., 10.1002/anie.201208285). However, this method suffers from the same operational drawbacks mentioned (i.e. high dilution and cryogenic temperatures) and the singular example of an asymmetric addition into acrolein proceeds with more modest (71%) ee.
- 11.For recent progress in the conjugate addition of 3-alkyloxindoles to βsubstituted enals, see: Galzerano P, Bencivenni G, Pesciaioli F, Mazzanti A, Giannichi B, Sambri L, Bartoli G, Melchiorre P. Chem. Eur. J. 2009;15:7846. doi: 10.1002/chem.200802466.Bravo N, Mon I, Companyó X, Alba A-N, Moyano A, Rios R. Tetrahedron Lett. 2009;50:6624.
- 12.For a larger-scale (67.5 mmol) synthesis of allylidene dipivalate, see Lombardo M, Licciulli S, Pasi F, Angelici G, Trombini C. Adv. Synth. Catal. 2005;347:2015.
- 13.When electron-deficient nucleophiles were reacted in THF/tBuOH, a decrease in regioselectivity was observed. As a result, these reactions were performed in PhMe/tBuOH (see Supporting Information for full experimental details and solvent selections for each nucleophile).
- 14.Attempts to extend this methodology to additional substrates including 3-alkyloxindoles are the subject of continued study.
- 15.Overman LE, Shin Y. Org. Lett. 2007;9:339. doi: 10.1021/ol062801y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.PhMe was used as the reaction solvent, and products 5k-5o were obtained in ca. 92-97% purity; see Supporting Information for full details.
- 17.Further experiments and mechanistic investigations to fully elucidate these interesting results are the subject of continued study.
- 18.a Node M, Hao X-J, Fuji K. Chem. Lett. 1991:57. [Google Scholar]; b Wilkerson WW, Kergaye AA, Tam SW. J. Med. Chem. 1993;36:2899. doi: 10.1021/jm00072a009. [DOI] [PubMed] [Google Scholar]; c Node M, Hao X-J, Nishide K, Fuji K. Chem. Pharm. Bull. 1996;44:715. [Google Scholar]; d Pellegrino S, Clerici F, Contini A, Leone S, Pilati T, Gelmi ML. Tetrahedron. 2009;65:1995. [Google Scholar]; e Chen G, Cushing TD, Faulder P, Fisher B, He X, Li K, Li Z, Liu W, Mcgee LR, Pattaropong V, Seganish JL, Shin Y, Wang Z. Alkynyl Alcohols as Kinase Inhibitors. Apr;14:2011. U.S. Patent Application 20110086834. [Google Scholar]
- 19.The antipode of aldehyde 9 has previously been synthesized by Maruoka and co-workers (see Scheme 1, ref. 10, and Supporting Information). The absolute stereochemistry of enol pivalates 2 and 5a-5o is assigned by analogy to 5k.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.